

Abstract
Colorectal cancer has emerged as a major cause of death in Western countries. Down-regulation of β-catenin expression has been considered a promising approach for cytotoxic drug formulation. Eight 4,9-friedodrimane-type sesquiterpenoids (1–8) were acquired using the oxidative potential of Verongula rigida on bioactive metabolites from two Smenospongia sponges. Compounds 3 and 4 contain a 2,2-dimethylbenzo[d]oxazol-6(2H)-one moiety as their substituted heterocyclic residues, which is unprecedented in such types of meroterpenoids. Gauge-invariant atomic orbital NMR chemical shift calculations were employed to investigate stereochemical details with support of the application of advanced statistics such as CP3 and DP4. Compounds 2 and 8 and the mixture of 3 and 4 suppressed β-catenin response transcription (CRT) via degrading β-catenin and exhibited cytotoxic activity on colon cancer cells, implying that their anti-CRT potential is, at least in part, one of their underlying antineoplastic mechanisms.
Approximately 140 000 people are diagnosed annually with colorectal cancer, and among them 50 000 patients have died of this malignant disease in the U.S. alone during the past decade.1 Molecular lesions in the adenomatous polyposis coli (APC) gene, the largest structural core protein of the β-catenin destruction complex, are observed frequently in colorectal cancer.2,3 This alteration results in stabilization of cytosolic β-catenin, a multifunctional protein capable of binding to the T-cell factor/lymphocyte enhancer factor (Tcf/LEF) family of transcription factors to induce gene expression.3 The gene targets of this expression, activated by β-catenin response regulated transcription (CRT), include c-Myc and cyclin D1, which play pivotal roles in colorectal tumorigenesis and malignancy.3 Since APC is a negative regulator directly upstream of β-catenin,3 only a few signaling components of the Wnt/β-catenin pathway can be targeted for development of clinical intervention for such oncogenicity. Among these candidates, an accelerated turnover of β-catenin has been recognized as a pertinent protocol for cytotoxic agent development.
Diverse pharmacophores have been derived from natural products, providing evidence for structural prerequisites for the disruption of the oncogenic β-catenin function. For instance, we demonstrated that galangin, a flavanone abundantly present in propolis, suppresses CRT via expediting degradation of cytosolic β-catenin, verifying the disruptive action on Tcf/β-catenin complexes as at least one of its cytotoxic mechanisms on certain human carcinomas.4 Moreover, ellagitannins (ETs) were shown to retard Tcf/LEF-dependent transcription activated by a mutant β-catenin construction, corroborating that the consumption of an ET-enriched diet results in a decline of colon cancer.5 Unlike terrestrial organisms,4,5 marine organisms producing effective cytotoxic secondary metabolites6 have been underutilized for exploration of potential modulators of the oncogenic Wnt/β-catenin pathway. For instance, sponge-derived 4,9-friedodrimane sesquiterpenoids exhibited potent antiproliferative activities via activation of DNA damage-inducible gene 153 and hypoxia-inducible factor-1.7,8 Yet, the inhibitory potential on β-catenin-associated cellular pathways as the underlying cytotoxic mechanisms of the meroterpenoids has rarely been investigated.9
Biotransformation is a rational approach to provide a diverse and new series of derivatives that could be employed for the generation of chemical libraries to implement preliminary lead optimization and SAR studies.10 Particularly, sponges in the order Verongida, featuring unusual biochemical profiles exemplified by the absence of terpenoids and the production of sterols and brominated compounds, exhibited strong oxidative potential. Such characteristics render those sponges as optimal sources to derivatize bioactive molecules for drug screening.
In ongoing efforts targeting the search for sponge-derived potent anticancer prototypes,9,11 the isolation and structural characterization of eight 4,9-friedodrimane-type sesquiterpenoids generated by biotransformation utilizing the oxidative potential of a Verongida sponge (Verongula rigida) on 4,9-friedodrimanes, typically isolated from Smenospongia sponges, are discussed herein. The potential of those meroterpenoids to be developed into colon cancer inhibitory agents capable of suppressing CRT via promoting the degradation of β-catenin is also addressed.
Results and Discussion
It was reported recently that ilimaquinone, a 4,9-friedodrimane-type sesquiterpenoid, and its analogues exhibited antiproliferative activity via suppressing β-catenin expression.9 These results initiated an exploration of new 4,9-friedodrimane derivatives capable of down-regulating expression of the oncogenic protein from marine resources. In an effort to generate new 4,9-friedodrimanes, the aggressive oxidative properties of a Verongida sponge, which are well established to undergo rapid oxidation and rearrangements when exposed to the atmosphere, were employed.12Smenospongia sponges producing 4,9-friedodrimanes (Smenospongia aurea and S. cerebriformis) and V. rigida were homogenized and incubated together at room temperature to facilitate the formation of new analogues from this series.13 This approach was successful in generating a number of new 4,9-friedodrimane derivatives for bioassay. The LC-MS analysis of the incubated mixtures of these three sponges exhibited unreported quasimolecular ions (m/z 384.2 and 398.2; Figure S1, Supporting Information) from identified friedodrimanes, which prompted further purification of the extract for identification of possible new inhibitors of β-catenin expression. Fractionation and purification of the extract afforded eight new 4,9-friedodrimane-type sesquiterpenoids (1–8) (Figure 1). This report reveals the unique utility that the Verongida sponges may have on metabolism, lead optimization, and SAR studies.
Figure 1.

Structures of compounds 1–8. The ethoxy groups in 5–8 could be derived from ethanol used for extraction.
