

Abstract
Bacterial persisters are cells with an impressive, yet transient, tolerance toward extraordinary concentrations of antibiotics. Persisters are believed to impose a significant burden on the healthcare system, due to their role in the proclivity of infections to relapse. During antibiotic challenge, these rare, phenotypic variants enter a dormant state where antibiotic primary targets are rendered inactive, allowing them to survive. Once the antibiotic is removed, persisters reawaken and resume growth, leading to repopulation of the environment. Metabolism plays a pivotal role in coordinating the entry, maintenance, and exit from the persister state. However, the low abundance, transient nature, and similarity of persisters to other cell-types have prevented their isolation, which is needed for direct metabolic measurements. In this unit, we describe a technique known as the aminoglycoside (AG) potentiation assay that can be used to rapidly and specifically measure the breadth of persister metabolism in heterogeneous populations.
Keywords: Persisters, Persister metabolism, Aminoglycoside
INTRODUCTION
Persisters comprise a small fraction of a bacterial population that is highly tolerant to antibiotics (Balaban et al., 2004; Bigger, 1944; Keren et al., 2004a). Persisters are major culprits of antibiotic failure in the case of recalcitrant infections (Lewis, 2010). They can be detected with persister assays, where biphasic killing kinetics indicates the existence of two or more subpopulations (Figure 1). In these biphasic kill curves, the death of normal, antibiotic-susceptible kin produces the initial rapid killing regime, whereas the existence of persisters is reflected in the second, slower killing regime (Balaban et al., 2004). In contrast to antibiotic-resistant mutants, which grow in the presence of elevated concentrations of antibiotics, persisters are phenotypic variants in an isogenic cell population that only subsist in the presence of antibiotics. Once the antibiotic is removed and persisters resume growth in a fresh medium, their tolerances are lost and they exhibit sensitivities similar to the original culture. Their low abundance, transient nature, and similarity to viable but non-culturable cells (VBNCs) have impeded their isolation for direct transcriptome, proteome, and metabolome measurements (Orman and Brynildsen, 2013b; Roostalu et al., 2008). In the absence of robust isolation techniques, methods that enable measurements of persister physiology are needed to improve understanding of this troublesome phenotype. Persister metabolism is of particular importance because it coordinates all stages of the phenotype (Amato et al., 2014; Amato et al., 2013; Orman and Brynildsen, 2013a). To survive antibiotics, persisters need to shutdown essential cell functions, which antibiotics target, maintain culturability during stasis, and resume growth once the treatment has concluded. Metabolism participates in each of these steps, and understanding the metabolic processes during each stage would enable us to develop therapeutic strategies to sabotage this multidrug tolerant state. This is exemplified by the number of persister elimination strategies developed to date that rely on persister metabolism to promote killing (Allison et al., 2011; Kim et al., 2011).
Figure 1. Biphasic kill curve during the antibiotic treatment.
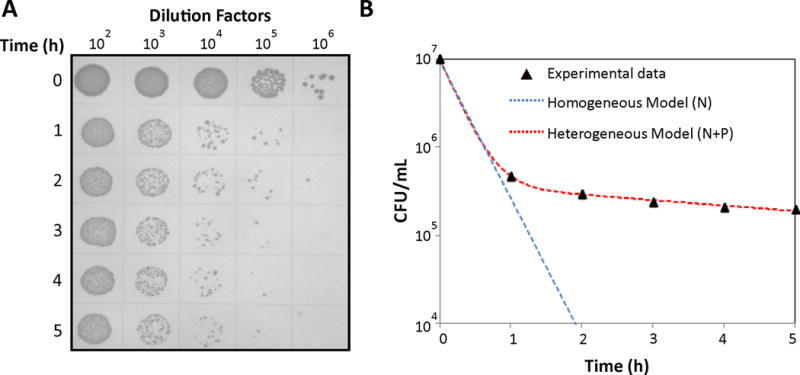
A. Overnight culture of E. coli MG1655 was diluted (100-fold) in fresh 50 mL LB and cultured for 2 h at 37 °C and 250 rpm to reach an exponential growth phase. Then, the culture was treated with OFL for 5 h. Culture samples were collected every hour during the course of antibiotic treatment, washed, serially-diluted, and plated on LB agar. After incubating the agar plate at 37°C for 16 h, CFUs were counted by taking the dilution factors into consideration. B. Biphasic kill curve was obtained when CFUs/mL were plotted on a logarithmic scale as a function of treatment time. If the culture were comprised of a single homogeneous population , a single killing rate would be observed (blue line). The biphasic nature of the kill curve demonstrates that the population was heterogeneous with at least two culturable cell-types (red line). N and P represent the abundance of normal cells and persisters, whereas kn and kp represent the net killing rates of normal and persister cells per unit time in the presence of antibiotics.
