

Abstract
The mechanisms of tolerance in the liver that limit susceptibility to food allergy and that mediate the acceptance of liver transplants even with a complete MHC mismatch remain poorly defined. Here we report that in a model of liver-directed gene transfer cytotoxic T lymphocyte (CTL) responses to non-self antigens are controlled by hepatic regulatory T cells (Tregs) that secrete the immunosuppressive cytokine interleukin (IL)-10 in response to the antigen. In addition, Kupffer cells (KCs), normally thought to initiate immune responses, are rendered tolerogenic in this context. The depletion of KCs results in a complete abrogation of IL-10 production by hepatic Tregs, indicating an interaction between Tregs and KCs in the induction of tolerance.
Conclusion
Our study suggests that hepatic Tregs together with KCs create a local suppressive microenvironment that prevents the establishment of the CTL response. These mechanisms provide pivotal insights and may prove instrumental in the tolerization toward non-self therapeutic proteins delivered to the liver.
Keywords: Liver tolerance, cytotoxic T lymphocyte suppression, hepatic regulatory T cells, Kupffer cells, interleukin-10
The liver serves as a shield between the gut and the systemic circulation. Pathogens, toxins and harmless food-derived antigens can enter the liver directly with arterial blood or from the gut lumen through the portal vein. About 30% of the total blood flow passes through the liver every minute (1). Despite such continuous exposure to microbial and non-self molecules, the liver is known to favor the induction of immune tolerance rather than immunity, best illustrated by examples of tolerization of food antigens in the liver and acceptance of liver allografts across full major histocompatibility complex (MHC) barriers. In addition, delivery of some foreign antigens into the liver has been reported to result in antigen-specific tolerance (2, 3). However, the precise mechanisms as to how the liver participates in the induction of systemic tolerance remain poorly understood. The liver's “tolerogenic” antigen-presenting cells, including hepatic dendritic cells, liver sinusoidal endothelial cells and Kupffer cells (KCs), have all been implicated in facilitating such tolerance induction (4-7).
Considerable evidence has been accumulated for the suppressive effects of CD4+CD25+Foxp3+ regulatory T cells (Tregs) on adaptive immune responses (8, 9). Tregs suppress proliferation and cytokine production by T cells and activation and antibody production by B cells. They are developmentally classified into natural Tregs that originate in the thymus and protect against autoimmunity, and induced/adaptive Tregs that are generated in the periphery and mediate tolerance to foreign antigens. Adaptive Tregs are further divided into type 1 regulatory T (Tr1) cells that do not express Foxp3 and mediate suppression via the production of interleukin (IL)-10, and Foxp3-expressing T helper (Th) 3 cells that suppress via a transforming growth factor (TGF)-β-dependent mechanism. Such Tregs have been described within various peripheral sites, including spleen, lymph nodes and peripheral blood. In contrast, little is known about the function of liver-resident Tregs. Increased CD4+ Tregs have been found in the liver of patients with hepatocellular carcinoma (10). Another study reported intrahepatic CD8+ T cells with regulatory activity in chronic hepatitis C virus (HCV) patients (11).
In the present study, we elucidated the role of hepatic regulatory pathways by which antigens targeting the liver escape from immune recognition and establish a state of tolerance. For this purpose, we employed a replication-defective adeno-associated virus (AAV) vector carrying the model antigen, human α-1 antitrypsin (hAAT). When administered systemically, this vector primarily targets the liver resulting in stable and long-term antigen expression in hepatocytes due to low or no activation of both innate and adaptive immune responses. Our data show that hepatic Tregs together with KCs that are rendered tolerogenic mediate systemic T cell tolerance to antigens expressed in the liver.
Materials and Methods
Vector production
All vectors were produced by PennVector at the University of Pennsylvania.
Mice
Male C57BL/6 mice (6–8 weeks of age) purchased from The Jackson Laboratory (Bar Harbor, ME), with three mice per group were injected via the tail vein with 1011 vector genome (VG) of AAV, or 1010 particles of adenovirus. Blood samples were collected by retro-orbital bleeding into heparinized capillary tubes. In vivo Treg depletion was performed by intraperitoneal (i.p.) injection of 0.75 mg PC61 (BioXCell, West Lebanon, NH) at day -5 prior to vector administration and then every 2 weeks. KCs were depleted by intravenous (i.v.) injection of 200 μl of clodronate or PBS liposomes (kindly provided by Dr. Nico van Rooijen, Vrije Universiteit Medical Center, Amsterdam, The Netherlands) at day -2 and -1 prior to vector administration and once a week thereafter. All animal procedure protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania.
Transgene detection
Plasma hAAT levels were measured by an ELISA as described (12). To examine expression of nuclear β-galactosidase, X-gal staining on snap frozen liver cryosections was performed according to standard protocols (13). In vivo bioluminescent imaging was performed with the Xenogen IVIS imaging system (Xenogen, Alameda, CA). The D-luciferin substrate (Caliper-Xenogen) was administered i.p. at a dose of 10 μl/g of body weight. After 5 minutes mice were anesthetized with ketamine/xylazine and imaged within 10 minutes of anesthesia. Signal intensity was calculated using the Living Image 3.0 software (Caliper-Xenogen).
