

Abstract
The era in which ROS (reactive oxygen species) were simply the ‘bad boys of biology’ is clearly over. High levels of ROS are still rightfully considered to be toxic to many cellular processes and, as such, contribute to disease conditions and cell death. However, the high toxicity of ROS is also extremely beneficial, particularly as it is used to kill invading micro-organisms during mammalian host defence. Moreover, a transient, often more localized, increase in ROS levels appears to play a major role in signal transduction processes and positively affects cell growth, development and differentiation. At the heart of all these processes are redox-regulated proteins, which use oxidation-sensitive cysteine residues to control their function and by extension the function of the pathways that they are part of. Our work has contributed to changing the view about ROS through: (i) our characterization of Hsp33 (heat-shock protein 33), one of the first redox-regulated proteins identified, whose function is specifically activated by ROS, (ii) the development of quantitative tools that reveal extensive redox-sensitive processes in bacteria and eukaryotes, and (iii) the discovery of a link between early exposure to oxidants and aging. Our future research programme aims to generate an integrated and system-wide view of the beneficial and deleterious effects of ROS with the central goal to develop more effective antioxidant strategies and more powerful antimicrobial agents.
Keywords: aging, chaperone, host defence, oxidative stress, protein unfolding
Reactive oxygen species: friends and foes in life
Living an aerobic lifestyle is treacherous. ROS (reactive oxygen species), including peroxide, superoxide and hydroxyl radicals, are constantly produced as (by-)products of respiration and other enzymatic reactions, continuously threatening cellular macromolecules with irreversible oxidative modifications [1,2]. It is therefore not surprising that cells have developed an elaborate system of small-molecule antioxidants and ROS-detoxifying enzymes to remove ROS before they can cause permanent damage [3]. Endogenous ROS accumulation beyond manageable levels triggers an oxidative stress response in cells, leading to the increased expression of proteins that detoxify ROS, repair oxidative damage and restore redox homoeostasis [4].
Accumulation of ROS occurs for different reasons and at different stages in an organismal life, and oxidative stress has been associated with and often blamed for a number of different physiological processes and pathological conditions, including aging, age-related neurodegenerative diseases (e.g. Parkinson’s disease), metabolic diseases (e.g. diabetes), inflammatory diseases and heart disease [5]. The free radical theory of aging, for instance, proposes that an age-mediated increase in oxidative damage, by increasing pro-oxidants and/or decreasing antioxidants, causes the physiological decline observed in aging organisms [6]. Indeed, numerous studies have confirmed the idea that oxidative damage accrues in aging tissues, revealed correlations between ROS accumulation and premature aging, and demonstrated that conserved longevity pathways affect ROS levels [7]. It was therefore rather unexpected when genetic-intervention studies in model systems failed to reproducibly support this theory. For instance, deletion of all five SODs (superoxide dismutases) in Caenorhabditis elegans, a genetic intervention that was expected to dramatically shorten lifespan, turned out to be modestly lifespan-extending [8]. Deletion of SOD in mice did not shorten mammalian lifespan either [9] and overexpression of SOD2, predicted to significantly extend mammalian lifespan, equally failed to yield the expected result [10]. These studies raised considerable doubts within the aging community concerning the validity of the free radical theory of aging [11]. However, given the mounting evidence that ROS, such as peroxide, also play important roles in a wide range of different signalling processes and with that affect growth, development and differentiation [12,13] (Figure 1), these results might not be as damaging for the free radical theory as first anticipated. In fact, although it has long been accepted that too much ROS is deleterious, too little ROS seems to be detrimental as well, providing a further example for the importance of maintaining homoeostasis [14]. Under these premises, we decided to take a different approach to assess the role of ROS in lifespan and aging.
Figure 1. Keeping the balance between oxidants and antioxidants.