In this unit, we will describe a method to measure persister metabolic activities that leverages the phenomenon of metabolite-enabled aminoglycoside (AG) potentiation in persisters (Allison et al., 2011; Orman and Brynildsen, 2013b). This technique, which our laboratory co-developed, can be used to study persister metabolism in heterogeneous populations, thereby circumventing the current technical challenges associated with isolating high purity persister samples (Allison et al., 2011; Orman and Brynildsen, 2013b). Performing this assay allows measurement of nutrient catabolism to drive respiratory activity in persisters, which generates knowledge of the metabolic pathways that can be targeted to devise novel anti-persister strategies.
BASIC PROTOCOL 1: PERSISTER ASSAY
The AG potentiation assay infers persister metabolic activity from losses in culturability, as measured by colony forming units (CFUs). Therefore, samples need to be preprocessed such that persisters comprise the only remaining cell-type capable of generating a colony. This is accomplished by treating cultures with antibiotics for a sufficient amount of time, such that biphasic killing is observed (Figure 1). These experiments have been termed “persister assays”, because they are used to enumerate persisters within bacterial cultures (Balaban et al., 2004; Keren et al., 2004a). Any bactericidal antibiotic can be used for this assay, but we note that the resulting persisters may be physiologically different (Amato et al., 2014). In addition, antibiotic concentrations and treatment time will vary based on strain, antibiotic, and culturing conditions (Balaban et al., 2004; Conlon et al., 2013; Keren et al., 2004a; Luidalepp et al., 2011). Once assay conditions have been identified to yield persisters as the only remaining culturable cells, the resulting samples will be ready for use in the AG potentiation assay.
Materials
Desired strain (E. coli MG1655 will be described here for demonstrative purposes)
Desired media (Luria-Bertani (LB) medium prepared from components: tryptone, yeast extract, NaCl is used in this study)
Antibiotic (here we use ofloxacin (OFL))
Phosphate buffered saline (PBS)
Agar
Test tubes (glass and/or 17×100 mm polypropylene tubes)
500 mL baffled flask
Micropipettor (single and multi-channel)
Sterile pipet tips
Syringes
0.22 μm filter units
Microcentrifuge tubes (1.5 mL)
96-well round-bottom plates
Disposable petri dishes (square petri dishes with 13×13mm grids can be used)
Bench top centrifuge
Shaker
Incubator
-
Prepare the overnight cultures by inoculating cells from a frozen stock stored in 25% glycerol at −80 °C into 2 mL LB medium in a test tube and incubate the sample at 37 °C with shaking (250 rpm) for 24 h.
Cell-stock storage and the overnight culture conditions can be adjusted.
-
Dilute the overnight cultures to a desired optical density (OD600) in 50 mL of fresh LB medium in a 500 mL baffled flask and incubate until a desired growth phase is achieved.
Note that one may use different media, volume, or flask type.
Under these conditions, 500 μl of overnight culture is sufficient to dilute in 50 ml of fresh LB to obtain an OD600 of ~0.04 to 0.05.
The volume can be adjusted according to the culture volume and desired initial OD600. If the volume that is to be added exceeds 2 ml, multiple cultures can be inoculated and pooled following overnight growth, to maintain consistency.
The researcher can choose whether to examine metabolism of cells isolated from exponential or stationary phase.
At the desired growth phase, add 50 μL of the OFL stock (5 mg/mL) into cell cultures, such that the final concentration is 5 μg/mL.
-
At desired time points during the course of treatment, transfer 1 mL of the cell cultures to a microcentrifuge tube and pellet the cells by centrifugation at 15,000 rpm for 3 minutes.
We usually collect the samples every hour during the antibiotic treatment, but it can be collected at different time intervals.
To wash the cells and dilute the antibiotics, remove 900 μL of supernatant and resuspend the pellets with 900 μL of PBS. Pellet the cells again by centrifugation.
-
Repeat step 5 until the antibiotic concentration is below the minimal inhibitory concentration (MIC) (Andrews, 2001).
Under these conditions, it is sufficient to wash samples twice.
After washing the cells, resuspend the pellet in the remaining 100 μL of supernatant, resulting in a 10x-concentrated sample.
-
Transfer 10 μL of the sample into 90 μL PBS in a 96-well round bottom plate. Serially dilute each sample, then plate 10 μL of each sample on LB agar.
We recommend using a 96-well round-bottom plate for easier mixing.
Incubate the plates at 37 °C for 16 h and count the CFUs. For each data point, 10 to 100 colonies should be counted (Figure 1).