Liver leukocyte and splenocyte isolation
Liver nonparenchymal cells and splenocytes were isolated as previously described (14, 15).
Interferon (IFN)-γ ELISPOT assay
The IFN-γ ELISPOT assay was performed according to the manufacturer's instructions (BD Biosciences, San Jose, CA). Liver leukocytes or splenocytes from individual mice were added to wells at a density of 105 or 5×105 cells/well along with 2 μg/ml of hAAT CD8 T cell epitope [FALVNYIFF; described in (16)] or β-galactosidase CD8 T cell epitope [ICPMYARV; described in (17)]. Cells were incubated at 37° C, 5% CO2 for 18 hours. For the phorbol myristate acetate/ionomycin positive control, cells were seeded at 2×103/well. Spots were detected with the AEC substrate set (BD Biosciences) and counted using the AID ELISPOT reader system (Cell Technology, Columbia, MD).
Cell culture and cytokine assays
For measurement of cytokines released in the media, liver leukocytes or splenocytes (2×105/well) from individual mice were cultured in 96-well plates pre-coated with anti-CD3 (BD Biosciences) in the presence of 1 μg/ml soluble anti-CD28 (BD Biosciences) and 100 U/ml IL-2 (Fitzgerald Industries International, Concord, MA). Supernatants were collected after 96 hours of culture. Where indicated, 5 μg/ml anti-GITR (BD Biosciences) was also added to the wells. IL-10 and TGF-β1 cytokine levels in the collected supernatants were measured using ELISA kits from BioSource (Camarillo, CA) following manufacturer's instructions. Levels of IL-4, IL-5, IL-13 and IL-10 in culture supernatants were measured using the 22-plex cytokine/chemokine Luminex bead immunoassay kit (Millipore, Bedford, MA) according to the manufacturer's instructions with a Luminex 100 System (Luminex Corporation, Austin, TX).
Flow cytometry
Fresh or cultured in the presence of monensin (eBioscience, San Diego, CA) liver leukocytes or splenocytes (106 cells) from individual mice were stained with antibodies from BD Biosciences, eBioscience or AbD Serotec (Raleigh, NC) and analyzed on a Cytomics FC500 flow cytometer (Beckman-Coulter, Miami, FL). Data were analyzed using FlowJo software (TreeStar, San Carlos, CA).
Statistics
Statistical analysis of the presented data was performed using a two-tailed Student's t test. A value of P < 0.05 was considered as statistically significant.
Results
Establishment of a model of systemic antigen-specific T cell tolerance
Antigen expression in the liver was achieved by using an AAV serotype-8 vector that is known for its high transduction efficiency of this organ. To ascertain that liver is the primary target for this AAV serotype, we injected C57BL/6 mice i.v. with AAV8 or AAV2 vector encoding firefly luciferase. Fig. 1A shows that AAV8 indeed resulted in high levels of luciferase expression that was observed predominantly in the liver. We then showed that AAV8-encoded hAAT, when injected i.v. in C57BL/6 mice, resulted in stable high serum expression levels of hAAT and failed to elicit a cytotoxic T lymphocyte (CTL) response to the mapped dominant epitope of the hAAT protein (Fig. 1C and 1D). To investigate whether such tolerant state was due to an active suppression mechanism, mice expressing hAAT for 2 weeks were challenged with an i.v. injection of a human adenovirus 5 vector expressing hAAT (Ad-hAAT; Fig. 1B). This vector alone results in a potent hAAT-specific CTL response and the subsequent elimination of hAAT expression (Fig. 1C and 1D). ELISPOT assay for IFN-γ on splenocytes and liver leukocytes demonstrated a hAAT-specific CTL response that was completely suppressed at the peak of the Ad immune response (Fig. 1C). hAAT expression was found to be unaffected at 8 weeks post Ad challenge, with levels equivalent to those achieved with AAV-hAAT alone (Fig. 1D). Taken together, these data indicated that AAV-encoded hAAT induces active systemic tolerance that suppresses the subsequent CTL response induced by Ad. Next, using the same experimental setup as shown in Fig. 1B, mice expressing hAAT were challenged with Ad expressing β-galactosidase (LacZ) in order to determine whether the observed suppression was specific to hAAT. Importantly, the Ad-LacZ challenge elicited a strong CTL response against LacZ and was followed by the elimination of β-galactosidase expression (Fig. 1E and 1F). These data indicated that the suppression induced by AAV-hAAT was specific to the hAAT antigen and not global.
Figure 1.