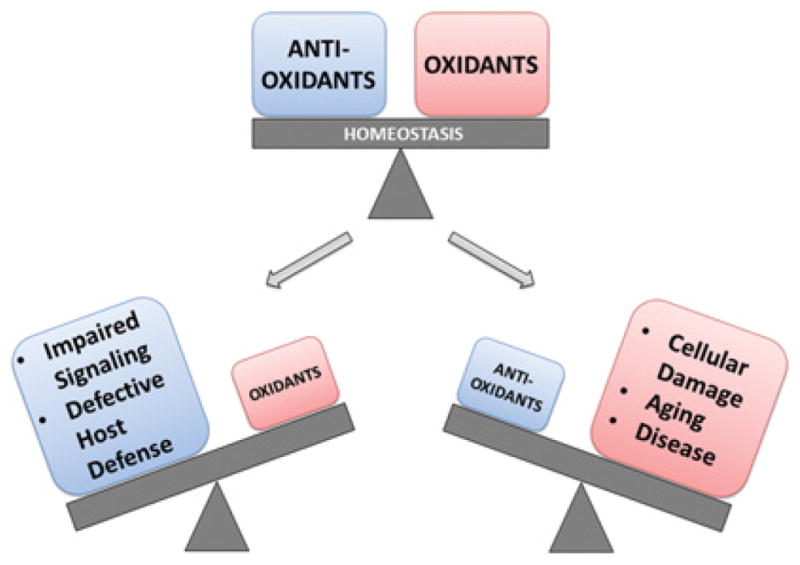
ROS, including superoxide, peroxide and hydroxyl radicals, are constantly produced as by-products of cellular processes. A number of highly conserved antioxidant systems work to detoxify excess ROS and maintain redox homoeostasis. Since ROS play important roles in signal transduction processes, too much detoxification or too little ROS production will affect growth, development and differentiation. Excess ROS production or diminished antioxidant capacity will cause widespread cellular damage and can lead to senescence and cell death.
Revisiting the free radical theory of aging
A widely used approach to assess oxidative damage in cells and organisms involves measuring the extent of protein carbonylation [15]. Protein carbonylation is a non-specific oxidative side-chain modification elicited by a range of different oxidants, including peroxide and HOCl (hypochlorous acid). It is a useful method to determine how fast treatment with select oxidants causes non-specific protein damage. However, it is still open for debate whether comparative analysis of the steady-state levels of protein carbonylation in different tissues, organisms or over time is a suitable in vivo readout for oxidative damage let alone for correlation analyses with aging or other phenotypes. Any observed increase in protein carbonylation as assessed by Western blot analysis can either be due to individual carbonylation reactions occurring on different molecules of a protein population or many carbonylation reactions taking place on one protein molecule. Deductions as to the physiological consequences of protein carbonylations are therefore almost impossible to make. Moreover, it is unclear how carbonylation affects the half-time of a given protein, implying that any increase in the steady-state levels of a carbonylated protein can reflect either an increase in oxidative damage or a decrease in the proteolytic activity of a cell. We therefore decided to take a different approach and use quantitative redox proteomics and in vivo ROS-sensing proteins to directly measure the oxidation of physiologically relevant target proteins and the levels of oxidants during the lifespan of C. elegans [16]. We applied OxICAT, a highly quantitative mass-spectrometric approach to determine the thiol oxidation status of redox-sensitive proteins in vivo [17,18]. This approach was based on the premise that most redox-regulated proteins contain one or more cysteine residues, whose thiol side chains are extremely sensitive towards even small alterations in oxidant levels [19]. They are rapidly oxidized when oxidant levels increase and re-reduced when oxidants are removed and reducing conditions restored. Since redox-sensitive cysteine residues commonly fulfil functional and/or structural roles in redox-regulated proteins, these oxidative modifications cause, either directly or indirectly, a change in protein activity [20]. Knowing the affected proteins, the location of their redox-sensitive cysteine residue thiols and the extent of thiol oxidation allows us therefore to predict the potential cellular adaptations that occur in response to changing in vivo oxidant levels. In a second independent approach, we made use of the peroxide-specific sensor protein HyPer [21], which provides a ratiometric readout of peroxide levels. We generated transgenic C. elegans strains with the chromosomally encoded HyPer sensor and directly monitored in vivo peroxide levels in live organisms over the complete lifespan. Both of our approaches led to the same unexpected result: we discovered that during its lifespan, C. elegans experiences two phases with significantly increased protein thiol oxidation and oxidant levels. The first phase occurs very early in life, encompassing all stages of larval development. The second phase, which was more expected based on the free radical theory of aging, occurred at a late stage in life. In fertile young adults, however, oxidant levels were found to be at a minimum and protein thiols are largely reduced [16]. Comparison of the peroxide levels in individual tissues revealed that this biphasic oxidation pattern is a system-wide event [16]. Comparative studies using long- and short-lived C. elegans mutants revealed a correlation between an organism’s ability to deal with increased developmental oxidant levels and lifespan [16]. The source of developmental peroxide remains to be investigated, as do the transcriptional and translational changes that allow for the accumulation of peroxide during development, and the rapid detoxification and re-establishment of redox homoeostasis during adulthood. To our astonishment, we also observed a very high variability in developmental peroxide levels when we compared individual worms of an isogenic population. On the basis of this observation and together with (i) the known lifespan variability of individuals within isogenic C. elegans populations [22], (ii) the previous findings that down-regulation of components of the respiratory chain only during development is sufficient to prolong lifespan [23], and (iii) the fact that worms show lifespan-predicting traits as early as on day 1 of adulthood [24], we propose that variances in developmental peroxide levels contribute to lifespan determination. Although our results might not fully agree with the quintessence of Harman’s free radical theory of aging [6], they do conform to the idea that ROS are extremely important players in the lifespan of organisms.