BASIC PROTOCOL 2: AMINOGLYCOSIDE (AG) POTENTIATION ASSAY
We have demonstrated that persisters can metabolize specific carbon sources to generate proton motive force (pmf), which promotes AG uptake and killing (Allison et al., 2011; Orman and Brynildsen, 2013b). This potentiation can be eliminated with KCN, which blocks cytochrome oxidoreductase activity and pmf generation (Allison et al., 2011; Orman and Brynildsen, 2013b). These properties form the basis of the AG assay. Since direct measurement of persister metabolism is not currently possible due to isolation difficulties, AG potentiation has become the standard method to measure persister catabolism (Allison et al., 2011; Amato et al., 2014; Orman and Brynildsen, 2013b). In this method, samples where persisters comprise the only culturable cells are incubated in defined media with AG and a metabolite. Potentiation of AGs is measured by CFU, and a reduction in CFUs in excess of the no metabolite control indicates persister catabolism (Figure 2). Elimination of the effect with KCN demonstrates that the mechanism of AG potentiation is consistent with previous studies (Allison et al., 2011; Orman and Brynildsen, 2013b) and that persisters catabolize the substrate to drive respiratory activity.
Figure 2. AG Potentiation of E. coli persisters.

A. Incubation of E. coli persisters from an exponential phase culture (as described in Figure 1A) with KAN and carbon sources for 1 h. Glycerol and glucose demonstrated AG potentiation as indicated by a loss in CFUs. KCN was used as a control to demonstrate that substrate catabolism generated NADH and respiratory activity to potentiate AGs. B. Survival fractions calculated from CFUs enumerated prior to and after the AG potentiation assay.
Materials
Desired strain (E. coli MG1655 will be described here for demonstrative purposes)
5x M9 minimal salts (33.9g/L dibasic sodium phosphate, 15 g/L monobasic potassium phosphate, 5 g/L ammonium chloride, and 2.5 g/L sodium chloride)
1 M calcium chloride
1 M magnesium sulfate
Aminoglycoside (we typically use kanamycin (KAN) or gentamicin (GENT) with E. coli)
β-Lactam antibiotic (Ampicillin (AMP) is used).
1 M potassium cyanide
Filter sterilized carbon source stock solutions (600 mM carbon)
PBS
LB medium for agar plates
Agar
Test tubes
Micropipettor (single and multi-channel)
Sterile pipet tips
Syringes
0.22 μm filter units
Microcentrifuge tubes (1.5 mL)
Sterile, gas-permeable sealing membranes (we typically use Breathe-Easy sealing membranes from Sigma-Aldrich)
96-well round-bottom plates
Disposable petri dishes (square petri dishes with 13×13mm grids can be used)
Benchtop centrifuge
Shaker
Incubator
Collect 1 mL of sample where persisters comprise the only remaining culturable cells (e.g., cultures treated with OFL for 5 h) and pellet the cells by centrifugation at 15,000 rpm for 3 min. Remove the supernatant and resuspend the pellet in 1 mL of sterile 1.25x M9 salt solution.
-
Wash the cells again in 1 mL of 1.25x M9. Adjust cell concentrations such that the final concentration is ~105 persisters/mL.
Depending on the persister levels in the culture under investigation, one might need to concentrate or dilute samples. Persister levels are enumerated from persister assays (Basic Protocol 1). We typically collect samples after 5 h of antibiotic treatment, because we have observed that for most E. coli strains that we have tested, 5 h is more than sufficient to reach the second phase of killing (Figure 1). If it is observed that 5 h is insufficient, antibiotic treatment time should be extended.
-
Serially-dilute 10 μL of the cell suspension and plate 10 μL diluted samples on LB agar to enumerate CFUs at t=0 of the assay.
Even though persister levels are enumerated from persister assays (Basic Protocol 1), we prefer to plate the cell suspension after performing the steps 1–2. This is necessary to count the exact levels of CFUs at t=0h which is used to calculate the survival fractions after the AG treatment.
-
Mix 80 μL of cell suspension, 10 μL of KAN solution (250 μg/mL) and 10 μL of carbon source solution (600 mM carbon) in each assay well of a 96-well plate. For the no carbon source control, add 10 μL of sterile water instead of carbon source.
This procedure result in ~104 persisters, 25 μg/mL KAN, and 60 mM carbon per well. We use minimal media and typically test carbon sources for AG potentiation. One might also perform the assay with nitrogen and sulfur sources or different media. It was previously demonstrated that AG potentiation is dependent on carbon concentration (Allison et al., 2011), therefore we typically normalize the carbon sources by their carbon content.
-
As controls, to determine if carbon sources stimulate persisters to awaken and resume cell division, mix 80 μL of cell suspension, 10 μL of AMP solution (1 mg/mL) and 10 μL of carbon source solution (600 mM carbon) in the AMP-control wells of a 96-well plate. For the no carbon source control, add 10 μL of sterile water instead of carbon source.
To confirm that persisters remain non-replicative and retain their antibiotic tolerance, a β-lactam antibiotic (such as AMP) is used, because they do not kill non-growing cells.