Establishment of a model of systemic antigen-specific T cell tolerance. (A) Mice were injected i.v. with 1010 VG of AAV2 or AAV8 expressing luciferase. At day 14 whole body bioluminescence was visualized. (B) Mice were injected i.v. with 1011 VG of AAV-hAAT, and then 2 weeks later challenged i.v. with 1010 particles of Ad-hAAT. Control mice were injected with PBS. (C) At 9 days after the Ad-hAAT injection, splenocytes or liver leukocytes were stimulated with the hAAT CD8 T cell epitope and subjected to the IFN-γ ELISPOT assay. Background spot-forming unit (SFU) values were subtracted prior to plotting. (D) At 8 weeks after the Ad-hAAT injection, hAAT plasma levels were measured using ELISA. (E) Mice were injected i.v. with 1011 VG of AAV-hAAT or PBS, and then 2 weeks later challenged i.v. with 1010 particles of Ad-LacZ. Livers were processed for LacZ expression 9 and 28 days later. (F) Mice were injected i.v. with 1011 VG of AAV-hAAT or PBS, and then 2 weeks later challenged i.v. with 1010 particles of Ad-LacZ. Nine days after the Ad-LacZ injection, splenocytes were stimulated with the LacZ CD8 T cell epitope and subjected to the IFN-γ ELISPOT assay. Background SFU values were subtracted prior to plotting. Data represent groups of three mice in three independent experiments that gave similar results.
Hepatic Tregs increase in numbers and produce IL-10 in response to hAAT
To explore the possible role hepatic Tregs play in the induction of tolerance to the hAAT antigen, liver leukocytes and splenocytes were isolated from mice administered AAV-hAAT and the frequencies of CD4+CD25+Foxp3+ Tregs were detected by flow cytometry. Interestingly, at 4 weeks post vector administration, hepatic Tregs exhibited a 2-fold relative increase in cell numbers when compared to mice receiving a sham injection (Fig. 2A). However, no such increase was observed in the splenic Treg cells. At 4 months post AAV, the numbers of hepatic Tregs remained elevated but did not increase further (data not shown). To gain further insight into the Treg response, we characterized their phenotypic changes following vector administration. Liver leukocytes and splenocytes from mice that received AAV-hAAT for 2 or 10 weeks were activated ex vivo. These data revealed a subset of CD4+CD25+ Tregs in the liver that persistently produced IL-10 at both early (2 weeks) and late (10 weeks) time points (Fig. 2B). Importantly, this population of hepatic Tregs was induced only in response to the hAAT antigen, since it was not present in mice administered with an AAV vector devoid of an expression cassette (AAV-Null). Again, no IL-10-secreting Tregs were detected in the mouse spleen.
Figure 2.

Hepatic Tregs increase in numbers and produce IL-10 in response to hAAT. (A) Mice were injected i.v. with 1011 VG of AAV-hAAT or PBS. At 4 weeks after injection, liver leukocytes or splenocytes were stained with antibodies to CD4, CD25 and Foxp3. Numbers within density plots indicate percent CD4+CD25+Foxp3+ cells. (B) Mice were injected i.v. with 1011 VG of AAV-hAAT, 1011 VG of AAV-Null or PBS. At 2 or 10 weeks after injection, liver leukocytes or splenocytes were cultured for 96 hours with anti-CD3 plus anti-CD28. Shown are density plots of cultured cells stained with antibodies to CD4, CD25 and IL-10. Percent CD4+CD25+IL-10+ cells is indicated. Data are representative of three mice per group in three independent experiments that gave similar results.
We next investigated whether hepatic Tregs were solely responsible for the observed IL-10 production. At week 2 post AAV-hAAT administration, liver leukocytes were isolated and activated ex vivo in the presence of an agonist anti-GITR (glucocorticoid-induced TNF-related protein) antibody that has previously been shown to abrogate the suppressive activity of Tregs (18) (Fig. 3A). Liver leukocytes from untreated mice displayed a complete abrogation of IL-10 production due to the inhibition of natural Tregs (Fig. 3B). Surprisingly, however, IL-10 production by liver leukocytes of vector-treated mice remained high despite the fact that Tregs were impaired, suggesting the presence of another IL-10-producing cell type in the liver.
Figure 3.

KCs increase in numbers in response to hAAT. (A) Mice (n = 3 per group) were injected i.v. with 1011 VG of AAV-hAAT or PBS. At 14 days after injection, liver leukocytes were cultured with anti-CD3 plus anti-CD28 in the presence of anti-GITR. (B and C) After 96 hours, the supernatants were collected and IL-10 (B) and TGF-β1 (C) levels determined by ELISA. (D) Mice (n = 3) were injected i.v. with 1011 VG of AAV-hAAT, 1011 VG of AAV-Null or PBS. After 2 or 10 weeks, liver leukocytes or splenocytes were stained with an antibody to CD68. Histograms show percent cells containing intracellular CD68. Data represent three independent experiments that gave similar results. A two-tailed Student's t test was used for statistical analysis; *P < 0.05, ***P < 0.001.
Since the production of the key immunosuppressive cytokines IL-10 and TGF-β is the basis for the classification of adaptive Tregs into Tr1 or Th3 Tregs, we examined the production of TGF-β in our experimental system. Liver leukocytes from mice administered AAV-hAAT for 2 weeks were assayed for the production of TGF-β1, which is considered to be the dominant TGF-β isoform produced by Treg cells. The data showed that hepatic Tregs produced low levels of TGF-β1, the production of which was abolished when Treg suppressive activity was inhibited by anti-GITR treatment (Fig. 3C). This provided direct evidence that hepatic Tregs are the sole source of this cytokine.