The HRFABMS of compound 1, which exhibited a quasimolecular ion at m/z 384.2540 (calcd [M + H]+, m/z 384.2539), and its 13C NMR data led to its molecular formula being established as C24H33NO3. The 1H and 13C NMR data (Table 1) exhibited resonances for a pentasubstituted aromatic moiety (δH 6.98; δC 98.8, 109.2, 132.3, 143.7, 144.6, 146.6, 162.0), an exomethylene (δH 4.36, 4.32; δC 102.7, 160.5), a methoxy (δH 3.90; δC 56.6), and four methyl groups (δH 0.91, 1.04, 1.04, 2.54; δC 14.6, 18.5, 17.7, 20.7). Inspection of the 2D NMR spectra indicated the presence of a 4,9-friedodrim-4(11)-ene-type sesquiterpenoid framework13 and a benzoxazole residue. The heterocyclic residue was identical to that of 5-epi-nakijinol C, a recently isolated sesquiterpenoid from the Indonesian sponge Dactylospongia metachromia,14 based on the deshielded 13C NMR chemical shift values of C-20 (δC 132.3), C-21 (δC 146.6), and C-22 (δC 162.0) and the HMBC correlation from H3-23 (δH 2.54) to C-22 (δC 162.0).14 HMBC correlations between H2-15 (δH 2.80, 2.89) and C-16, C-17, and C-21 (δC 109.2, 143.7, 146.6) indicated that the benzoxazole moiety is connected to C-15. The relative configuration of the core framework was established on the basis of analysis of the NOESY spectrum. Key NOESY correlations were detected from H-1ax (δH 1.51) to H3-12 (δH 1.04) and H3-14 (δH 0.91), from H-2ax (δH 1.22) to H-10 (δH 0.92), and from H3-12 (δH 1.04) and H3-13 (δH 1.04) to H3-14 (δH 0.91), demonstrating that compound 1 is related structurally to the trans-4,9-friedodrimanes (Figure 2).13 This NOESY-based configurational analysis was corroborated by the shielded 13C NMR chemical shift value of C-12 (δC 20.7), in view of previous studies verifying that 13C NMR chemical shift values of C-12 in trans-decalins are more shielded than those in cis-decalins (Δ ca. 10 ppm).14−16 The absolute configuration of 1 was deduced from the consistent absolute configuration of trans-4,9-friedodrimane meroterpenoids obtained from natural sources and confirmed based on the similar specific rotation value of 1 with those of reported meroterpenoids (Figure 3).13 The trivial name (−)-nakijinol E was assigned to 1.
Table 1. 1H (600 MHz) and 13C NMR (150 MHz) Data for Compounds 1–8 in CDCl3.
| |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| position | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz)b | δC | |
| 1 | ax | 1.51, m | 23.4 | 1.94, m | 23.0 | 1.42, m | 23.0 | 1.85, m | 22.7 | 1.40, m | 22.1 | 1.81, m | 21.6 | 1.91, m | 21.5 | 1.84, m | 22.7 |
| eq | 2.27, m | 2.30, m | 2.13, m | 2.19, m | 2.09, m | 1.54, m | 2.14, m | 2.10, tda | |||||||||
| 2 | ax | 1.22, m | 28.9 | 1.82, m | 25.3 | 1.20, m | 28.8 | 1.77, m | 25.2 | 1.29, m | 28.5 | 1.72, m | 24.7 | 1.74, m | 24.8 | 1.65, m | 25.1 |
| eq | 1.87, m | 1.68, m | 1.84, m | 1.65, m | 1.85, m | 1.61, m | 1.66, m | 1.76, m | |||||||||
| 3 | ax | 2.33, t (7.5) | 33.2 | 2.44, dd (14, 5.4) | 32.2 | 2.29, dt (14, 5.2) | 33.2 | 2.42, dt (14, 6.6) | 32.2 | 2.27, m | 33.2 | 2.44, m | 32.1 | 2.45, m | 32.0 | 2.10, tda | 32.1 |
| eq | 2.02, m | 2.11, dt (14, 6.6) | 2.04, m | 2.10, dd (14, 4.8) | 2.06, m | 2.09, m | 2.11, m | 2.41, td (13.2, 6.0) | |||||||||
| 4 | 160.5 | 153.9 | 161.0 | 153.7 | 160.7 | 153.5 | 153.9 | 153.7 | |||||||||
| 5 | 40.6 | 39.6 | 40.5 | 39.6 | 40.3 | 39.4 | 39.5 | 39.7 | |||||||||
| 6 | ax | 1.19, m | 36.5 | 0.93, dt (14, 3.3)a | 37.8 | 1.30, m | 36.9 | 1.01, dt (14, 3.0)a | 38.2 | 1.46, m | 37.0 | 1.19, m | 38.3 | 1.19, m | 38.2 | 1.07, td (14.1, 3.0)a | 38.1 |