Fighting bacteria with oxidants and the ways bacteria are fighting back
One clearly very beneficial role of ROS for humans is their role in host defence. Activated by the invading pathogens, NADPH oxidases present in macrophages produce high concentrations of superoxide, which rapidly dismutes to peroxide [25]. In neutrophils, the enzyme myeloperoxidase then converts peroxide into HOCl, the active ingredient of household bleach [26]. With the same efficacy with which household bleach disinfects our countertops, endogenous HOCl kills micro-organisms within the phagosome. HOCl appears to also be produced at mucosal barrier epithelia (i.e. lining of the gut), in a process catalysed by the dual oxidase DUOX [27]. Much of our recent research has dealt with the question about what makes bleach such an effective antibacterial, and what are potential strategies that bacteria might employ to fend off this toxic insult? Once we know these strategies, we will be a major step closer to identifying alternative antimicrobial targets.
Although bleach has been used in households for more than 200 years, the molecular mechanism(s) behind its extremely effective antimicrobial power have been largely unknown. Research in our laboratory revealed that the major targets of bleach-mediated cell damage are proteins [28]. With reaction rates up to one million-fold faster than peroxide [29], HOCl reacts with and damages the side chains of proteins, leading to irreversible unfolding and protein aggregation [30]. One confounding factor under these stress conditions is that many molecular chaperones, the cellular lifeguards of proteins, also fall victim to bleach-mediated damage, either directly through bleach-mediated protein unfolding or indirectly due to the oxidative stress-mediated decrease in intracellular ATP, which leaves these chaperones devoid of their essential energy source [31]. We have now discovered that bacteria use at least two different strategies to protect their proteins against bleach-induced protein unfolding. One immediate defence mechanism involves the HOCl-induced activation of Hsp33 (heat-shock protein 33), an ATP-independent chaperone [28] (Figure 2). Intriguingly, activation of Hsp33 requires precisely the stress conditions that HOCl elicits: an oxidizing protein-unfolding environment [32]. Under reducing non-stress conditions, Hsp33 is not active as chaperone. The four absolutely conserved cysteine residues, which are located in the C-terminal redox switch domain of Hsp33, are reduced and engaged in zinc co-ordination. Upon exposure to HOCl, two intramolecular disulfide bonds form and zinc is released. The loss of stabilizing zinc binding together with structural constraints imposed by the formation of two disulfide bonds that connect near-neighbour cysteine residues appear to contribute to the unfolding of Hsp33’s C-terminus and the exposure of a high-affinity-binding platform for unfolding proteins [28,33]. Once activated by oxidative profein unfolding, Hsp33 effectively prevents protein aggregation and increases bacterial HOCl resistance. Once cells recover from the stress conditions and ATP levels are restored, ATP-dependent chaperones reactivate, supporting the refolding of client proteins released from Hsp33 [34]. Subsequently, Hsp33 returns into its chaperone-inactive state ready to jump into action when oxidative stress conditions re-occur.
Figure 2. Hsp33: a chaperone specialized to protect against bleach stress.

Exposure of organisms to the highly effective antimicrobial hypochlorous acid (HOCl) causes the oxidative unfolding of numerous cellular proteins. Hsp33, an ATP-independent redox-regulated chaperone, also undergoes oxidative protein unfolding upon bleach treatment. In the case of Hsp33, however, this unfolding is highly co-ordinated and instigated by intramolecular disulfide bond formation and zinc release. The concomitant conformational changes convert Hsp33 into an active chaperone. Once activated, Hsp33 binds to other unfolding proteins and prevents the irreversible loss of protein function (i.e. protein aggregation). This mode of action substantially increases bacterial bleach resistance.