KCs are critical for hepatic IL-10 production
Although KCs play an important role in the first-line of defense against invading pathogens, they have also been implicated in the induction of tolerance (3, 6). A dramatic relative increase in KC numbers was evident at 2 weeks following AAV-hAAT administration (Fig. 3D). This was in stark contrast to splenic macrophages, which did not exhibit any significant changes in their numbers. Interestingly, the observed increase in KC numbers was transient and the numbers returned to baseline at 10 weeks post antigen administration. Furthermore, at 2 weeks following AAV-hAAT KCs also produced IL-10 (Supplementary Fig. 1). Consistent with Treg IL-10 production, such activation of KCs was found in response to the hAAT antigen and not the AAV capsid.
To investigate the functional role of KCs, mice were depleted of KCs by i.v. administration of clodronate-loaded liposomes on days -2 and -1 prior to AAV-hAAT administration and then once a week until the end of the experiment. This treatment resulted in a transient depletion of both KCs and splenic macrophages (Fig. 4A). Strikingly, we found a complete abrogation of total IL-10 production by liver leukocytes from clodronate-treated mice (Fig. 4B). Here, KC depletion abrogated the production of IL-10 by hepatic Tregs, suggesting a possible cross-talk between the two cell types in the induction of tolerance.
Figure 4.

KCs are critical for hepatic IL-10 production. (A) Mice received two consecutive i.v. injections of 200 μl of clodronate liposomes or PBS liposomes. Representative density plots of F4/80 expression on splenocytes or liver leukocytes on day 2 post treatment are shown. (B) Mice were injected i.v. for two consecutive days with 200 μl of clodronate liposomes, followed one day later by 1011 VG of AAV-hAAT. Fourteen days after vector administration, liver leukocytes were cultured with anti-CD3 plus anti-CD28; 96 hours later supernatants were collected and analyzed for IL-10 production by ELISA. (C) Mice received an i.p. injection of 0.75 mg of PC61 or PBS. Representative density plots show percentage of CD4+CD25+ cells expressing Foxp3 on day 5 post treatment. (D) Mice were injected i.p. with 0.75 mg of PC61, followed 5 days later by 1011 VG of AAV-hAAT. Fourteen days after vector administration, liver leukocytes were cultured with anti-CD3 plus anti-CD28; 96 hours later supernatants were collected and analyzed for IL-10 production by ELISA. Groups consisted of three mice and the data are representative of three independent experiments that gave similar results.
Depletion of Tregs breaks tolerance
To further characterize the role of Tregs in the induction and maintenance of tolerance, we depleted Tregs by a single i.p. administration of PC61 anti-CD25 antibody. Paradoxically, flow cytometric analysis of splenocytes harvested 5 days later revealed that while we failed to detect CD4+CD25+ Tregs after PC61 treatment, the numbers of CD4+Foxp3+ Tregs remained unchanged (Fig. 4C). These results are in agreement with recent reports demonstrating that PC61 antibody does not induce the physical depletion of CD4+CD25+ Tregs but rather downregulates CD25 cell surface expression leading to a functional inactivation of Tregs (19, 20). Thus, our inability to detect CD4+CD25+ Treg may result from lower expression levels of CD25 on the cells rather than from the absence of Tregs. In the liver, the numbers of CD4+CD25+Foxp3+ Tregs were significantly reduced after PC61 treatment. It should be noted that cultured liver leukocytes from AAV-treated mice that were depleted of Tregs still produced IL-10, albeit at significantly lower levels compared to non-depleted mice (Fig. 4D). To gain further information on the functional role Tregs play in suppression of the antigen-specific CTL response, mice were depleted of Tregs 5 days prior to AAV-hAAT administration and every 2 weeks thereafter until the end of the experiment (Fig. 5A). Next, 2 weeks post AAV these mice were challenged with Ad-hAAT. As a result, a detectable hAAT-specific CTL response was found in the spleen (Fig. 5B). In the liver, this CTL response became even more pronounced, reaching up to 600 spot-forming units (SFU) per million cells. Moreover, these mice displayed a dramatic reduction in hAAT expression levels (Fig. 5C), indicating that AAV-induced tolerance was broken by the depletion of Tregs.
Figure 5.