| eq | 1.43, m | 1.92, m | 1.47, m | 1.97, br d (14) | 1.55, ma | 2.06, m | 2.06, m | 1.99, br d (13.8) | |||||||||
| 7 | ax | 1.39, m | 28.2 | 1.50, m | 28.0 | 1.36, m | 28.1 | 1.46, m | 28.0 | 1.40, m | 27.6 | 1.52, m | 27.7 | 1.52, m | 27.6 | 1.19, m | 28.0 |
| eq | 1.17, m | 1.15, m | 1.45, m | 1.24, m | 1.24, m | 1.48, m | |||||||||||
| 8 | 1.40, m | 37.4 | 1.33, m | 38.7 | 1.22, m | 38.1 | 1.11, m | 39.1 | 1.40, m | 37.0 | 1.29, m | 38.6 | 1.39, m | 38.2 | 1.19, m | 39.7 | |
| 9 | 43.1 | 44.5 | 42.9 | 44.2 | 39.7 | 41.1 | 41.2 | 45.0 | |||||||||
| 10 | 0.92, m | 49.4 | 1.40, d (6.6) | 47.6 | 0.80, m | 50.0 | 1.17, d (6.0)a | 47.8 | 0.87, br d (10.2) | 49.0 | 1.43, d (6.0) | 47.0 | 1.25, d (6.0)a | 47.0 | 1.19, m | 48.7 | |
| 11 | a | 4.36, br s | 102.7 | 4.69, br s | 105.8 | 4.40, br s | 102.4 | 4.68, br s | 105.8 | 4.45 br s | 102.6 | 4.70, br s | 106.1 | 4.70, br s | 105.9 | 4.68, br s; 4.65, br s | 105.9 |
| b | 4.32, br s | 4.66, br s | 4.38, br s | 4.65, br s | 4.68, br s | 4.68, br s | |||||||||||
| 12 | 1.04, s | 20.7 | 0.98, s | 33.2 | 1.01, s | 20.7 | 1.01, s | 33.3 | 1.03, s | 20.6 | 1.12, s | 33.0 | 1.09, s | 33.2 | 1.04, s | 33.4 | |
| 13 | 1.04, da | 18.5 | 1.01, d (6.6) | 18.8 | 0.97, d (6.6) | 18.0 | 0.95, d (6.6) | 18.4 | 0.72, da | 16.5 | 0.90, d (6.6) | 16.6 | 0.71, d (6.6) | 16.7 | 0.92, d (6.0) | 18.5 | |
| 14 | 0.91, s | 17.7 | 0.95, s | 19.0 | 0.81, s | 17.5 | 0.85, s | 19.0 | 0.72, s | 17.8 | 0.80, s | 19.2 | 0.79, s | 19.3 | 0.86, s | 18.8 | |
| 15 | 2.80, d (14) | 34.6 | 2.83, d (14) | 34.7 | 2.37, d (14) | 32.7 | 2.47, d (14) | 32.5 | 1.00, dd (15.6, 4.2) | 33.9 | 1.25, dd (16, 4.8)a | 33.9 | 1.05, dd (16, 3.6) | 33.9 | 2.47, d (13.2) | 33.0 | |
| 2.89, d (14) | 2.96, d (14) | 2.53, d (14) | 2.51, d (14) | 2.04, dd (15.0, 3.6)a | 1.99, dd (16, 3.6) | 2.07, dd (16, 4.8)a | 2.57, d (13.2) | ||||||||||
| 16 | 109.2 | 109.4 | 110.3 | 110.1 | 2.61, t (4.2) | 54.9 | 2.68, t (4.8)a | 54.8 | 2.67, t (4.2) | 54.7 | 117.7 | ||||||
| 17 | 143.7 | 143.6 | 181.6 | 181.57 | 80.6 | 80.6 | 80.5 | 153.3 | |||||||||
| 18 | 144.6 | 144.5 | 158.9 | 158.9 | 5.92, s | 121.9 | 5.93, s | 122.0 | 5.93, s | 121.9 | 182.7 | ||||||
| 19 | 6.98, s | 98.8 | 6.98, s | 98.8 | 6.14, s | 96.9 | 6.16, s | 96.89 | 158.9 | 159.0 | 159 | 5.82, s | 102.4 | ||||
| 20 | 132.3 | 132.5 | 155.6 | 155.6 | 198.5 | 198.4 | 198.7 | 161.2 | |||||||||
| 21 | 146.6 | 146.7 | 161.4 | 161.5 | 174.4 | 174.4 | 174.4 | 182.3 | |||||||||
| 22 | 162.0 | 162.0 | 116.2 | 116.19 | 4.15, m | 63.4 | 4.17, m | 63.4 | 4.17, m | 63.4 | 4.04, q (7.2) | 66.1 | |||||
| 4.26, m | 4.26, m | 4.26, m | |||||||||||||||
| 23 | 2.54, s | 14.6 | 2.54, s | 14.6 | 1.58, s | 25.9 | 1.61, s | 25.8 | 1.21, t (7.2) | 14.3 | 1.21, ta | 14.3 | 1.21, ta | 14.3 | 1.49, t (7.2) | 14.0 | |
| 24 | 1.60, s | 26.0 | 1.61, s | 25.8 | |||||||||||||
| OH | 5.87, s | 5.85, s | 3.93, s | 3.95, br s | 3.95, br s | 7.42, s | |||||||||||
| OCH3 | 3.90, s | 56.6 | 3.91, s | 56.7 | 3.83, s | 56.4 | 3.84, s | 56.5 | 3.72, s | 57.4 | 3.73, s | 57.4 | 3.73, s | 57.4 | |||
Partially overlapped.
Diastereotopic protons may be interchangeable.
Figure 2.
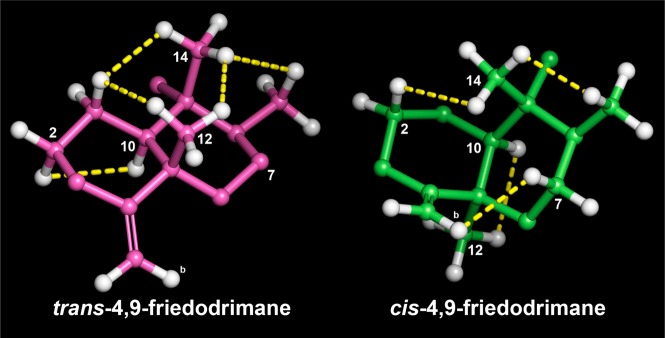
NOESY analysis to establish trans- and cis-4,9-friedodrim-4(11)-ene moieties found in this study. Minimization was implemented at the MMFF level employing MacroModel (Schrodinger LLC), and yellow dotted lines indicate NOE correlations.
Figure 3.