The second defence strategy involves the universally conserved and very ancient molecule polyP (polyphosphate) [35,36] (Figure 3). This long polymer of phosphoanhydride-linked Pi (inorganic phosphate) has long been known to protect prokaryotes and eukaryotes against a variety of different stress conditions, yet its mode of action has remained enigmatic. We have discovered that, under bleach-like stress, bacteria re-direct large amounts of ATP into long chains of polyP [37]. Our data suggest that this reallocation of ATP is largely responsible for the observed oxidative stress-mediated depletion of cellular ATP. Absence of polyP [i.e. deletion of PPK (polyphosphate kinase)] makes bacteria highly sensitive to bleach treatment, indicating that polyP plays a significant role in bacterial bleach resistance [37]. Indeed, our studies have revealed that polyP also works by protecting a large variety of different proteins against stress-induced protein aggregation. We have demonstrated that polyP has protein chaperone-like properties, binding to unfolding proteins with high affinity and releasing them to other ATP-dependent chaperones once non-stress conditions are restored and polyP is re-converted into ATP [37]. It remains now to be tested how polyP stabilizes proteins, recognizes unfolding proteins and maintains proteins in a soluble state. Our finding that longer chains of polyP work significantly more efficiently than shorter chains excludes the idea of a ‘simple’ stabilization-by-binding mechanism, but suggests a more complicated mode of interaction. Mechanistic studies will elucidate the working mode of polyP, whose chaperone function has probably influenced protein folding since early in evolution.
Figure 3. Bacterial defence against bleach-induced protein unfolding.
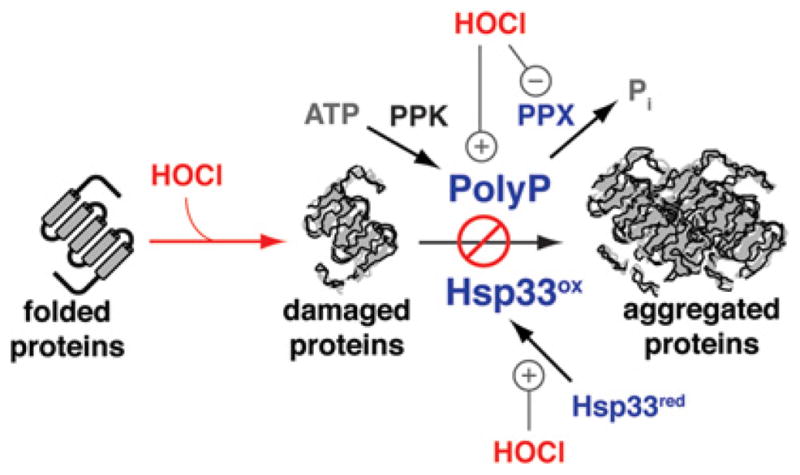
Proteins are the major targets of bleach damage in bacteria. To prevent bleach-induced protein unfolding and aggregation, bacteria employ at least two distinct chaperone systems: (i) the redox-regulated protein chaperone Hsp33, which is specifically activated by bleach, and (ii) the chemical chaperone polyP, which specifically accumulates during bleach stress conditions. PolyP is produced by PPK, which converts ATP into polyP. Bleach-induced accumulation of polyP is caused by the reversible oxidative inactivation of polyP phosphatase (PPX), which fails to hydrolyse the polymer into Pi. This mechanism explains the ATP-depletion phenotype observed in bleach-treated bacteria. red, reduced; ox, oxidized.
Acknowledgments
Funding
Our own work was supported by the National Institutes of Health (NIH) [grant numbers GM065318 and R21-AI097893 (to U.J.)], and by an NIH Ruth Kirschstein Award [grant number F32-GM096613 (to M.J.G)]. D.R. was funded by a fellowship of the Human Frontiers in Science agency. W.-Y.W. was supported by a Cellular and Molecular Biology Training Grant [grant number T32-GM007315].