-
As additional controls, add 1 M KCN to the 1.25x M9 salt solution to obtain a final concentration of 1.25 mM KCN, and perform the steps 1–3. Then mix 80 μL of cell suspension, 10 μL of KAN solution (250 μg/mL) and 10 μL of carbon source solution (600 mM carbon) in the KCN-control wells of a 96-well plate. For the no carbon source control, add 10 μL of sterile water instead of carbon source.
This introduces 1 mM KCN to each sample, which can block respiration and proton motive force generation, thereby eliminating AG potentiation.
-
Cover the plates with sterile, gas-permeable sealing membranes and incubate at 37 °C with shaking at 250 rpm for 2 h.
Duration of AG potentiation assay can be adjusted.
After incubation, transfer 100 μL cell cultures from each well to a microcentrifuge tube and add 900 μL of PBS.
Centrifuge the samples at 15,000 rpm for 3 min. Remove 900 μL of supernatant and wash the pellets with 900 μL of PBS.
Repeat step 5 until the antibiotic concentration is below the MIC. After washing the cells, resuspend the pellet in 100 μL of the remaining supernatant.
Transfer 10 μL of the sample into 90 μL PBS and serially dilute the samples in a 96-well round-bottom plate.
Plate 10 μL of the serially-diluted samples on LB agar. The remaining 90 μL sample should be plated onto LB agar as well in order to improve the limit of detection.
-
Incubate the plates at 37°C for 16 h before counting CFUs. 10–100 CFUs should be counted for each data point. Calculate the survival fractions for cells treated with each metabolite by taking the ratio of persisters counted at t=2 h after AG treatment to the number of persisters counted at t=0 h (before the AG treatment).
Using a statistical test, such as a two-tailed t-test, the carbon sources that significantly reduce survival fractions compared to control groups (no carbon source, KCN and AMP controls) can be identified.
With this technique we typically test approximately 12 carbon sources at a time. We note that this method can be modified for high-throughput (HT) screening using phenotype microarrays (Orman and Brynildsen, 2013b). Modifications for the HT version include higher cell density, one-step dilution followed by plating, and the absence of wash steps as described previously (Orman and Brynildsen, 2013b). We suggest that phenotype microarrays can be used as a pre-screen to identify carbon sources with the potential to be catabolized by persisters, and the AG potentiation assay as described above should be performed for confirmation.
BASIC PROTOCOL 3: COMPETITION ASSAY
Dead cells and VBNCs are highly abundant in antibiotic-treated cultures, and we have shown that VBNCs are responsible for metabolic activity measured from these cultures using standard techniques (Orman and Brynildsen, 2013b). Consumption of metabolites and generation of by-products by VBNCs may influence the ability of persisters to generate pmf and take up AGs (Figure 3). For instance, AG potentiation assay with WT cells demonstrated that persisters could consume glycerol to drive aerobic respiration, and this potentiation was lost when the ΔgldAΔglpK strain, which cannot metabolize glycerol, was assayed (Orman and Brynildsen, 2013b). However, in the WT assays VBNCs could have consumed glycerol and released a by-product that persisters catabolized to potentiate AGs, and monoculture assays with ΔgldAΔglpK would not discount this possibility, since all bacteria would be unable to consume glycerol. Therefore, to definitively attribute AG potentiation to persister catabolism, free from VBNC interference, we perform competition assays. In these assays, antibiotic-treated WT and mutant cells are mixed to achieve an approximate 50/50 proportion of persisters for each strain. The mutant strain should contain genetic perturbations that prevent catabolism of the metabolite under consideration (e.g., ΔgldAΔglpK for glycerol in E. coli). An AG potentiation assay with the metabolite of interest is performed on the mixed culture, and if persisters are not influenced by surrounding cells WT persister levels should be drastically reduced, whereas mutant persisters will remain unchanged (Figure 3). If interference is occurring as evidenced by reduced persister levels of the mutant (Figure 3), we drop the density of the assay conditions and perform the competition assay again. By reducing the cell density in assays, we decrease the impact of surrounding cell-types and obtain a more accurate measurement of persister catabolism.
Figure 3. Overview of persister competition assay.

A. Antibiotic-treated cultures contain persisters, dead cells, and VBNCs. To ensure that VBNCs do not interfere with measurements of persister metabolism, WT E. coli is mixed with a mutant strain that cannot catabolize a specific metabolite and the mixture is subjected to an AG potentiation assay. B. Before the addition of AG and a metabolite that can only be catabolized by WT, WT and mutant persisters are mixed in 50/50 proportions and approximately 104 persisters are present in the well. C. If VBNCs do not affect measurements, AG potentiation by the metabolite used in the assay would only occur in WT persisters and the mutant persisters would not lose abundance. D. Without interference, all remaining persisters would be of the mutant and their abundance would not change. E. If VBNCs interfere with the assay (e.g., VBNCs catabolize the metabolite and produce a by-product that potentiates AG killing in persisters) loss of both WT and mutant persisters would be observed. F. This interference would present as a loss of abundance of mutant persisters.
