

Abstract
The synthesis and biological analysis of a number of novel congeners of the aminocyclopentitol pactamycin is described. Specific attention was paid to the preparation of derivatives at crucial synthetic branch points of the parent structure, and biological assays revealed a number of insights into the source of pactamycin’s biological activity. Additionally, the encapsulation of pactamycin and select derivatives into the PRINT© nanoparticle technology was investigated as a proof-of-concept, and evidence of bioactivity modulation through nanoparticle delivery is demonstrated. This work has provided heretofore unrealized access to a large number of novel compounds for further evaluation.
Keywords: Pactamycin, Nanoparticles, Structure activity relationships, Structural derivatization
1. Introduction
Pharmaceutical development through organic synthesis remains a critical feature of the drug discovery process.1 Upon identification of an initial hit via high-throughput screening, a significant amount of structural modification is often required before a lead candidate can be advanced to clinical trials. Natural molecules are often identified as initial hits in these screenings; however, later modification of their complex structures toward the preparation of useful drug molecules can be hindered by the deficiency of a practical and flexible chemical synthesis.2 As a result, the continued advancement of synthetic organic methodology is critical for facile and flexible drug discovery and development.
Pactamycin (1, Fig. 1) is an example of a valuable natural target that has yet to reach its full medicinal potential, at least in part due to its structural complexity. Isolated in 1961 from a fermentation broth of Streptomyces pactum var. pactum by scientists at the former Upjohn Chemical Co.,3 pactamycin represents the most complex aminocyclitol antibiotic ever discovered. Researchers at Upjohn showed it to be active against Gram-positive and Gram-negative bacteria as well as against a number of cancer cell lines in vitro.4 More recent biological studies have demonstrated pactamycin to have potent antiviral (complete inhibition of polio-infected HeLa cells at 10−7 M) and antiprotozoal qualities (P.f. K1: IC50 = 14.2 nM).5,4b Unfortunately, this promising biological profile is hindered by pactamycin’s high cytotoxicity against human eukaryotic cell lines (MRC-5: IC50 = 95 nM).5,4b X-ray crystallographic studies have shown that the source of this activity stems from pactamycin’s ability to bind to the 30S ribosomal subunit acting as an RNA dinucleotide mimic.6 A complex array of H-bonding interactions within the 30S site enables pactamycin to act as a universal inhibitor of translocation. Its impressive biology has attracted the attention of a multidisciplinary field in hopes of transforming pactamycin into a suitable therapeutic (Fig. 1).
Figure 1.

Structures of pactamycin (1) and natural, synthetic, and biosynthetic congeners.
In addition to 1, a number of naturally-occurring structural congeners have been isolated from related Streptomyces bacteria, displaying varied bioactivities. 7-Deoxypactamycin (2) and jogyamycin (3) have shown increased antiprotozoal activity relative to 1.7 A third natural analog, pactamycate (4), has also been reported.8 Alternatively, biosynthetic engineering studies pioneered by Mahmud and co-workers have provided researchers with the first series of unnatural structural analogs such as TM-025F (5) and TM-026F (6), which display comparable activities to pactamycin against Plasmodium falciparum.8b,9 These data have renewed promise for pactamycin analogs in drug development.
Moreover, encapsulation of natural cytotoxic agents into nanoparticles (NPs) has also shown improved clinical benefits, the most germane of these being reduction of undesired toxic side effects and increased therapeutic delivery to the target of interest. This approach has been successfully implemented in the case of doxorubicin (Doxil©),10 paclitaxel (Abraxane©)11 and others.12 More recently, Bind Therapeutics13 and Cerulean14 have ongoing clinical trials in NP formulations of cancer therapeutics (docetaxel, irinotecan, and camptothecin). DeSimone and co-workers have demonstrated the use of the Particle Replication in Non-Wetting Templates (PRINT®) technology to modulate the activity of cytotoxic agents such as docetaxel, reducing unwanted side-effects and increasing therapeutic activity in vivo.15 To the best of our knowledge, however, the incorporation of pactamycin or its congeners into NPs of any type with the goal of bioactivity attenuation has not yet been explored.
While an efficient chemical synthesis of 1 might provide the most flexibility in structural derivatization, the inherent complexity of the molecule has rendered this a difficult undertaking. The heavily-compacted and heteroatom-rich functionality in pactamycin presents a number of challenges toward selective structural modification. Additionally, while the unique functional groups present in the molecule (salicylate, dimethylurea, aniline) offer novel branch points for structural diversification, methods with which to install these moieties are underexplored in the literature.16 To these aims, a number of synthetic studies have been reported by Isobe, Knapp, Looper, Nishikawa, and our group in the past decade.17 In 2011, Hanessian and co-workers described the first total synthesis of pactamycin in 32 steps from L-threonine, enabling previously unrealized access to synthetic congeners.18 Since this initial publication, Hanessian has demonstrated the efficacy of his route to deliver pactamycin derivatives at the C1-dimethylurea and the C3 aniline positions such as compounds 7 and 8.19
Our group began work on the total synthesis of 1 in 2009, and this work culminated in 2013 with a 15-step, asymmetric synthesis from commercially available 2,4-pentanedione (Fig. 2).20 Critical to our approach was to assemble the molecule in a fashion such that key functional groups were installed both in their native form and in a late-stage fashion; we surmised that this approach would provide the greatest possible flexibility, facilitating investigations of structure-activity relationships at all critical branch points. To this end, we envisaged a synthon such as 9 in which exploitation of appropriate functional handles at the correct stage would install the requisite functionalities.
Figure 2.
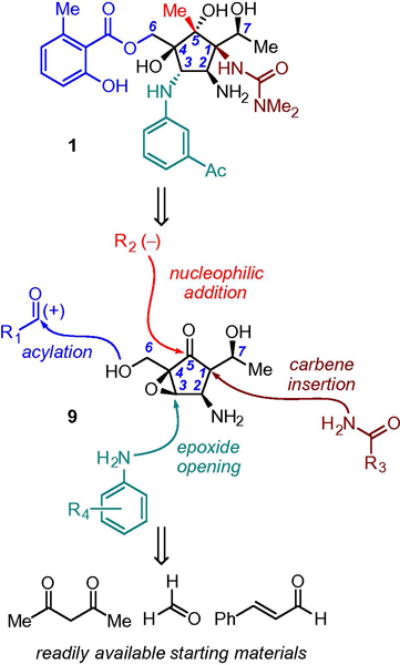
Synthon analysis of 1 showing key branch points for structural derivatization.
A summary of our disclosed synthesis endgame is described in Figure 3, wherein ketone intermediate 10 (synthesized in ten steps from 2,4-pentanedione in gram quantities) would serve as our first point of derivatization.20 Nucleophilic methylation of 10 proceeded in good yield to provide carbinol 11 in 75% yield of a single diastereomer at C5. Sc(OTf)3-promoted addition of m-acetylaniline installed the substituted C3-aniline necessary for elaboration to 1, upon which silyl deprotection afforded tetraol 12. Introduction of the remaining salicylate moiety to the C6-hydroxymethylene of 12 was accomplished via reaction with the previously reported acyl electrophile 13, which upon hydrogenative removal of the Cbz protecting group delivered pactamycin in 15 steps and 1.9% overall yield. With this strategy established, we shifted our focus to examining the route’s flexibility toward analog preparation.
Figure 3.