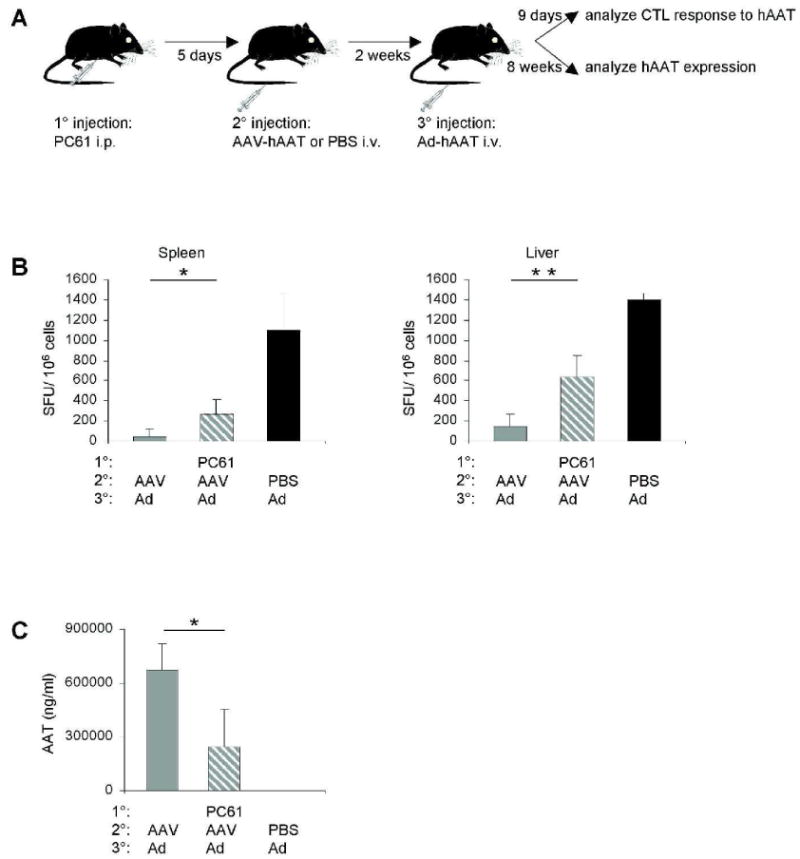
Depletion of Tregs breaks tolerance. (A) Mice (n = 3) were injected i.p. with 0.75 mg of PC61, followed 5 days later by an i.v. injection of 1011 VG of AAV-hAAT or PBS. Two weeks later these mice were challenged i.v. with 1010 particles of Ad-hAAT. (B) At 9 days after the Ad-hAAT injection, splenocytes or liver leukocytes were stimulated ex vivo with the hAAT CD8 T cell epitope and subjected to the IFN-γ ELISPOT assay. Background spot-forming unit (SFU) values were subtracted prior to plotting. (C) At 8 weeks after the Ad-hAAT administration, hAAT plasma levels were measured using ELISA. Data represent three independent experiments that gave similar results. A two-tailed Student's t test was used for statistical analysis; *P < 0.05, **P < 0.01.
IL-4, IL-5 and IL-13 are induced in the liver
We also examined whether cytokines other than IL-10 and TGF-β1 were involved in tolerance induction to the hAAT antigen. Liver leukocytes or splenocytes isolated from mice treated with AAV-hAAT for 2 weeks were cultured for 96 hours and subsequently analyzed for cytokine production by using a multiplex cytokine-chemokine cytometric bead array. As a result, we found that liver leukocytes produced strongly elevated levels of IL-4, IL-5 and IL-13 (Fig. 6A). Production of IL-13 was particularly significant, with levels that were 20-fold higher than in control mice. This was in dramatic contrast to splenocytes, which did not show any significant change in the production of any of the cytokines. To determine the kinetics of cytokine production by liver leukocytes, we measured the production of IL-5, IL-13 and IL-10 after 48, 72 and 96 hours of culture. The results demonstrated that all cytokines had similar kinetics profiles, with production occurring at 72 hours and reaching its peak at 96 hours (Fig. 6B).
Figure 6.

IL-4, IL-5 and IL-13 are induced in the liver. (A and B) Liver leukocytes or splenocytes of mice injected i.v. with 1011 VG of AAV-hAAT or PBS were cultured with anti-CD3 plus anti-CD28 at 14 days after the injection; 96 hours later (A) and 48, 72 or 96 hours later (B) supernatants were collected and analyzed for the production of IL-4, IL-5, IL-13 and IL-10 using a multiplex cytokine-chemokine cytometric bead array. Groups consisted of 3 mice and the data are representative of three independent experiments that gave similar results.
Discussion
Throughout life, the entire blood volume carrying pathogens, dietary antigens and commensal bacteria passes through the liver 360 times per day. Remarkably, while mounting an immune response to harmful antigens, the liver is able to induce tolerance to innocuous ones. Further highlighting liver's tolerogenic ability are reports that the application of non-self antigens via the portal vein leads to immune tolerance (2, 3). It has been suggested that liver-resident antigen-presenting cells can present antigen in a tolerogenic fashion (4), however, the precise mechanisms by which the liver implements such tolerance are as yet poorly understood. Surprisingly, little attention has been paid to the role of hepatic Tregs in liver tolerance. Hepatic CD8+ T cells with suppressive capacity were found in human chronic HCV infection (11). In addition, an increased number of CD4+ Tregs was seen in transplanted murine liver (21).
To address the mechanisms of liver tolerance, we first developed a model of tolerance induction to antigens targeting the liver. As an experimental antigen, we chose a secreted protein, human α-1 antitrypsin (hAAT), which was administered systemically by using a replication-defective AAV serotype-8 vector. Such AAV vector treatment resulted in long-term high-level antigen expression due to the absence of the CTL response. To establish that the active suppression mechanism was responsible for such tolerance, mice were challenged systemically with an Ad vector, a potent activator of CTLs, expressing the same antigen. Strikingly, AAV was able to completely suppress the CTL response induced by Ad, thus leaving the hAAT protein expression unaffected. The suppression of CTL activity was observed in both spleen and liver, indicating systemic tolerance.