Materials
WT strain (we use E. coli MG1655::CMR which has a chromosomal copy of a chloramphenicol resistance marker between lacA and cynX genes to allow for quantification on chloramphenicol-selective media)
Mutant strain (mutations depend on metabolic pathways, but we will use E. coli MG1655ΔgldAΔglpK::GENTR for demonstrative purposes because it cannot consume glycerol and can be quantified by plating on gentamicin-selective media. Note that although both GENT and KAN are AGs, cross resistance is not conferred by the resistance genes we use.)
KAN, Chloramphenicol (CM) and Gentamicin (GENT)
1 M potassium cyanide
5x M9 minimal salts (33.9g/L dibasic sodium phosphate, 15 g/L monobasic potassium phosphate, 5 g/L ammonium chloride, and 2.5 g/L sodium chloride)
1 M calcium chloride
1 M magnesium sulfate
Filter sterilized carbon source stock solutions (600 mM carbon)
PBS
LB medium for agar plates
Agar
Test tubes
Micropipettors (single and multi-channel)
Sterile pipet tips
Syringes
0.22μm filter units
96-well flat-bottom plates
Sterile, gas-permeable sealing membranes
Microcentrifuge tubes (1.5mL)
96-well round-bottom plates
Disposable square petri dishes with 13×13mm grids
Bench top centrifuge
Shaker
Incubator
Determine the persister levels of WT and mutant strains as described in Basic Protocol 1.
Pellet 1 mL of cell cultures treated with antibiotics by centrifugation at 15,000 rpm for 3 min. Remove the supernatant and resuspend the pellet in sterile 1.25x M9 salt solution.
Mix WT and mutant to obtain an approximate 50/50 proportion of persisters. Dilute the cell mixture with sterile 1.25x M9 salt solution so as to obtain ~105 persisters/well.
Serially dilute and plate 10 μL of the samples to count CFUs at t=0 of the assay.
Perform AG potentiation assay for the cell mixtures as described in steps 4–11 in Basic Protocol 2.
-
Plate 10 μL of serially diluted samples on antibiotic-selective media to enumerate WT and mutant persisters, and on non-selective media to check for consistency. Plate the remaining undiluted 90 μL sample on antibiotic-selective media at equal volumes to improve the limit of detection.
LB agar with 25 μg/mL CM to select for WT (E. coli MG1655::CMR) persisters, LB agar with 10 μg/mL GENT to select for mutant MG1655ΔgldAΔglpK::GENTR persisters, and LB agar to enumerate the total number persisters (both wild type and mutant) for internal consistency are used.
If significant reduction in the persister levels of ΔgldAΔglpK is observed (Figure 3EF), dilute the cell mixture in step 3 (10-fold) and repeat steps 4–5 until no reduction in ΔgldAΔglpK persister levels following the AG assay is observed.
REAGENTS AND SOLUTIONS
LB Medium
Dissolve 10 g of tryptone, 5 g of yeast extract, and 10 g of NaCl in 1L deionized (DI) water.
Autoclave for 30 min at 121°C.
Store the autoclaved LB in the dark and at room temperature.
LB Agar
Add 15 g pure agar powder to 1 L LB medium as described above.
Autoclave for 30 min at 121 °C.
Allow the media to cool to 50–60 °C, and add antibiotics if necessary.
Pour approximately 30 mL of LB agar into each square petri dish with grids.
Leave the plates on the bench at room temperature about one day to dry and store at 4°C.
5X M9 Salt Solution
-
Dissolve 64 g of dibasic sodium phosphate heptahydrate (Na2HPO4.7H2O), 15 g of monobasic potassium phosphate (KH2PO4), 5 g of ammonium chloride (NH4Cl), and 2.5 g of sodium chloride (NaCl) in DI water. Adjust the volume to 1 L.
Alternatively, a 5X M9 salt mixture can be used.
Autoclave for 30 min at 121°C.
Store in the dark and at room temperature.
1.25X M9 Salt Solution
Mix 2.5mL 1M MgSO4 and 125μL 1M CaCl2 with 747.5mL DI water.
Add 250 mL 5X M9 salt solution.
Filter-sterilize with 0.22μm filter unit and store in the dark at room temperature.
Antibiotic Solutions
AMP, KAN, and GENT: To prepare 1000x concentrated solutions of each antibiotic, dissolve 100 mg of AMP, 25 mg of KAN, and 10 mg of GENT in 1 mL DI water. Filter-sterilize the solutions with 0.22 μm filter units and store at 4°C.
CM: Dissolve 25 mg of CM in 1 mL of ethanol to prepare a 1000x solution.
OFL: Dissolve 5 mg of OFL in 1 mL of DI water and titrate with sodium hydroxide (1M, dissolved in sterile DI H2O) until the OFL is fully in solution. Filter-sterilize and store at 4 °C.