Pactamycin: endgame strategy and synthesis completion.
Herein, we delineate our efforts in the synthesis and biological evaluation of novel pactamycin congeners. Additionally, we report the first studies incorporating pactamycin and select derivatives into polymeric NPs, fabricated using the PRINT® technology, with the goal of enhancing activity and selectivity while mitigating unwanted toxicities.
2. Results and discussion
2.1. C3-aniline
We first pursued the preparation of pactamycin congeners at the C3-aniline position, inspired by a related epoxide-opening strategy by Hanessian and co-workers (Table 1).18,19 We were encouraged by the aniline flexibility demonstrated by Hanessian in their earlier report and hoped that epoxide 11 would participate in related transformations with functionally and electronically diverse anilines.
Table 1.
Addition of substituted anilines and other nitrogen nucleophiles to epoxide 11

| |||
|---|---|---|---|
|
| |||
| Entry | H–Nu | Product | % Yielda |
| 1 |
|
14a | 83 |
| 2 |

|
14b | 43 |
| 3 |
|
14c | 95 |
| 4 |
|
14d | 71 |
| 5 |
|
14e | 86 |
| 6 | 2-Fluorenyl aniline | 14f | 59 |
| 7 |

|
14g | 87 |
| 8 |
|
14h | 47 |
| 9 |
|
– | –b |
| 10 | BnNH2 | – | –c |
| 11 | NaN3 | – | –d |
Isolated yield.
TBS deprotection was observed as the sole product.
No reaction was observed.
Conditions: NaN3 (1.1 equiv), oxone (0.5 equiv), CH3CN/H2O (9:1), rt; only TBS deprotection was observed in this reaction.
Indeed, we were pleased to find that epoxide 11 reacted readily in the presence of a number of substituted anilines in moderate to excellent yields, providing anilines 14a–h. Notably, while the installation of m-acetylaniline to 11 necessitated superstoichiometric amounts of Sc(OTf)3, all subsequent anilines examined in the reaction proceeded to completion using 50 mol % Sc(OTf)3. We suspect that this is an outgrowth of both low nucleophilicity and poor solubility of the parent m-acetylaniline under the reaction conditions. With the aniline tolerance established, we turned our attention to alternative nitrogen nucleophiles. Unfortunately, all non-aromatic nitrogen sources (RNH2, R2NH, (−)N3) failed to react with 11, giving either no reaction or starting material decomposition. Addition products 14a–h were carried through the endgame sequence described previously, delivering derivatives 15a–h (Fig. 4). In the case of addition product 14g, the aryl bromide was reduced during hydrogenolysis, delivering α-naphthyl anilide 15g.
Figure 4.