One of the peripheral tolerance mechanisms is the induction of Tregs. We found that the percentage of CD4+CD25+Foxp3+ Tregs increased approximately 2-fold in the liver of mice administered with AAV encoding hAAT. Moreover, ex vivo activated hepatic CD4+CD25+ Tregs produced persistently high levels of the immunosuppressive cytokine IL-10, whose production was observed as early as 7 days post vector administration and lasted for at least 2 months. IL-10 is a known inhibitor of Th1 effector functions (22) and has been shown to induce antigen-specific unresponsiveness in both CD4 and CD8 T cells (23, 24) and to inhibit production of most inflammatory cytokines and chemokines by monocytes/macrophages (25, 26) and natural killer cells (27). Importantly, hepatic Tregs were found to produce IL-10 only in response to the hAAT antigen and not the components that make up the AAV viral capsid. It is also interesting to note that hepatic IL-10 production by regulatory CD8+ T cells is observed in patients with chronic HCV (11), a virus that exploits tolerance-promoting properties of the liver to evade the immune system. This points to a generalized mode of suppression against chronically expressed antigens in the liver. Following systemic administration of AAV-hAAT, hepatic Tregs produced another key immunosuppressive cytokine, TGF-β1. Since this CD4+CD25+ Treg subset produced high levels of IL-10 and low levels of TGF-β1, it falls into the category of Tr1 Tregs (28). However, because the frequency of IL-10+Foxp3+ Tregs was too low to be detected by flow cytometric analysis, the exact classification of hepatic Tregs identified in this study will need to be further elucidated. It is probable that these Tregs belong to the as-yet-unknown liver Tr1-like cells or represent a novel Treg subset.
In vivo depletion of Tregs partially broke tolerance to the hAAT antigen, resulting in the generation of CTLs against hAAT, which was particularly pronounced in the liver, and a strong reduction in hAAT expression. This finding implicated Tregs in playing a critical role in controlling the CTL response to systemic antigens. Depletion of Tregs in vivo by the anti-CD25 antibody or blocking their function ex vivo by anti-GITR antibody resulted in only partial inhibition of hepatic IL-10 production. The possibilities that could explain these observations include incomplete depletion of Foxp3+ Tregs, existence of non-GITR-dependent pathways of Treg activation and finally the existence of an additional cell type in the liver that secretes IL-10. Indeed, we found that KCs undergo a transient dramatic increase in their numbers and produce IL-10 following administration of the hAAT antigen. This, combined with the observation that KC depletion completely abrogated hAAT-induced IL-10 production ex vivo, strongly suggests a role for these cells in the induction of liver tolerance. This is in agreement with previous data showing that both splenic Tregs and KCs mediated tolerance to concanavalin A via production of IL-10 (3). Given the evidence that depletion of KCs completely inhibited the production of IL-10 by hepatic Tregs, it is possible that KCs interact with Tregs in the induction of tolerance. The fact that KC activation was only transient while Tregs persistently produced IL-10 raises the possibility that KCs undergo a transient activation in response to AAV-encoded antigens and induce hepatic Tregs via the action of IL-10 (Fig. 7).
Figure 7.

Potential interactions between AAV delivered antigens and immune cells in the liver. Sinusoids are lined with highly fenestrated sinusoidal endothelial cells (LSEC) and bounded circumferentially by hepatocytes. The space of Disse contains Ito cells. Human α-1 antitrypsin protein (hAAT), B cell (B), cytotoxic T lymphocyte (CTL), dendritic cell (DC), Kupffer cell (KC), lymph node (LN), natural killer cell (NK), T cell (T), regulatory T cell (Treg).
Previous studies have shown the induction of splenic Tregs to AAV2-encoded antigens delivered to the liver (29-31). Although we did not detect any changes in the frequency or phenotype of splenic Tregs, it is likely that these Tregs do play a role, thus contributing to the multiple layers of regulation of hepatic tolerance. In such a scenario, hepatic-derived cytokines induced in response to the antigen expressed in the liver may stimulate the generation of Tregs in the spleen through conversion of naive T cells or expansion of pre-existing natural Tregs.
We further found a substantial upregulation of IL-4, IL-5 and IL-13 production in the liver of mice administered AAV-hAAT, with the greatest increase observed in IL-13 levels. It is interesting to note that these cytokines are known to predominantly skew the immune response towards a Th2 pathway and away from a Th1 pathway that mediates the development of a strong CTL response. It has been reported that intravenous high dose antigen administration leads to inhibition of IFN-γ-producing Th1 cells and promotes the development of IL-4-expressing Th2 cells in the liver, thereby causing tolerance (32). However, because of the absence of an antibody response to the hAAT antigen (data not shown), it is also possible that hepatic Tregs themselves may be secreting the Th2-like cytokines by analogy with the recently discovered ability of splenic Tregs to tolerize blood monocytes via the production of IL-10, IL-4 and IL-13 (33). IL-4 and IL-13 have also recently been described to convert naïve CD4+CD25- T cells into CD4+CD25+ Tregs (34). Moreover, KCs themselves could be the source of Th2-like cytokines, since these cells have been shown to produce IL-4 and IL-13 following infection with Schistosoma mansoni, a portal vein-residing helminth (35).