KCN Solution
-
Dissolve 65.12 mg KCN in 1 mL DI water, and filter sterilize with 0.22 μm filter unit and store at 4°C.
CAUTION: A mask and other appropriate PPE should be worn for this step. Dispose of KCN contaminated waste following proper hazardous waste disposal protocols.
Carbon Source Solutions
Dissolve required amounts of carbon sources in DI water yielding 600 mM carbon in each stock solution. Filter sterilize with 0.22 μm filter unit and store at 4 °C.
COMMENTARY
Background
Bacterial persisters have long been recognized as a clinically significant phenotype (Fauvart et al., 2011; Lewis, 2007), but many aspects of persister physiology remain elusive (Balaban et al., 2013). The major roadblock in studying persister physiology is the inability to isolate highly pure persister samples (Orman and Brynildsen, 2013b; Roostalu et al., 2008). This technical challenge arises from their low abundance (e.g., 0.001%–1% of bacterial populations), transient nature (persister cannot be gathered and propagated for further analysis), and similarities to the more highly-abundant VBNC phenotype. Similar to persisters, VBNCs are non-growing under antibiotic stress, stain as live cells, and retain metabolic activity, yet they do not readily resume growth on standard culture media following antibiotic treatment (Orman and Brynildsen, 2013b). Unfortunately, this distinguishing feature, resumption of growth following antibiotic treatment, cannot be used to segregate persisters from VBNCs in real time, because upon reawakening persisters revert to normal physiology. Approaches for single cell analysis offer the possibility of interrogating rare cells in a heterogeneous population (Iino et al., 2012; Iino et al., 2013), but the ability to differentiate persisters from VBNCs before they overcome antibiotic treatment and resume cell division has not yet been achieved.
Several methods have tried to isolate persisters from other cell-types based on different physiological characteristics. One approach relies on the lysis of normal antibiotic susceptible cells with AMP (Keren et al., 2004b) and collecting the survivors by centrifugation. It was hypothesized that the survivors are comprised largely of persisters as they cannot be lysed by AMP (Keren et al., 2004b); however, subsequent studies have shown that VBNCs also remain intact following AMP treatment. Therefore, this method produces samples with 100-fold more VBNCs than persisters (Orman and Brynildsen, 2013b). A different method used a growth-rate dependent reporter and fluorescence-activated cell sorting (FACS) to segregate fast growing cells from their dormant kin (Shah et al., 2006). Although persisters were enriched in the non-growing subpopulation, the majority of non-growing cells were of other cell types, thus only a fraction of the FACS-generated samples were persisters (Orman and Brynildsen, 2013b). In the absence of an isolation technique, the distinguishing feature between persister and VBNC- culturability on standard media- must be used to differentiate between persister and VBNC physiologies. Since AG potentiation infers metabolic activity from culturability changes, it is an adept method to measure catabolic abilities of persisters.
Metabolic stimulation has been shown to generate pmf in persisters, enabling transport of AGs and subsequent killing (Allison et al., 2011; Davis, 1987). Since isolation of persisters from other cell types is not possible, the AG potentiation assay provides a direct connection between metabolic activity and loss of culturability, which can be used to probe persister metabolism. With this strategy, libraries of metabolites can be screened simultaneously to deduce the breadth of persister metabolism and identify adjuvants that improve the activity of AG in persisters.
Studying persister metabolism is expected to yield a rich resource of targets for anti-persister therapeutics, because strong evidence suggests that metabolism is intimately connected to the persister phenotype (reviewed in (Amato et al., 2014)). Indeed, persistence can be framed as a metabolic program, where the shut-down of metabolic processes promotes entry into the low energy persister state; maintenance of basal metabolism ensures viability during persistence; and reactivation of metabolic pathways allows for reawakening and growth once antibiotic treatment is over. The potential for corrupting the persister metabolic program to eliminate persisters is best demonstrated by the number of anti-persister strategies reported to date that are based on the reactivation of metabolism that renders persisters sensitive to a co-administered antibiotic (Allison et al., 2011; Kim et al., 2011; Pan et al., 2012; Pan et al., 2013). Knowledge of the metabolic activities retained in persisters would allow us to elucidate the changes governing each stage of the persister metabolic program and improve the rational design of strategies that can perturb these processes. The AG potentiation assay described in this unit offers one path toward the discovery of the metabolic capabilities and enzymatic activities of bacterial persisters.
Critical Parameters and Troubleshooting
Persister levels are sensitive to culture and antibiotic-treatment conditions. It has been observed that persister levels with longer overnight culturing were enhanced due to prolonged cell dormancy (Luidalepp et al., 2011). To ensure reproducibility of results, we recommend measuring persister levels of a bacterial strain under the chosen culturing conditions, including media type, culturing time, media volume, flask type, aeration and growth stage, and keeping those parameters consistent across experiments. Under any condition, biphasic kill curves should be obtained to establish that the metabolic activities of persisters are being measured (Basic Protocol 1). Otherwise, results may be reflective of normal cells that have not yet been killed by antibiotics.