Synthesis of C3 aniline pactamycin derivatives. aHydrogenation of the of bromide functionality was observed.
2.2. C1-dimethylurea
Hanessian’s approach toward preparation of pactamycin analogs at the C1 dimethylurea position substituent relied on the trapping of an in situ generated isocyanate electrophile late in the synthesis.18 This tactic proved effective in the preparation of a series of functionalized ureas in good yields.19 By contrast, our synthesis of 1 utilized an early-stage N–H insertion reaction to install the urea.20 Synthetic diversification from this early intermediate would be a significant challenge. Consequently, we envisaged a similar isocyanate formation/trapping strategy from carbinol intermediate 11 via the acid-catalyzed elimination of dimethylamine (Fig. 6).
Figure 6.

Failed isocyanate formation/trapping strategy from 11.
After some experimentation, treatment of 11 with NH4Cl in H2O and MeOH resulted in complete elimination of dimethylamine and convergence to a single product. However, 1H NMR analysis revealed that the intermediate isocyanate had undergone intramolecular ring closure with the C2-carbamateto furnish imidazolidinone 17 in 73% yield. Although this result rendered our intermolecular isocyanate trapping strategy unfeasible, we postulated that this reactivity might be exploited toward analog preparation of the naturally-occurring congener pactamycate 4 (Fig. 5).
Figure 5.

Preparation of de-6-MSA pactamycate and pactamycate analogs. Conditions: (a) oxone, CH3CN/H2O (9:1), rt; (b) NH4Cl, MeOH/H2O (8:1), 85 °C; (c) TBAF, THF, 0 °C; (d) H2 (1 atm), Pd(OH)2/C (0.5 mass equiv), MeOH, rt; (e) aniline (10 equiv), Sc(OTf)3 (50 mol %), PhMe, 50 °C; (f) K2CO3,13, DMA, rt.
Selective deprotection of the C7-TBS ether in 16 with oxone furnished the corresponding secondary alcohol 18 in 84% yield, which we postulated would serve as a more nucleophilic trapping agent for the intermediate isocyanate. Indeed, upon treatment of 18 with NH4Cl in H2O/MeOH, the in situ generated isocyanate underwent trapping by the unprotected C7 hydroxyl to afford oxazolidinone 19 in 73% yield. Removal of the TBDPS protecting group with TBAF provided triol 20, which upon Cbz deprotection, afforded De-6-MSA pactamycate 21, which has been previously characterized.18b This result confirmed that the intermediate isocyanate generated from 18 had indeed been engaged by the C7 hydroxyl (and not the C2 carbamate). With this route established, we resolved to prepare a subset of varied C3 pactamycate structures. Beginning with anilides 14b, 14c, and 14e already in hand from the C3 derivatization studies, TBS deprotection followed by oxazolidinone formation gave the corresponding diols, which upon desilylation, acylation and hydrogenolysis, provided pactamycate derivatives 22a–c.
2.3. C5-tertiary alcohol
We next began examining reactivity of the ketone in 10 with the goal of diversification at C5 (Table 2). To this end, we were pleased to find this ketone reacted readily with ethyl, nhexyl, and vinyl magnesium bromide to give the corresponding carbinols in good yields. Unfortunately, when larger alkyl (entries 4, 5 and 8) and aryl nucleophiles (entries 6 and 7) were examined, we observed only un-productive side reactions21 or complete starting material recovery. With this limitation established, we speculated that hydride might also be a suitable nucleophile. In the event, treatment of 10 with NaBH4 in MeOH at −45 °C provided alcohol 23c in good yield with analogous stereofidelity to that observed in the addition of carbon nucleophiles.
Table 2.
Addition of nucleophiles to ketone 10a

| |||
|---|---|---|---|
|
| |||
| Entry | R | Product | % Yieldb |
| 1 | Et | 23a | 75 |
| 2 | nHexyl | 23b | 73 |
| 3 |
|
– | 43b |
| 4 |
|
– | –c |
| 5 |
|
– | –c |
| 6 |
|
– | –d |
| 7 |
|
– | –d |
| 8 | PhCH2 | – | –e |
| 9 | H | 23c | 88f |
Isolated yields.
Product could not be elaborated further.
No reaction was observed.
Undesired side products isolated.
Complex mixture.
Conditions: NaBH4, MeOH, −45 °C.
With addition products 23a–c in hand, we proceeded in the synthesis to complete C5 analog preparation (Fig. 7). We were surprised to find, however, that subjection of these intermediates to the optimized conditions for C3 m-acetylaniline installation gave only significant amounts of recovered starting material. Increasing the loading of Sc(OTf)3 or the reaction time/temperature had seemingly no effect. It seems reasonable that this addition is sluggish either due to poor coordination of the Lewis acid or by an unfavorable substrate conformation for addition relative to the parent C5-methyl compound (11). In order to circumvent this issue, we turned to the strategy of Hannesian and coworkers wherein the required C3 m-acetylaniline was incorporated via an 2-(prop-1-en-2-yl)aniline surrogate.18 The acetophenone was later revealed via oxidation. In our system, this strategy also proved effective, providing 2-(prop-1-en-2-yl)aniline anilines 24a–c in good yields. Johnson–Lemieux oxidation of the resulting alkenes provided the desired m-acetylanilines 25a–c in good yields over the three-step sequence. Completion of the remaining synthetic sequence provided C5 pactamycin derivatives 26a–c.
Figure 7.