Establishment of absolute configuration of optically pure compounds in this study (i.e., compounds 1, 2, 5, and 8). The trans-friedodrimanes (−)-nakijinol E (1) and (−)-dactylospongenone E (5) displayed negative specific rotations in accord with those of (−)-ilimaquinone8,25 and (−)-dactylospongenone A.17 Inversion of the C-5 absolute configuration with formation of the cis-friedodrimane core structure led to a significant conformational change that is exemplified by a consistent change in the sign of the optical rotation. Thus, the specific rotations of (+)-5-epi-nakijinol E (2) and (+)-5-epi-ethylsmenoquinone (8) are consistent with those of the analogous (+)-5-epi-ilimaquinone.15,26
The molecular formula of compound 2 was established as C24H33NO3 based on 13C NMR and HRFABMS data (obsd [M + H]+, m/z 384.2542; calcd [M + H]+, m/z 384.2539), implying that compound 2 is an isomer of 1. The 1D NMR spectra were similar to those of 1 except for the more deshielded C-12 resonance (δC 33.2) compared to 1 (δC 20.7). Such deshielding of the methyl carbon in 4,9-friedodrim-4(11)-ene sesquiterpenoids is indicative of inversion of configuration of the C-5 stereogenic center, validating that compound 2 is the C-5 epimer of 1. Key NOESY correlations were detected from H3-14 (δH 0.95) to H-2ax (δH 1.82) and H3-13 (δH 1.01), from H-7ax (δH 1.50) to H-11b (δH 4.66), and from H3-12 (δH 0.98) to H-10 (δH 1.40), confirming the presence of a cis-4,9-friedodrimane architecture in 2 (Figure 2).13 The absolute configuration of compound 2 was established as for 1 (Figure 3), and the trivial name (+)-5-epi-nakijinol E was assigned to 2.
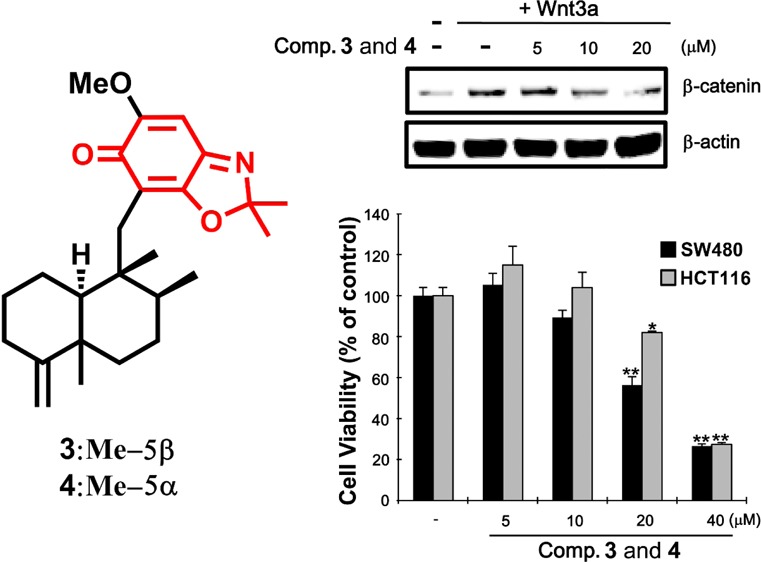
Compounds 3 and 4 were obtained as a 1:2 mixture, with the ratio being established via the integration of corresponding 1H NMR resonances for each compound. The HRFABMS data of this mixture displayed a quasimolecular ion at m/z 398.2696, which, in conjunction with the 13C NMR data, led to assignment of their same molecular formula as C25H35NO3 (calcd [M + H]+, m/z 398.2695). The 1H and 13C NMR data of compounds 3 and 4 exhibited several common features to those of 1 and 2 aside from the presence of a resonance for an additional methyl group (δH 1.58, 1.61; δC 25.8, 25.9) in 3 and 4. This suggested that compounds 3 and 4 possess 4,9-friedodrim-4(11)-ene scaffolds with different C-15 substituents. The HMBC correlations from H2-15 to C-16, C-17, and C-21 and from H3-23 and H3-24 to C-22 (Figure 4) revealed that the C-23 methyl groups of the benzoxazole moieties in 1 and 2 are replaced with gem-dimethyl moieties in 3 and 4. The 13C NMR chemical shift values of C-12 were observed as δC 20.7 and 33.3, establishing trans- and cis-junctions of the decalin rings in 3 and 4, respectively. This was validated by the NOESY analysis shown in Figure 2. When considered in conjunction with the consistent biosynthetic pathways toward the 4,9-friedodrim-4(11)-ene-type meroterpenoids derived from natural sources,13 the absolute configurations of compounds 3 and 4 are presumably identical to those of the trans- and cis-4,9-friedodrim-4(11)-enes, respectively. Thus, these substances, based on 4,9-friedodrimanes with a 2,2-dimethylbenzo[d]oxazol-6(2H)-one moiety, possess unique scaffolds, and the trivial names nakijinone A and 5-epi-nakijinone A were assigned to compounds 3 and 4, respectively.
Figure 4.

Selected HMBC correlations for 3 and 4.