The experiments described in this unit were conducted in LB media, which is commonly used for culturing E. coli, but it can also introduce variability. We have observed batch-to-batch variations with tryptone and yeast extract, which can contribute to changes in persister levels. Furthermore, other groups have observed that variability in autoclave temperatures and duration can change the composition of the media and have recommended sterilizing LB by filtration instead of autoclaving (Luidalepp et al., 2011). To ensure the reproducibility and robustness of results, we perform persister and AG potentiation assays using different batches of LB components and/or different media preparation methods.
Cell density during AG assays is a critical parameter that should be taken into consideration. We previously demonstrated that cell cultures after antibiotic treatment have many more VBNCs than persisters (Orman and Brynildsen, 2013b). We have further shown that VBNCs may influence AG potentiation in persisters when high cell density cultures (~106–107 persisters/mL) were used (Orman and Brynildsen, 2013b). Therefore, cell cultures should be diluted sufficiently before the AG assay so that non-persisters cells do not affect the AG activity in persisters. With competition assays (Basic Protocol 3), we previously determined that diluting cell cultures to ~104–105 persisters/mL prevents this phenomenon in our experimental setup. However, since the number of non-persisters and their influence on persister metabolism can be affected by the culture conditions, we recommend that competition assays should be performed when the conditions have changed.
For the AG assay, gentamicin can be used instead of kanamycin. The resistance markers in WT and mutant strains, which are necessary for differential plating in the competition assay, should be chosen accordingly. If plating on antibiotic-selective media is not preferred for competition assays, a different strategy can be employed to determine the proportion of WT or mutant surviving cells in the mixed populations. For instance, a number of colonies can be randomly selected after propagation of the survivors from the mixed samples on LB agar. Colonies can then be grown in M9 media with the non-utilizable carbon source (for instance, glycerol for ΔgldAΔglpK) using 96-well plates, and this will allow quantification of the proportion of surviving cells that were mutants (e.g., ΔgldAΔglpK mutants cannot grow in minimal-glycerol media, whereas WT cells can) (Orman and Brynildsen, 2013b). Another method is to perform PCR for selected colonies to determine the ratio of colonies that lack the gene(s) of interest (e.g., gldA and glpK).
Persisters should remain non-replicative and retain their antibiotic tolerance during the AG assay; otherwise results may indicate reawakening rather than metabolic activity while in the persister state. To determine whether metabolites stimulate reawakening, cells are treated with a β-lactam and carbon sources. In addition to β-lactam control, KCN treatment, which inhibits cytochrome activity, should be included in the assay. We use 1 mM KCN to block the respiration (Orman and Brynildsen, 2013b), however one should test various KCN concentrations and measure the oxygen utilization rate of cells to select the most appropriate concentration.
Anticipated Results
If persisters can catabolize a carbon source, incubating cultures with the substrate and AG should significantly reduce CFUs compared to a control that does not contain any substrate. Although this potentiation indicates catabolism of the metabolite to cytochrome activity, it does not exclude the possibility of catabolism through other pathways. One can expect that different persisters may exhibit different metabolic activities. For example, persisters from stationary phase cultures may catabolize different substrates than persisters from exponential phase cultures, and the same may be true for AMP persisters compared to OFL persisters. The no carbon source control provides the baseline from which the substrates that are significantly catabolized by persisters are quantified. KCN treatment should significantly reduce the AG activity. Furthermore, CFUs should not be significantly altered in AMP-treated cultures if persisters remain non-replicative during 2 h incubation period.
Time considerations
We generally incubate the overnight cultures for 16 or 24 h. These cultures are diluted in fresh media and further cultured until the desired growth stage is reached. In our previous study, we diluted the overnight culture 1000-fold in fresh LB media and cultured cells for ~2.5 h to obtain exponential phase cells (OD600 0.1) (Orman and Brynildsen, 2013a). After the desired growth stage is achieved, cell cultures are treated with antibiotics. Treating the exponential phase cultures (OD600 ~0.1) with antibiotics for 5 h was sufficient to produce a biphasic kill curve under our experimental conditions (Orman and Brynildsen, 2013a); however this period may vary depending on the experimental conditions and the bacterial strains used. The AG assay takes approximately 30 min to set up, and 2 h of incubation with AG and carbon sources have been found to be adequate for potentiation and reduction in CFUs to be observed. Within this time frame, we have found persisters to remain non-replicative as well. Following incubation, the washing, serial dilution, and plating of samples treated with carbon sources and AG or AG with KCN generally takes approximately 1 h. Finally, we incubate the plates for 16 h to allow for CFU formation before counting the colonies.