Preparation of C5 pactamycin analogs. Conditions: (a) m-propenylaniline (10 equiv), Sc(OTf)3 (50 mol %), C7H8, 50 °C; (b) OsO4 (10 mol %), NMO (5 equiv), THF/acetone/H2O (5:5:1), 0 °C–rt; (c) NalO4 (3.5 equiv), THF/H2O (1:1), rt; (d) TBAF, THF, 0°C; (e) K2CO3, 13, DMA, rt; (f) H2 (1 atm), Pd(OH)2/C (0.5 mass equiv), MeOH, rt.
2.4. C6,C7-hydroxyl
The final point of diversification centered on manipulation of the salicylate-bearing C6 ester in 1 (Table 3). Gratifyingly, efficient monoacylation of tetraol 12 was accomplished with a variety of aliphatic and aromatic acyl electrophiles in good yields (entries 1–5). The resulting monoesters were subjected to the previously employed conditions for Cbz deprotection, providing derivatives 27b–e. However, in the case of the differentiated methoxyphenol 27a (entry 1), only decomposition was observed upon hydrogenolysis.
Table 3.
Functionalization of C6 (C7) alcohols

| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | R1 | R2 | Product | Yield 1a | Yield 2a |
| 1 |

|
H | 27ab | 57 | –c |
| 2 |

|
H | 27bb | 43 | 62 |
| 3 |

|
H | 27cd | 60 | 73 |
| 4 |
|
H | 27dd | 83 | 76 |
| 5 |
|
H | 27ed | 87 | 61 |
| 6 |
|
Ac | 27fe | 86 | 38 |
| 7 |
|
Piv | 27gf | 92 | 53 |
| 8 |
|

|
27hf | 76 | 72 |
Isolated yields.
X = OCH2CN; conditions: K2CO3, DMA, rt.
Only decomposition was observed.
X = Cl; conditions: 2,4,6-collidine, CH2Cl2, −78 °C to rt.
X = OAc; conditions: NEt3, DMAP (10 mol %), CH2Cl2, 0 °C–rt.
X = Cl; conditions: NEt3, DMAP (10mol%), CH2Cl2, 0°C–rt. See Supporting information for details on electrophile preparation.
Cognizant of the documented bioactivity difference across multiple cell lines observed between 1 and its 7-deoxy congener (2),7 reduction of the C7-hydroxyl to its corresponding methylene was also probed. Unfortunately, all conditions explored (from a number of different intermediates in our route) failed to deliver the desired C7-methylene. As an alternative strategy, we envisaged masking of the C7 hydroxyl via its ester might serve the same purpose (i.e., removal of the H-bonding interaction at C7).6 To this end, C6–C7 bis-acylated derivatives (entries 6–8) were synthesized with varying degrees of steric encumbrance. Cbz hydrogenolysis provided the diesters 27f–h.
3. Results and discussion
3.1. Biological evaluation
Having prepared a library of novel compounds, we set out to examine their varied biological profiles. Specifically, compounds were tested against breast, ovarian, and lung carcinoma cell lines. Additionally, the human embryonic cell line for which pactamycin’s toxicity has been established (MRC-5) was assayed for comparison.5,4b The results for all derivatives are summarized in Table 4. As anticipated, pactamycin (entry 1) displayed exceptional potency, showing nanomolar inhibition against all three carcinoma cell lines. For comparison, the penultimate intermediate in our synthesis of pactamycin (28) bearing Cbz protection at the C2-aminomethine (entry 2) showed a dramatic decrease in activity relative to 1. In order to better understand the effect of chirality on the parent pactamycin structure, ent-pactamycin (ent-(1)) (entry 3) was synthesized in high enantiomeric purity via a slight modification of the previously published route (see Supporting information) and assayed in our study. As illustrated in Table 4, ent-(1) showed a threefold order of magnitude decrease in bioactivity, illustrating the impact of the natural enantiomer of 1 to effective cell-growth inhibition.
Table 4.
Biological examination of pactamycin and synthetic analogsa
| Entry | Structure | Code number | A549 EC50 (lung cancer) |
MDA-MB-231 EC50 (breast cancer) |
SK-OV-3 EC50 (ovarian cancer) |
MRC-5 EC50 (human lung fibroblast) |
|---|---|---|---|---|---|---|
| 1 |

|
1 | 160 nM | 124 nM | 129 nM | 53 nM |
| 2 |

|
28 | 11.8 μM | 10.4 μM | 12 μM | n.t.b |
| 3 |

|
ent-(1) | 2.1 μM | 1.2 μM | 1.6 μM | 933 nM |
| 4 |
|
15a | 800 nM | 659 nM | 1.4 μM | 380 nM |
| 5 |