The HRFABMS data of compound 5 displayed an [M + H]+ ion at m/z 405.2638 (calcd [M + H]+, m/z 405.2641), which, in conjunction with the 13C NMR data, led to assignment of the molecular formula, C24H36O5. The 1D NMR data were similar to those of compounds 1–4, substantiating the presence of a 4,9-friedodrimane framework. The substituted moieties in 5 showed close similarities with the cyclopentenone moieties in dactylospongenones,15,17 similar merosesquiterpenoids from the Palau and Fijian sponges Dactylospongia spp., based on the observed resonances for an isolated olefinic methine group (δH 5.92, δC 121.9), keto and ester carbonyl groups (δC 198.5, 174.4), a methoxy group (δH 3.72, δC 57.4), and an oxygenated tertiary carbon (δC 80.6). Compound 5 exhibited resonances corresponding to an ethoxy group (δH 1.21, 4.15, 4.26; δC 14.3, 63.4) instead of the methoxy groups in dactylospongenones, implying that the substituted heterocyclic moiety in 5 carries an ethyl ester group. This was verified by HMBC correlations from H2-22 (δH 4.15, 4.26) to C-21 (δC 174.4) and C-23 (δC 14.3) and from H-18 (δH 5.92) and H-16 (δH 2.61) to C-21 (δC 174.4). The HMBC and COSY spectra indicated that the planar structure of 5 is identical with those of dactylospongenones A–D,15,17 except for the ethyl ester moiety. The shielded C-12 resonances (δC 20.6) indicated that the new meroterpenoid has a trans-decalin moiety, and this was corroborated further by the NOESY correlations shown in Figure 2. The similar specific rotation and NMR data of 5 to those of dactylospongenone A (Figure 3), except for resonances derived from the ethoxy moiety, imply that 5 is the ethyl ester equivalent of the methyl ester, dactylospongenone A. Thus, compound 5 was assigned the trivial name (−)-dactylospongenone E.
Compounds 6 and 7 were obtained as a 2:3 mixture based on the integration of associated 1H NMR resonances for each compound. The HRFABMS data exhibited a single quasimolecular ion at m/z 405.2637 (calcd [M + H]+, m/z 405.2641). In conjunction with the 13C NMR data, this indicated that these components are isomers with a C25H36O5 molecular formula. The 1D NMR spectra showed close similarities with those of 5 except for the deshielded 13C NMR chemical shift values of the C-12 resonances in compounds 6 and 7 (δC 33.0, 33.2), indicating that they are structurally based on cis-friedodrim-4(11)-ene with C-15 cyclopentenone moieties.15,17
The stereochemical assignment of the C-15 cyclopentenone moieties was attempted by utilizing gauge-invariant atomic orbital (GIAO) NMR chemical shift calculations coupled with advanced statistics CP3 and DP4 analyses,18,19 given characteristically variable chemical shift values of diastereomers associated with a cyclopentenone moiety. Prior to application of the computational approach, the method was verified for the investigation of stereochemical details of avarane-type sesquiterpenoids. Each pair of epimers addressed in this study (i.e., 1 vs 2, 3 vs 4) was used for the validation process employing GIAO NMR chemical shift calculations coupled with CP3. Major conformers were generated by conformational searches using Macromodel (Schrodinger LLC) and subjected to GIAO shielding constant calculations at the B3LYP/6-31G(d,p) level employing the Gaussian 09 package (Gaussian Inc.) (computational details and Table S1, Supporting Information). These calculated chemical shift values were Boltzmann-averaged according to their relative MMFF94 potential energy, and the averaged values were provided for the calculation of CP3 analysis (computational details, Tables S1–S3, Supporting Information). CP3 supported the NMR-based assignment made for each pair of epimers with 100% probability (Figures S2 and S3, Supporting Information), validating that the application of such advanced statistics is a relevant tool in establishing stereochemical details of sesquiterpenoids addressed in this study. With the in silico method corroborated, the four cis-4,9-friedodrimane-based diastereomers associated with the C-15 cyclopentenone moieties of 6 and 7 (I: 16R, 17S; II: 16S, 17R; III: 16R, 17R; IV: 16S, 17S, Figure 5) were considered and subjected to chemical shift calculation coupled with DP4 analysis because experimental data were available for only two diastereomers. DP4 application with calculated chemical shift values of diastereomers I–IV (Table S4, Supporting Information) supported the structural equivalence of diastereomer I with 7 with 100% probability (Figure S4, Supporting Information), and the consecutive application with the remaining candidates (II–IV) suggested with 100% certainty (Figure S6, Supporting Information) that the absolute configuration of 6 was identical to diastereomer II. These new merosesquiterpenoids were assigned the trivial names 5-epi-dactylospongenones E (6) and F (7).
Figure 5.

Plausible diastereomers of four cis-4,9-friedodrimane-based diastereomers associated with the C-15 cyclopentenone moieties of 6 and 7.
The molecular formula of compound 8 was deduced as C23H32O4 based on 13C NMR data and HRFABMS analysis (obsd [M + H]+, m/z 373.2381; calcd [M + H]+, m/z 373.2379). The NMR data displayed the presence of a 5α-methyl-4,9-friedodrimane moiety (δC 33.4) carrying a C-15 dioxygenated-1,4-benzoquinone moiety (δH 5.82; δC 102.4, 117.7, 153.3, 161.2, 182.3, 182.7) and, hence, showing structural resemblance with 5-epi-ilimaquinone.15 The NMR data of these two compounds differ in resonances of an ethoxy group (δH 1.49, 4.04; δC 14.0, 66.1) in 8 instead of a methoxy moiety in 5-epi-ilimaquinone. This suggests that compound 8 is an ethyl ether homologue of 5-epi-ilimaquinone, which is confirmed by the HMBC correlations from H2-22 (δH 4.04) to C-20 (δC 161.2). The specific rotation of compound 8 is similar to that of 5-epi-ilimaquinone. This permitted the definition of an absolute configuration identical to that of 5-epi-ilimaquinone (Figure 3). The trivial name (+)-5-epi-20-O-ethylsmenoquinone was assigned to 8.