It is important to note that similar levels of IL-10, IL-5 and IL-13 were found in the liver in response to two other antigens (canine coagulation factor IX and mouse ovalbumin) delivered in the context of AAV (Supplementary Fig. 2), suggesting that the established tolerance was not unique to hAAT. Nonetheless, since these antigens were not analyzed for all of the tolerogenic effects described here, some contribution of anti-inflammatory properties of the hAAT protein cannot be completely ruled out.
The establishment of hepatic immune tolerance is of crucial importance for food antigens constantly passing through the liver, transplanted liver grafts and therapeutic proteins used to treat liver diseases. Moreover, the abnormal state of tolerance acquired by HCV, Plasmodium, hepatocellular carcinoma and tumors that metastasize to the liver is undesirable and harmful to the host, and warrants the study of its induction and maintenance. The importance of liver tolerance is further underscored by a recent report demonstrating that systemic delivery of myelin basic protein to the liver in an experimental autoimmune encephalomyelitis mouse model of human multiple sclerosis can transfer tolerance to the brain, which results in protection against experimental autoimmune encephalomyelitis (36). In the current study, we provide the first evidence for a key role of hepatic Tregs and KCs in suppressing the CTL response to antigens targeting the liver (Fig. 7). We show that hAAT encoded by AAV promotes an intrahepatic expansion of Tregs that produce high levels of IL-10. This antigen also ‘deactivates’ KCs, which, instead of initiating innate immune responses, become tolerogenic, undergo a transient increase in numbers and regulate IL-10 production. Importantly, we show for the first time the previously unknown interplay between Tregs and KCs in the liver by demonstrating that IL-10 production by hepatic Tregs is critically dependent on KCs. Finally, the cytokine milieu in the liver following interaction of Tregs and KCs would skew the immune response to a Th2 pathway or suppress proinflammatory cytokines, in turn adversely affecting a CTL response. We conclude that these mechanisms of tolerance induction together with the unique anatomical characteristics of the liver are ideally suited to induce systemic tolerance to non-self antigens.
Supplementary Material
Acknowledgments
We thank Deirdre McMenamin and Regina Munden for their assistance with animal studies, PennVector Core at the University of Pennsylvania for providing the vectors and Divya Nadkarni for her assistance with assays.
This work was funded by the following grants to JMW: NIDDK P30 DK47757, NHLBI P01 HL59407, NICHD P01 HD57247, and GlaxoSmithKline, Inc.
Abbreviations
- MHC
major histocompatibility complex
- KC
Kupffer cell
- Treg
regulatory T cell
- Tr1
type 1 regulatory
- IL
interleukin
- Th
T helper
- TGF
transforming growth factor
- HCV
hepatitis C virus
- AAV
adeno-associated vector
- hAAT
human α-1 antitrypsin
- VG
vector genome
- IFN
interferon
- CTL
cytotoxic T lymphocyte
- Ad
adenovirus vector
- GITR
glucocorticoid-induced TNF-related protein
- SFU
spot-forming unit
Contributor Information
Ekaterina Breous, Email: breous@mail.med.upenn.edu.
Suryanarayan Somanathan, Email: somanath@mail.med.upenn.edu.
Luk H. Vandenberghe, Email: lucv@mail.med.upenn.edu.