|
15b | 141 nM | 556 nM | 434 nM | 314nM |
| 6 |
|
15c | 1μM | n.t.b | 600 nM | 582 nM |
| 7 |
|
15d | 777 nM | 4μM | 4 μM | 682 nM |
| 8 |
|
15e | 884 nM | 3.3 μM | 1.6 μM | 2326 nM |
| 9 |
|
15f | 324 nM | 376 nM | 145 nM | 431 nM |
| 10 |

|
15g | 2.21 μM | 1.84 μM | 2.44 μM | 860 nM |
| 11 |
|
15h | 760 nM | 800 nM | 436 nM | 366 nM |
| 12 |

|
21 | 6μM | n.t.b | 3.8 μM | 2933 nM |
| 13 |

|
22a | n.t.b | n.t.b | n.t.b | n.t.b |
| 14 |
|
22b | n.t.b | n.t.b | n.t.b | n.t.b |
| 15 |
|
22c | n.t.b | n.t.b | n.t.b | n.t.b |

|
||||||
| 16 |

|
26a | n.t.b | n.t.b | n.t.b | 2063 nM |
| 17 |

|
26b | n.t.b | n.t.b | n.t. b | 10.9 μM |
| 18 |

|
26c | 32 nM | 50 nM | 7 nM | 6.5 nM |
| 19 |

|
29 | 83 nM | 356 nM | 91 nM | 49 nM |
| 20 |

|
27b | 88 nM | 203 nM | 103 nM | 129 nM |
| 21 |
|
27c | 114 nM | 79 nM | 80 nM | 105 nM |
| 22 |
|
27d | 118 nM | 300 nM | 75 nM | 100 nM |
| 23 |

|
27e | 194 nM | 352 nM | 436 nM | 366 nM |
| 24 | R = Ac | 27f | 137 nM | 458 nM | 123 nM | 132 nM |
| 25 |