Over the past two decades, many natural products with hydroquinone/benzoquinone-type moieties appended to a sesquiterpene or diterpene skeleton have been reported from marine sponges.13 The isolation of 4,9-friedodrimane-type sesquiterpenoids 5, and 6 and 7 indicate that a competitive intramolecular Michael addition might be associated in the formation of these secondary metabolites. As illustrated in Figure 6, the intramolecular addition of enolate B onto the C-20 carbonyl would result in the generation of 5 and 7 or 6, depending on re- or si-face addition (path A in Figure 6). Alternatively, the addition of the enolate onto the C-21 carbonyl group and tautomerization followed by transetherification would lead to the formation of 8 (path B in Figure 6). In addition to these oxidative metabolites, natural products such as nakijiquinones, smenospongine, and smenospongidine, featuring hydroquinones attached to a sesquiterpene or diterpene scaffold, are also known to be generated from marine organisms.13 In this regard, reductive amination of D followed by Schiff’s base formation with acetone or acetaldehyde and consecutive isomerization would generate F and G, respectively. Finally, the oxidation of these two intermediates would lead to the more stable benzoxazole moieties in 1–4, which might be facilitated by the oxidative properties of V. rigida utilized in the current biotransformation study.
Figure 6.

Plausible biosynthetic pathways involved in the generation of 1–8.
Aberrant activation of the Wnt/β-catenin pathway accompanied by accumulation of intracellular β-catenin and subsequent activation of CRT is important in the development and progression of colorectal cancer.2−4 Owing to limited amounts of isolated compounds, only compounds 2, 3, 4, and 8 were evaluated for their potential to serve as prototypes of new anticolorectal cancer agents capable of tuning the Wnt/β-catenin pathway. To evaluate their inhibitory activities on the target pathway, HEK293-firefly luciferase (FL) reporter cells stably transfected were constructed with TOPflash, a synthetic β-catenin/Tcf-dependent FL reporter, and human Frizzled-1 (hFz-1) expression plasmids.20 Incubation of HEK293-FL reporter cells with different concentrations of compounds 2 and 8 and the mixture of 3 and 4 decreased the CRT levels that had been activated upon Wnt3a-conditioned medium (Wnt3a-CM) treatment (Figure 7A).
Figure 7.

(A) HEK293-FL reporter cells were incubated with the active molecules in the presence of Wnt3a-CM for 15 h, and luciferase activity was determined. (B) Cytosolic proteins were prepared from HEK293-FL reporter cells treated with the vehicle or the active meroterpenoids in the presence of Wnt3a-CM for 15 h and subjected to Western blotting. (C) Two CRT-positive colon cancer cell lines were incubated with different concentrations of the active meroterpenoids, and cell viability was determined by Cell-Titer-Glo assay. *p < 0.05 and **p < 0.01, compared with the vehicle control group.
Based on previous studies corroborating that CRT is predominantly dependent on expression levels of intracellular β-catenin,4 Western blot analysis with anti-β-catenin antibody in HEK293 reporter cells treated with the active compounds 2, 3, 4, and 8 was conducted to examine whether these secondary metabolites can reduce intracellular β-catenin levels. Consistent with previous results,4 β-catenin expression was increased upon treatment with Wnt3a-CM (Figure 7B). Incubation of HEK293 cells with those active molecules mitigated up-regulated cytosolic β-catenin expression (Figure 7B). Interestingly, the inhibitory potency of the compounds on β-catenin expression was in accord with their suppressing effects on Wnt3a-CM-activated CRT, suggesting that the active compounds may inhibit the Wnt/β-catenin pathway via promoting cytosolic β-catenin degradation.
The specific reduction of β-catenin has been shown to suppress the proliferation of CRT-positive colon cancer cells.4 To validate if the active sesquiterpenoids retard proliferation of colon cancer cells via the observed inhibition on the Wnt/β-catenin pathway, two CRT-positive colon cancer cell lines, SW480 and HCT116, were treated with various concentrations of the compounds. Compound 2 and the mixture of compounds 3 and 4 started to exert antiproliferative activity at concentrations more than 20 μM, while compound 8 began to inhibit tumor growth at 0.75 and 1.5 μM against SW480 (IC50 = 3.24 μM) and HCT116 cells (IC50 = 2.95 μM), respectively (Figure 7C), demonstrating that 8 displayed more potent cytotoxic activity on the CRP-positive cancer cells than the other compounds. It is thus plausible that the underlying mechanism of the antineoplastic properties of the tested meroterpenoids is closely involved in their mitigating activities on the Wnt/β-catenin pathway considering that the observed cytotoxic potency on the colon cancer cells is consistent with their down-regulating potential against β-catenin expression elevated by Wnt3a-CM.
In conclusion, marine invertebrates could be a promising resource in providing drug frameworks modulating the oncogenic Wnt/β-catenin pathway, which may warrant further exploration of such marine organisms for the discovery of colon cancer inhibitory leads. The current study demonstrates the potential of chemical oxidation of the cell contents of a Verongida sponge to expand the diversity of 4,9-friedodrimane metabolites capable of suppressing the Wnt/β-catenin signaling pathway. Such an approach may also be applicable in similar studies with other marine organisms.
The current findings show that the 4,9-friedodrimane chemotype could serve as a drug template capable of exerting antineoplastic properties via inhibition of β-catenin expression. Further structural refinements in selective anticancer drug design can build on this study, presumably in conjunction with insights acquired from well-characterized biochemical mechanisms of colorectal tumorigenesis.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a JASCO DIP-1000 automatic digital polarimeter (Tokyo, Japan). UV–vis spectra were measured on an Agilent 1100 series diode array and multiple-wavelength detectors. The NMR spectra were recorded on a Varian 600 MHz (VNS 600) spectrometer equipped with a 5 mm direct detection pulsed field gradient probe. All NMR experiments were conducted at 294 K using CDCl3 as a solvent and referenced by residual solvent signals for CHCl3 (δH = 7.24, δC = 77.16). HRFABMS data were obtained from the Korea Basic Science Institute (Daegu) on a JEOL JMS 700 high-resolution mass spectrometer, and LC-MS profiling was performed utilizing an Agilent 1100 HPLC system with a Phenomenex Luna 5 μm C18 column (4.6 × 250 mm) eluted with an MeOH–H2O gradient solvent system and a Bruker Daltonics microTOF mass spectrometer. Vacuum-liquid chromatography (VLC) was performed on Merck silica gel (70–230 mesh). Medium-pressure liquid chromatography (MPLC) was carried out using a Biotage Isolera equipped with a reversed-phase C18 SNAP cartridge KP-C18-HS (120 g, Biotage, Charlotte, NC, USA). High-performance liquid chromatography (HPLC) was implemented on a Gilson system using Luna C18 (250 × 21.20 mm, 10 μm), Luna C18 (250 × 4.60 mm, 5 μm), YMC C18 (250 × 4.60 mm, 5 μm), and Mightysil C18 columns (150 × 4.60 mm, 5 μm). Thin-layer chromatography (TLC) was executed on glass plates precoated with silica gel F254 (20 × 20 cm, 200 μm, 60 Å, Merck).