References
- 1.Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 2.Cantor HM, Dumont AE. Hepatic suppression of sensitization to antigen absorbed into the portal system. Nature. 1967;215:744–745. doi: 10.1038/215744a0. [DOI] [PubMed] [Google Scholar]
- 3.Erhardt A, Biburger M, Papadopoulos T, Tiegs G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology. 2007;45:475–485. doi: 10.1002/hep.21498. [DOI] [PubMed] [Google Scholar]
- 4.Crispe IN. Hepatic T cells and liver tolerance. Nat Rev Immunol. 2003;3:51–62. doi: 10.1038/nri981. [DOI] [PubMed] [Google Scholar]
- 5.Schildberg FA, Hegenbarth SI, Schumak B, Scholz K, Limmer A, Knolle PA. Liver sinusoidal endothelial cells veto CD8 T cell activation by antigen-presenting dendritic cells. Eur J Immunol. 2008;38:957–967. doi: 10.1002/eji.200738060. [DOI] [PubMed] [Google Scholar]
- 6.You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology. 2008;48:978–990. doi: 10.1002/hep.22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology. 2008;47:296–305. doi: 10.1002/hep.21965. [DOI] [PubMed] [Google Scholar]
- 8.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 9.Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 10.Unitt E, Rushbrook SM, Marshall A, Davies S, Gibbs P, Morris LS, et al. Compromised lymphocytes infiltrate hepatocellular carcinoma: the role of T-regulatory cells. Hepatology. 2005;41:722–730. doi: 10.1002/hep.20644. [DOI] [PubMed] [Google Scholar]
- 11.Abel M, Sene D, Pol S, Bourliere M, Poynard T, Charlotte F, et al. Intrahepatic virus-specific IL-10-producing CD8 T cells prevent liver damage during chronic hepatitis C virus infection. Hepatology. 2006;44:1607–1616. doi: 10.1002/hep.21438. [DOI] [PubMed] [Google Scholar]
- 12.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 13.Bell P, Limberis M, Gao G, Wu D, Bove MS, Sanmiguel JC, et al. An optimized protocol for detection of E. coli beta-galactosidase in lung tissue following gene transfer. Histochem Cell Biol. 2005;124:77–85. doi: 10.1007/s00418-005-0793-2. [DOI] [PubMed] [Google Scholar]
- 14.Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110:4077–4085. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Limberis MP, Figueredo J, Calcedo R, Wilson JM. Activation of CFTR-specific T Cells in cystic fibrosis mice following gene transfer. Mol Ther. 2007;15:1694–1700. doi: 10.1038/sj.mt.6300210. [DOI] [PubMed] [Google Scholar]
- 16.Breous E, Somanathan S, Wilson JM. Identification of the immunodominant cytotoxic T-cell epitope of human alpha-1 antitrypsin. Gene Ther. 2009 doi: 10.1038/gt.2009.100. Letter to the Editor. in press. [DOI] [PubMed] [Google Scholar]
- 17.Hoerr I, Obst R, Rammensee HG, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol. 2000;30:1–7. doi: 10.1002/1521-4141(200001)30:1<1::AID-IMMU1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3:135–142. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 19.Kohm AP, McMahon JS, Podojil JR, Begolka WS, DeGutes M, Kasprowicz DJ, et al. Cutting Edge: Anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J Immunol. 2006;176:3301–3305. doi: 10.4049/jimmunol.176.6.3301. [DOI] [PubMed] [Google Scholar]
- 20.Couper KN, Blount DG, de Souza JB, Suffia I, Belkaid Y, Riley EM. Incomplete depletion and rapid regeneration of Foxp3+ regulatory T cells following anti-CD25 treatment in malaria-infected mice. J Immunol. 2007;178:4136–4146. doi: 10.4049/jimmunol.178.7.4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W, Kuhr CS, Zheng XX, Carper K, Thomson AW, Reyes JD, et al. New insights into mechanisms of spontaneous liver transplant tolerance: the role of Foxp3-expressing CD25+CD4+ regulatory T cells. Am J Transplant. 2008;8:1639–1651. doi: 10.1111/j.1600-6143.2008.02300.x. [DOI] [PubMed] [Google Scholar]
- 22.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–2095. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groux H, Bigler M, de Vries JE, Roncarolo MG. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. J Exp Med. 1996;184:19–29. doi: 10.1084/jem.184.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groux H, Bigler M, de Vries JE, Roncarolo MG. Inhibitory and stimulatory effects of IL-10 on human CD8+ T cells. J Immunol. 1998;160:3188–3193. [PubMed] [Google Scholar]
- 25.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 27.D'Andrea A, Aste-Amezaga M, Valiante NM, Ma X, Kubin M, Trinchieri G. Interleukin 10 (IL-10) inhibits human lymphocyte interferon gamma-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J Exp Med. 1993;178:1041–1048. doi: 10.1084/jem.178.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 29.Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, et al. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110:1132–1140. doi: 10.1182/blood-2007-02-073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobrzynski E, Fitzgerald JC, Cao O, Mingozzi F, Wang L, Herzog RW. Prevention of cytotoxic T lymphocyte responses to factor IX-expressing hepatocytes by gene transfer-induced regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:4592–4597. doi: 10.1073/pnas.0508685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klugewitz K, Blumenthal-Barby F, Schrage A, Knolle PA, Hamann A, Crispe IN. Immunomodulatory effects of the liver: deletion of activated CD4+ effector cells and suppression of IFN-gamma-producing cells after intravenous protein immunization. J Immunol. 2002;169:2407–2413. doi: 10.4049/jimmunol.169.5.2407. [DOI] [PubMed] [Google Scholar]
- 33.Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skapenko A, Kalden JR, Lipsky PE, Schulze-Koops H. The IL-4 receptor alpha-chain-binding cytokines, IL-4 and IL-13, induce forkhead box P3-expressing CD25+CD4+ regulatory T cells from CD25-CD4+ precursors. J Immunol. 2005;175:6107–6116. doi: 10.4049/jimmunol.175.9.6107. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi N, Matsui K, Tsutsui H, Osada Y, Mohamed RT, Nakano H, et al. Kupffer cells from Schistosoma mansoni-infected mice participate in the prompt type 2 differentiation of hepatic T cells in response to worm antigens. J Immunol. 1999;163:6702–6711. [PubMed] [Google Scholar]
- 36.Luth S, Huber S, Schramm C, Buch T, Zander S, Stadelmann C, et al. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J Clin Invest. 2008 doi: 10.1172/JCI32132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.