|
27h | 175 nM | 1.93 μM | 86 nM | 396 nM |
| 26 |

|
27g | 588 nM | 2.44 μM | 593 nM | 778 nM |
Assays were carried out as triplicates.
Not toxic.
Generally, all C3-aniline derivatives (entries 4–11) showed a marginal to significant decrease in activity relative to 1 across all cell lines, although 15b (entry 5) showed comparable activity against A549 (EC50 = 141 nM) with a marginal decrease in MRC5 activity. With regard to the pactamycate series of analogs, De-6-MSA pactamycate 21 (entry 12) showed only minor cell-growth inhibition. This was not an altogether unexpected result, however, as biological assays of 21 conducted by Hanessian and co-workers also showed little promising activity.19,6c Altering the C3 aniline position of the pactamycate parent structure (entries 13–15) resulted in complete loss of biological activity. These results, in combination with those of the pactamycin C3 analogs, speak to the importance of the m-acetyl functionality in 1 to its bioactivity.22
The results of compounds bearing diversity at C5 are shown in entries 16–19. In combination with the C5 derivatives previously described (vide supra), an additional derivative 29 (entry 19) was prepared bearing alternate functionality at the C3 aniline for comparison (see Supporting information). Extending the length of the carbon chain at C5 (entries 16 and 17) had significantly deleterious effects to bioactivity as a complete loss of carcinoma activity was observed, leaving only low inhibition of MRC-5. However, removing alkyl functionality altogether at C5 (entries 18 and 19) had the opposite effect, as these C5 protio analogs displayed the greatest activity across all cell lines of any compound tested in our study (including pactamycin). We speculate that these results are primarily a function of adjusting the lipophilicity of the structure relative to 1.23
The results of our diversification of the C6 hydroxymethylene (entries 22–25) are in agreement with Hanessian’s earlier findings.19,6c Namely, no significant gain (or loss) of biological activity was observed when the salicylate ester was altered relative to the parent pactamycin structure. These results further support the hypothesis that the C6 ester side chain has a limited role in the key binding event of 1 in the 30S ribosome.19,6c The three prepared C6,C7 bis-acylated derivatives (entries 24–26) showed a linear decrease in activity with steric encumbrance of the ester group. These results suggest that the C7 hydroxyl in 1 plays a larger role in the bioactivity of the structure than the C6 hydroxymethylene.
Upon collection of these initial data, derivatives 15f, 26c, 27f, 27c, and ent-(1) were identified as the most promising compounds and were assayed via the NCI 60 human tumor cell line screen. Upon initial one-dose screening, all five compounds were found to have sufficient activity to merit the subsequent five-dose assay. These derivatives were evaluated to determine GI50 (50% growth inhibition) values. The results of these assays are summarized in Table 5. Additionally, the previously documented cell data for 1 is shown for comparison.
Table 5.
Summary GI50 values from NCI-60 cell line screeninga
| GI50 (μM) | 1b | (ent)-1 | 26c | 27c | 15f | 27f |
|---|---|---|---|---|---|---|
| MOLT-4 | <0.10 | 1.19 | 0.046 | 0.12 | 0.78 | 0.33 |
| NCI-H322M | 0.12 | 3.72 | 0.016 | 0.33 | 1.07 | 0.48 |
| HCT-15 | 0.03 | 20.0 | 0.16 | 0.65 | 1.46 | 10.2 |
| SNB-19 | <0.10 | 3.07 | 0.52 | 0.19 | 1.40 | 0.57 |
| M14 | 0.12 | 3.01 | 0.10 | 0.19 | 0.88 | 0.68 |
| OVCAR-3 | <0.10 | 2.50 | 0.041 | 0.20 | 0.73 | 0.53 |
| RXF 393 | <0.10 | 1.50 | 0.064 | 0.12 | 0.61 | 0.61 |
| DU-145 | <0.01 | 7.26 | 0.15 | 0.26 | 1.37 | 0.34 |
| MCF7 | <0.01 | 2.04 | 0.051 | 0.17 | 0.73 | 7.31 |
Data obtained from NCI-60 screening. See Supporting information for comprehensive results. MOLT-4, leukemia cell line; NCI-H322 M, nonsmall-cell lung cancer cell line; HCT-15, colon cancer cell line; SNB-19, CNS tumor cell lines; M14, melanoma; OVCAR-3, ovarian cancer cell line; RXF 393, renal cancer cell line; DU-145, prostate cancer cell line; MCF7, breast cancer cell line.
Data can be accessed from the CAS: 23668-11-3 at the following website: http://dtp.cancer.gov/dtpstandard/dwindex/index.jsp.
As expected based on our initial screen, ent-(1) showed multiple orders of magnitude loss in activity across the entire assay. By contrast, compound 26c bearing a secondary hydroxyl at C5 demonstrated exceptional activity, showing nM inhibition throughout the screen and outperforming pactamycin in multiple cell lines. Derivatives 27c (modified salicylate ester) and 27f (C6, C7 diace-toxypactamycin) also demonstrated general nM activity in the assay. The final derivative 15f bearing a fluorenyl aniline at C3 showed a general decrease in biological activity relative to 1 by factors of 10–100.
3.2. Nanoparticle fabrication-biological evaluation