Abbreviations
- Hsp33
heat-shock protein 33
- polyP
polyphosphate
- PPK
polyphosphate kinase
- ROS
reactive oxygen species
- SOD
superoxide dismutase
References
- 1.Stadtman ER. Protein oxidation in aging and age-related diseases. Ann NY Acad Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x. [DOI] [PubMed] [Google Scholar]
- 2.Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem. 2008;283:16961–16965. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 4.Chiang SM, Schellhorn HE. Regulators of oxidative stress response genes in Escherichia coli and their functional conservation in bacteria. Arch Biochem Biophys. 2012;525:161–169. doi: 10.1016/j.abb.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 7.Salmon AB, Richardson A, Perez VI. Update on the oxidative stress theory of aging: does oxidative stress play a role in aging or healthy aging? Free Radic Biol Med. 2010;48:642–655. doi: 10.1016/j.freeradbiomed.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Raamsdonk JM, Hekimi S. Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci USA. 2012;109:5785–5790. doi: 10.1073/pnas.1116158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Ikeno Y, Qi W, Chaudhuri A, Li Y, Bokov A, Thorpe SR, Baynes JW, Epstein C, Richardson A, et al. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J Gerontol A Biol Sci Med Sci. 2009;64:1212–1220. doi: 10.1093/gerona/glp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 2009;8:73–75. doi: 10.1111/j.1474-9726.2008.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y, Richardson A. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790:1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 13.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species, (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 15.Cabiscol E, Tamarit J, Ros J. Protein carbonylation: proteomics, specificity and relevance to aging. Mass Spectrom Rev. 2014;33:21–48. doi: 10.1002/mas.21375. [DOI] [PubMed] [Google Scholar]
- 16.Knoefler D, Thamsen M, Koniczek M, Niemuth NJ, Diederich AK, Jakob U. Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Mol Cell. 2012;47:767–776. doi: 10.1016/j.molcel.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Strahler JR, Andrews PC, Jakob U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci USA. 2008;105:8197–8202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumsta C, Thamsen M, Jakob U. Effects of oxidative stress on behavior, physiology, and the redox thiol proteome of Caenorhabditis elegans. Antioxid Redox Signal. 2011;14:1023–1037. doi: 10.1089/ars.2010.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groitl B, Jakob U. Thiol-based redox switches. Biochim Biophys Acta. 2014;1844:1335–1343. doi: 10.1016/j.bbapap.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV, Lukyanov S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 22.Kirkwood TB, Feder M, Finch CE, Franceschi C, Globerson A, Klingenberg CP, LaMarco K, Omholt S, Westendorp RG. What accounts for the wide variation in life span of genetically identical organisms reared in a constant environment? Mech Ageing Dev. 2005;126:439–443. doi: 10.1016/j.mad.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Dillin A, Hsu AL, Arantes-Oliveira NA, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 24.Rea SL, Wu D, Cypser JR, Vaupel JW, Johnson TE. A stress-sensitive reporter predicts longevity in isogenic populations of Caenorhabditis elegans. Nat Genet. 2005;37:894–898. doi: 10.1038/ng1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal. 2013;18:642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- 26.Winterbourn CC. Biological reactivity and biomarkers of the neutrophil oxidant, hypochlorous acid. Toxicology. 2002;181–182:223–227. doi: 10.1016/s0300-483x(02)00286-x. [DOI] [PubMed] [Google Scholar]
- 27.Ha EM, Oh CT, Bae YS, Lee WJ. A direct role for dual oxidase in Drosophila gut immunity. Science. 2005;310:847–850. doi: 10.1126/science.1117311. [DOI] [PubMed] [Google Scholar]
- 28.Winter J, Ilbert M, Graf PC, Ozcelik D, Jakob U. Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell. 2008;135:691–701. doi: 10.1016/j.cell.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 30.Gray MJ, Wholey WY, Jakob U. Bacterial responses to reactive chlorine species. Annu Rev Microbiol. 2013;67:141–160. doi: 10.1146/annurev-micro-102912-142520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winter J, Linke K, Jatzek A, Jakob U. Severe oxidative stress causes inactivation of DnaK and activation of the redox-regulated chaperone Hsp33. Mol Cell. 2005;17:381–392. doi: 10.1016/j.molcel.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 32.Ilbert M, Horst J, Ahrens S, Winter J, Graf PC, Lilie H, Jakob U. The redox-switch domain of Hsp33 functions as dual stress sensor. Nat Struct Mol Biol. 2007;14:556–563. doi: 10.1038/nsmb1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reichmann D, Xu Y, Cremers CM, Ilbert M, Mittelman R, Fitzgerald MC, Jakob U. Order out of disorder: working cycle of an intrinsically unfolded chaperone. Cell. 2012;148:947–957. doi: 10.1016/j.cell.2012.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffmann JH, Linke K, Graf PC, Lilie H, Jakob U. Identification of a redox-regulated chaperone network. EMBO J. 2004;23:160–168. doi: 10.1038/sj.emboj.7600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kornberg A, Rao NN, Ault-Riche D. Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem. 1999;68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- 36.Rao NN, Gomez-Garcia MR, Kornberg A. Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem. 2009;78:605–647. doi: 10.1146/annurev.biochem.77.083007.093039. [DOI] [PubMed] [Google Scholar]
- 37.Gray MJ, Wholey WY, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, Krieger AG, Smith EM, Bender RA, Bardwell JC, et al. Polyphosphate is a primordial chaperone. Mol Cell. 2014;53:689–699. doi: 10.1016/j.molcel.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]