Animal Material
Smenospongia aurea (08FL-20-B), Smenospongia cerebriformis (08FL-20), and Verongula rigida (08FL-20-A) were collected from Key Largo, FL, in October 2008. The sponges were collected from a shallow coral reef habitat between 3 and 24 m depth11 and identified by Dr. Michelle Kelly (National Institute of Water and Atmospheric Research, Auckland, New Zealand). Voucher specimens under the aforementioned codes have been deposited in the University of Mississippi.
Extraction and Isolation
Smenospongia aurea and S. cerebriformis were homogenized and incubated with V. rigida in ethanol for a week. The dried ethanol extract (3.6 kg) of the mixture of three sponges was subjected to silica gel VLC (36 kg, 14 (H) × 17.5 (D) cm) and eluted with a stepwise gradient of hexanes (100%), hexanes–acetone (80:20, 60:40, 50:50, 40:60, 20:80), acetone (100%), acetone–MeOH (80:20, 60:40, 50:50), MeOH (100%), MeOH–H2O (50:50), and H2O (100%) to give 13 fractions (Fr. 1–13). Fraction 10 (39.3 g) was further divided into nine fractions (Fr. 10-1–10-9) using silica gel VLC (12 (H) × 17.5 (D) cm) with a mixture of hexanes–acetone (95:5, 90:10, 85:15, 80:20), MeOH (100%), and MeOH–H2O (50:50). Fraction 10-7 (3.7 g) was applied to C18 MPLC (15.5 × 4 cm) with an isocratic condition of MeOH–H2O (85:15) to yield six subfractions (Fr. 10-7-1–10-7-6). Fraction 10-7-3 (115.8 mg) was chromatographed over C18 HPLC (250 × 21.20 mm, 10 μm) with MeOH–H2O (83:17) to produce three fractions (Fr. 10-7-3-1–10-7-3-3). Fraction 10-7-3-2 (12.4 mg) was purified using C18 HPLC (250 × 4.60 mm and 150 × 4.60 mm, 5 μm, connected in line) with MeOH–H2O (75:25) to obtain 2 (2.2 mg, tR = 223 min) and a mixture of 3 and 4 (4.6 mg, tR = 208 min). Fraction 10-7-3-3 (9.7 mg) was subjected to C18 HPLC (250 × 4.60 mm and 150 × 4.60 mm, 5 μm, connected in line) with MeOH–H2O (78:22) followed by C18 HPLC (250 × 4.60 mm, 5 μm) with gradient conditions of MeOH–H2O (80:20 → 100:0) for 120 min to afford 1 (1.8 mg, tR = 151 min). Fraction 10-7-1 was chromatographed over C18 HPLC (250 × 21.20 mm, 10 μm) with MeOH–H2O (78:22) to afford a mixture of 6 and 7 (1.6 mg, tR = 92 min) as well as 5 (1.5 mg, tR = 100 min). Fraction 10-5 was combined with Fr. 10-6 based on their similar TLC patterns and fractionated to generate seven subfractions (Fr. 10-6-1–10-6-7) by C18 MPLC (15.5 × 4 cm) with an isocratic condition of MeOH–H2O (85:15). Fraction 10-6-2 was sequentially purified using C18 HPLC (250 × 21.20 mm, 10 μm) with MeOH–H2O (78:22), C18 HPLC (250 × 4.60 mm, 5 μm) with MeOH–H2O (75:25), and C18 HPLC (250 × 4.60 mm, 5 μm) with MeOH–H2O (73:27) to give 8 (1.1 mg).
(−)-Nakijinol E: 18-methoxy-22-methyl-16-[{(5S,8S,9R,10S)-5,8,9-trimethyl-4-methylenedecahydronaphthalen-9-yl}methyl]benzo[d]oxazol-17-ol (1):
white, amorphous solid; [α]D25 −29 (c 0.1, MeOH); UV (MeOH) λmax 295 nm; 1H and 13C NMR, see Table 1; HRFABMS m/z 384.2540 [M + H]+ (calcd for C24H34NO3, 384.2539), 406.2357[M + Na]+ (calcd for C24H33NO3Na, 406.2358).
(+)-5-epi-Nakijinol E: 18-methoxy-22-methyl-16-[{(5R,8S,9R,10S)-5,8,9-trimethyl-4-methylenedecahydronaphthalen-9-yl}methyl]benzo[d]oxazol-17-ol (2):
pale yellow, amorphous solid; [α]D25 +22 (c 0.1, MeOH); UV (MeOH) λmax 294 nm; 1H and 13C NMR, see Table 1; HRFABMS m/z 384.2542 [M + H]+ (calcd for C24H34NO3, 384.2539).
Mixture of nakijinone A and 5-epi-nakijinone A: 18-methoxy-22,22-dimethyl-16-[{(5R,8S,9R,10S)- and (5S,8S,9R,10S)-5,8,9-trimethyl-4-methylenedecahydronaphthalen-9-yl}methyl]benzo[d]oxazol-17(2H)-one (3 and 4):
yellow, amorphous solid; [α]D25 +21 (c 0.1, MeOH); UV (MeOH) λmax 297 nm; 1H and 13C NMR, see Table 1; HRFABMS m/z 398.2696 [M + H]+ (calcd for C25H36NO3, 398.2695), 420.2509 [M + Na]+ (calcd for C25H35NO3Na, 420.2515).