With these studies completed, we set out to examine the efficacy of pactamycin and select analogs to activity modulation via nanoparticle encapsulation. Polymeric PRINT® nanoparticles were fabricated by encapsulating compounds 1, 15e, and 26c, in poly(D,L-lactide) using previously described methods.15b,24 Compounds 15e and 26c were selected on the basis of observing the effect of nanoformulation on derivatives both more and less bioactive than 1. PRINT NPs containing 1 and derivatives 15e and 26c all showed similar hydrodynamic radii and PDI as determined by dynamic light scattering (DLS).25 Scanning electron microscopy (SEM) analysis confirmed uniform particle size and shape regardless of compound identity, and drug loading of each sample was found to be ~10% as determined by HPLC.26 NP-encapsulated compounds NP-1, NP-15e, and NP-26c, were then examined in our assay, and the results are given in Table 6 where the baseline toxicity values for each compound are restated for comparison.
Table 6.
Cell-based assay comparison for compounds 1, 15e, 26c and NP counterpartsa
| Compound | A549 EC50 (nM) |
MDA-MB-231 EC50 |
MRC-5 EC50 |
|---|---|---|---|
| 1 | 160 | 124 nM | 53 nM |
| NP-1 | 52 | 117 nM | 52 nM |
| 15e | 884 | 3.3 μM | 2.3 μM |
| NP-15e | 693 | 5.5 μM | 1.8 μM |
| 26c | 32 | 50 nM | 6.5 nM |
| NP-26c | 6.5 | 724 nM | 18 nM |
Assays were carried out as triplicates
In vitro cytotoxicity analysis of the derivative NP formulations showed bimodal effects on therapeutic activity. In the A549 assay, nanoparticle delivery increased the cytotoxicity of the therapeutic cargo. NP-1 demonstrated an EC50 threefold more potent than pactamycin itself (52–160 nm, respectively). NP-26c showed a near fivefold increase in potency when compared to the unadulterated small molecule (6.5–32 nM, respectively). Even compounds 15e, a less active drug in comparison to 1, showing a nominal reduction in EC50 value for the A549 cell line. Of significant interest was the increase in selectivity observed for 26c, wherein the EC50 for A549 decreased while the EC50 for MDA-MB-231 and MRC5 increased.
4. Conclusions
In summary, we have demonstrated the efficacy of our synthetic approach to efficient and modular preparation of a number of varied analogs of the complex aminocyclitol pactamycin. These results have provided additional insight into the roles that each functional group plays in providing the observed activity of the parent structure. Additionally, we have established a heretofore undocumented proof-of-concept for the modulation of the pactamycin structure via the use of the PRINT® nanoparticle delivery vehicle. A wider range of cell-based assays and the further examination of pactamycin derivatives using the PRINT technology is planned and will be reported in due course.
Supplementary Material
Acknowledgments
The project described was supported by Award No. R01 GM084927 from the National Institute of General Medical Sciences and by the University Cancer Research Fund. RJ.S. acknowledges an NSF Graduate Research Fellowship. RJ.S. and J.T.M. acknowledge an ACS Division of Organic Chemistry graduate fellowship. F.S. acknowledges BI Research Italia S.a.s. di BI IT S.r.l. and University of Camerino (Italy) for a doctoral fellowship. Maribel Portillo is acknowledged for experimental assistance and preparation of select derivatives.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2015.02.022.
References and notes
- 1.MacCoss M, Baillie TA. Science. 2004;303:1810. doi: 10.1126/science.1096800. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber S. Science. 2000;287:1964. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 3.Argoudelis AD, Jahnke HK, Fox JA. Antimicrob Agents Chemother. 1962:191. [PubMed] [Google Scholar]
- 4.(a) Bhuyan BK, Dietz A, Smith CG. Antimicrob Agents Chemother. 1962:184. [Google Scholar]; (b) Iwatsuki M, Nishihara-Tsukashima A, Ishiyama A, Namatame M, Watanabe Y, Handasah S, Pranamuda H, Marwoto B, Matsumoto A, Takahashi Y, Otoguro K, Omura S. J Antibiot. 2012;65:169. doi: 10.1038/ja.2011.136. [DOI] [PubMed] [Google Scholar]
- 5.Taber R, Rekosh D, Baltimore D. J Virol. 1971;8:395. doi: 10.1128/jvi.8.4.395-401.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Dinos G, Wilson DN, Teraoka Y, Szaflarski W, Fucini P, Kalpaxis D, Nierhaus KH. Mol Cell. 2004;13:113. doi: 10.1016/s1097-2765(04)00002-4. [DOI] [PubMed] [Google Scholar]; (b) Carter AP, Clemons WM, Jr, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Nature. 2000;407:340. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]; (c) Tourigny DS, Fernández IS, Kelley AC, Vakiti RR, Chattopadhyay AK, Dorich S, Hanessian S, Ramakrishnan V. J Mol Biol. 2013;425:3907. doi: 10.1016/j.jmb.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurley TR, Smitka TA, Wilton JH, Bunge RH, Hokanson GC, French JC. J Antibiot. 1986;39:1086. doi: 10.7164/antibiotics.39.1086. [DOI] [PubMed] [Google Scholar]
- 8.(a) Ito T, Roongsawang N, Shirasaka N, Lu W, Flatt PM, Kasanah N, Miranda C, Mahmud T. ChemBioChem. 2009;10:2253. doi: 10.1002/cbic.200900339. [DOI] [PubMed] [Google Scholar]; (b) Lu W, Roongsawang N, Mahmud T. Chem Biol. 2011;18:425. doi: 10.1016/j.chembiol.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Almabruk KH, Lu W, Li Y, Abugreen M, Kelly JX, Mahmud T. Org Lett. 2013;15:1678. doi: 10.1021/ol4004614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Brien MER. Ann Oncol. 2004;15:440. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 11.Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. J Clin Oncol. 2005;23:7794. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 12.(a) Wang AZ, Langer R, Farokhzad OC. Annu Rev Med. 2012;63:185. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]; (b) Petros RA, DeSimone JM. Nat Rev Drug Disc. 2010;9:615. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]; (c) Caron WP, Morgan KP, Zamboni BA, Zamboni WC. Clin Cancer Res. 2013;19:3309. doi: 10.1158/1078-0432.CCR-12-3649. [DOI] [PubMed] [Google Scholar]