Acknowledgments
Financial support was provided by the Department of Army under award number W81XWH-12-2-0138 and the NIAID of the NIH under award number R21AI105342. The content is the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
LITERATURE CITED
- Allison KR, Brynildsen MP, Collins JJ. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature. 2011;473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato SM, Fazen CH, Henry TC, Mok WW, Orman MA, Sandvik EL, Volzing KG, Brynildsen MP. The role of metabolism in bacterial persistence. Front Microbiol. 2014;5:70. doi: 10.3389/fmicb.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato SM, Orman MA, Brynildsen MP. Metabolic Control of Persister Formation in Escherichia coli. Molecular cell. 2013;50:475–487. doi: 10.1016/j.molcel.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Andrews JM. Determination of minimum inhibitory concentrations. J Antimicrob Chemother. 2001;48(Suppl 1):5–16. doi: 10.1093/jac/48.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- Balaban NQ, Gerdes K, Lewis K, McKinney JD. A problem of persistence: still more questions than answers? Nat Rev Microbiol. 2013;11:587–591. doi: 10.1038/nrmicro3076. [DOI] [PubMed] [Google Scholar]
- Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- Bigger JW. The bactericidal action of penicillin on Staphylococcus pyogenes. The Irish Journal of Medical Science. 1944;19:553–568. [Google Scholar]
- Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature. 2013;503:365–370. doi: 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BD. Mechanism of bactericidal action of aminoglycosides. Microbiol Rev. 1987;51:341–350. doi: 10.1128/mr.51.3.341-350.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvart M, De Groote VN, Michiels J. Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J Med Microbiol. 2011;60:699–709. doi: 10.1099/jmm.0.030932-0. [DOI] [PubMed] [Google Scholar]
- Iino R, Hayama K, Amezawa H, Sakakihara S, Kim SH, Matsumono Y, Nishino K, Yamaguchi A, Noji H. A single-cell drug efflux assay in bacteria by using a directly accessible femtoliter droplet array. Lab Chip. 2012;12:3923–3929. doi: 10.1039/c2lc40394c. [DOI] [PubMed] [Google Scholar]
- Iino R, Matsumoto Y, Nishino K, Yamaguchi A, Noji H. Design of a large-scale femtoliter droplet array for single-cell analysis of drug-tolerant and drug-resistant bacteria. Front Microbiol. 2013;4:300. doi: 10.3389/fmicb.2013.00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. Persister cells and tolerance to antimicrobials. FEMS Microbiol Lett. 2004a;230:13–18. doi: 10.1016/S0378-1097(03)00856-5. [DOI] [PubMed] [Google Scholar]
- Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol. 2004b;186:8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Heo P, Yang TJ, Lee KS, Cho DH, Kim BT, Suh JH, Lim HJ, Shin D, Kim SK, Kweon DH. Selective killing of bacterial persisters by a single chemical compound without affecting normal antibiotic-sensitive cells. Antimicrob Agents Chemother. 2011;55:5380–5383. doi: 10.1128/AAC.00708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. Persister cells, dormancy and infectious disease. Nature Reviews Microbiology. 2007:48–56. doi: 10.1038/nrmicro1557. [DOI] [PubMed] [Google Scholar]
- Lewis K. Persister Cells. In: Gottesman S, Harwood CS, editors. Annual Review of Microbiology. Vol. 64. Annual Reviews; Palo Alto: 2010. pp. 2010pp. 357–372. vol. 64. [DOI] [PubMed] [Google Scholar]
- Luidalepp H, Jõers A, Kaldalu N, Tenson T. Age of inoculum strongly influences persister frequency and can mask effects of mutations implicated in altered persistence. J Bacteriol. 2011;193:3598–3605. doi: 10.1128/JB.00085-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orman MA, Brynildsen MP. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob Agents Chemother. 2013a;57:3230–3239. doi: 10.1128/AAC.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orman MA, Brynildsen MP. Establishment of a method to rapidly assay bacterial persister metabolism. Antimicrob Agents Chemother. 2013b;57:4398–4409. doi: 10.1128/AAC.00372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JC, Bahar AA, Syed H, Ren DC. Reverting Antibiotic Tolerance of Pseudomonas aeruginosa PAO1 Persister Cells by (Z)-4-bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one. Plos One. 2012;7 doi: 10.1371/journal.pone.0045778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JC, Xie X, Tian W, Bahar AA, Lin N, Song FC, An J, Ren DC. (Z)-4-Bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one sensitizes Escherichia coli persister cells to antibiotics. Applied Microbiology and Biotechnology. 2013;97:9145–9154. doi: 10.1007/s00253-013-5185-2. [DOI] [PubMed] [Google Scholar]
- Roostalu J, Jõers A, Luidalepp H, Kaldalu N, Tenson T. Cell division in Escherichia coli cultures monitored at single cell resolution. BMC Microbiol. 2008;8:68. doi: 10.1186/1471-2180-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 2006;6:53. doi: 10.1186/1471-2180-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]