- 13.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, Low S, McDonnell K, Peeke E, Retnarajan B, Sabnis A, Schnipper E, Song JJ, Song YH, Summa J, Tompsett D, Troiano G, Van Geen Hoven T, Wright J, LoRusso P, Kantoff PW, Bander NH, Sweeney C, Farokhzad OC, Langer R, Zale S. Sci Transl Med. 2012;4:12839. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 14.(a) Batist G, Gelmon Ka, Chi KN, Miller WH, Chia SKL, Mayer LD, Swenson CE, Janoff AS, Louie AC. Clin Cancer Res. 2009;15:692. doi: 10.1158/1078-0432.CCR-08-0515. [DOI] [PubMed] [Google Scholar]; (b) Svenson S, Wolfgang M, Hwang J, Ryan J, Eliasof S. J Controlled Release. 2011;153:49. doi: 10.1016/j.jconrel.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 15.(a) Rolland JP, Maynor BW, Euliss LE, Exner AE, Denison GM, DeSimone JM. J Am Chem Soc. 2005;127:10096. doi: 10.1021/ja051977c. [DOI] [PubMed] [Google Scholar]; (b) Chu KS, Schorzman AN, Finniss MC, Bowerman CJ, Peng L, Luft JC, Madden AJ, Wang AZ, Zamboni WC, DeSimone JM. Biomaterials. 2013;34:8424. doi: 10.1016/j.biomaterials.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chu KS, Finniss MC, Schorzman AN, Kuijer JL, Luft JC, Bowerman CJ, Napier ME, Haroon ZA, Zamboni WC, Desimone JM. Nano Lett. 2014;14:1472. doi: 10.1021/nl4046558. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gharpure KM, Chu KS, Bowerman C, Miyake T, Pradeep S, Mangala LS, Han H-D, Rupaimoole R, Armaiz-Pena GN, Rahhal TB, Wu SY, Luft C, Napier ME, Lopez-Berestein G, Desimone JM, Sood AK. Mol Cancer Ther. 2014 doi: 10.1158/1535-7163.MCT-13-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Summary methods for synthesis of unsymmetrical dialkylureas:; (a) Ozaki S. Chem Rev. 1972;72:457. [Google Scholar]; (a) Gallou I, Eriksson M, Zeng X, Senanayake C, Farina V. J Org Chem. 2005;70:6960. doi: 10.1021/jo0507643. [DOI] [PubMed] [Google Scholar]; (b) Matsumura Y, Satoh Y, Onomura O, Maki T. J Org Chem. 2000;65:1549. doi: 10.1021/jo991076k. [DOI] [PubMed] [Google Scholar]; (c) Gastaldi S, Weinreb SM, Stien D. J Org Chem. 2000;65:3239. doi: 10.1021/jo9919714. [DOI] [PubMed] [Google Scholar]; (d) Han C, Porco JA. Org Lett. 2007;9:1517. doi: 10.1021/ol0702728. [DOI] [PubMed] [Google Scholar]; (e) Liu Q, Luedtke NW, Tor Y. Tetrahedron Lett. 2001;42:1445. [Google Scholar]; (f) Peterson SL, Stucka SM, Dinsmore C. J Org Lett. 2010;12:1340. doi: 10.1021/ol100259j. [DOI] [PubMed] [Google Scholar]; (g) Dubé P, Nathel NFF, Vetelino M, Couturier M, Aboussafy CL, Pichette S, Jorgensen ML, Hardink M. Org Lett. 2009;11:5622. doi: 10.1021/ol9023387. [DOI] [PubMed] [Google Scholar]
- 17.(a) Tsujimoto T, Nishikawa T, Urabe D, Isobe M. Synlett. 2005;433 [Google Scholar]; (b) Knapp S, Yu Y. Org Lett. 2007;9:1359. doi: 10.1021/ol0702472. [DOI] [PubMed] [Google Scholar]; (c) Haussener TJ, Looper RE. Org Lett. 2012;14:3632. doi: 10.1021/ol301461e. [DOI] [PubMed] [Google Scholar]; (d) Matsumoto N, Tsujimoto T, Nakazaki A, Isobe M, Nishikawa T. RSC Adv. 2012;2:9448. [Google Scholar]; (e) Malinowski JT, McCarver SJ, Johnson JS. Org Lett. 2012;14:2878. doi: 10.1021/ol301140c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Hanessian S, Vakiti RR, Dorich S, Banerjee S, Lecomte F, Del Valle JR, Zhang J, Deschênes-Simard B. Angew Chem, Int Ed. 2011;50:3497. doi: 10.1002/anie.201008079. [DOI] [PubMed] [Google Scholar]; (b) Hanessian S, Vakiti R, Dorich S, Banerjee S, Deschênes-Simard B. J Org Chem. 2012;77:9458. doi: 10.1021/jo301638z. [DOI] [PubMed] [Google Scholar]
- 19.Hanessian S, Vatiki RR, Chattopadhyay AK, Dorich S, Lavallée C. Bioorg Med Chem. 2013;21:1775. doi: 10.1016/j.bmc.2013.01.037. [DOI] [PubMed] [Google Scholar]
- 20.(a) Malinowski JT, Sharpe RJ, Johnson JS. Science. 2013;340:180. doi: 10.1126/science.1234756. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sharpe RJ, Malinowski JT, Johnson JS. J Am Chem Soc. 2013;135:17990. doi: 10.1021/ja409944u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.In these cases, the product was identified as nucleophile addition followed by Payne rearrangement of the resulting epoxy-alcohol.
- 22.(a) Weller DD, Rinehart KL., Jr J Am Chem Soc. 1978;100:6757. [Google Scholar]; (b) Rinehart KL, Jr, Potgieter M. J Am Chem Soc. 1981;103:2099. [Google Scholar]
- 23.Waring MJ. Expert Opin Drug Discov. 2010;5:235. doi: 10.1517/17460441003605098. [DOI] [PubMed] [Google Scholar]
- 24.Chu KS, Hasan W, Rawal S, Walsh MD, Enlow EM, Luft JC, Bridges AS, Kuijer JL, Napier ME, Zamboni WC, DeSimone JM. Nanomedicine. 2013;9:686. doi: 10.1016/j.nano.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.See Table S1, Supplementary data.
- 26.See Figure S1, Supplementary data.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.