(−)-Dactylospongenone E: (16R,17S)-17-hydroxy-19-methoxy-20-oxo-16-[{(5S,8S,9R,10S)-5,8,9-trimethyl-4-methylenedecahydronaphthalen-9-yl}methyl]cyclopent-18-ene-17-carboxylate (5):
yellow, amorphous solid; [α]D25 −91 (c 0.1, MeOH); UV (MeOH) λmax 246, 283 nm; 1H and 13C NMR, see Table 1; HRFABMS m/z 405.2638 [M + H]+ (calcd for C24H37O5, 405.2641), 427.2464 [M + Na]+ (calcd for C24H36O5Na, 427.2460).
Mixture of 5-epi-dactylospongenones E and F: ethyl (16R,17S)- and (16S,17R)-17-hydroxy-19-methoxy-20-oxo-16-[{(5R,8S,9R,10S)-5,8,9-trimethyl-4-methylenedecahydronaphthalen-9-yl}methyl]cyclopent-18-ene-17-carboxylate (6 and 7):
yellow, amorphous solid; [α]D25 +5 (c 0.1, MeOH); UV (MeOH) λmax 247, 287 nm; 1H and 13C NMR, see Table 1; HRFABMS m/z 405.2637 [M + H]+ (calcd for C24H37O5, 405.2641), 427.2463 [M + Na]+ (calcd for C24H36O5Na, 427.2460).
(+)-5-epi-20-O-Ethylsmenoquinone: 20-ethoxy-17-hydroxy-16-[{(5R,8S,9R,10S)-5,8,9-trimethyl-4-methylenedecahydronaphthalen-9-yl}methyl]cyclohexa-16,19-diene-18,21-dione (8):
dark yellow, amorphous solid; [α]D25 +39 (c 0.1, MeOH); UV (MeOH) λmax 288 nm; 1H and 13C NMR, see Table 1; HRFABMS m/z 373.2381 [M + H]+ (calcd for C23H33O4, 373.2379), 395.2203 [M + Na]+ (calcd for C23H32O4Na, 395.2198).
Computational Details18,19,21
All conformational searches were implemented using the Macromodel (version 9.9, Schrodinger LLC) program with “mixed torsional/low mode sampling” in the MMFF force field. The searches were conducted in the gas phase with a 50 kJ/mol energy window limit and 10 000 maximum number of steps to thoroughly examine all low-energy conformers. The Polak–Ribiere conjugate gradient method was utilized for minimization processes with 10 000 maximum iterations and a 0.001 kJ (mol Å)−1 convergence threshold on the rms gradient. Conformers within 10 kJ/mol of each global minimum of compounds (i.e., compounds 1–4 in Figure 1 and diastereomers I–IV in Figure 5) were used for GIAO shielding constant calculations without geometry optimization employing the Gaussian 09 package (Gaussian Inc.) at the B3LYP/6-31G(d,p) level in the gas phase. Calculated chemical shift values were acquired based on the following equation: δcalcdx = (σ0 – σx)/(1 – σ0/106), in which δcalcd is the calculated chemical shift value for nucleus x (e.g., 1H or 13C) and σx and σ0 are the calculated isotropic constants for nucleus x and tetramethylsilane (TMS), respectively. In particular, the geometry of TMS was optimized using the B3LYP/6-31G(d,p) level in the gas phase using the Gaussian 09 package (Gaussian Inc.) for unbiased and consistent comparison of calculated chemical shift values with those of other molecules in this study according to the original authors’ recommendation.19 These calculated chemical shift values of compounds 1–4 and diastereomers I–IV were averaged based on their Boltzmann populations (Tables S1–S4, Supporting Information) and used for calculations of CP3 and DP4 analysis employing applets available at http://www-jmg.ch.cam.ac.uk/tools/nmr/ and http://www-jmg.ch.cam.ac.uk/tools/nmr/DP4/, respectively. In application of the statistical analyses, calculated and experimental chemical shift values of diastereotopic protons were compared with their closest matches based on the original author’s recommendation.19 The 3D images in Figure 2 were illustrated utilizing Pymol 1.6.x (Schrödinger LLC.).
Cell Culture, Reporter Assays, and Chemicals
HEK293 (human embryonic kidney cell), HCT116 (human colon carcinoma), and SW480 (human colon adenocarcinoma) cells were acquired from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 120 μg/mL penicillin, and 200 μg/mL streptomycin. HEK293-FL reporters (TOPFlash) and controls (FOPFlash) were established as described previously.22,23 The Wnt3a-CM was prepared as described previously.22 The luciferase assay was implemented using the Dual Luciferase Assay kit (Promega, Fitchburg, WI, USA).
Plasmid Constructs, siRNA, and Transfection
The hFz-1 cDNA was constructed with reference to a previous protocol.20 The pTOPFlash and pFOPFlash reporter plasmids were purchased from Millipore Corporation (Billerica, MA, USA).
Western Blots
The cytosolic fraction was generated as described previously.24 Proteins were separated utilizing SDS-polyacrylamide gel electrophoresis in a 4% to 12% gradient gel (Invitrogen, Grand Island, NY, USA) and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked with 5% nonfat milk and probed with anti-β-catenin (BD Transduction Laboratories, Lexington, KY, USA) and anti-actin antibodies (Cell Signaling Technology, Danvers, MA, USA). The membranes were incubated with horseradish-peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG and visualized employing the enhanced chemiluminescence (ECL) system (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Cell Viability Assay
The HCT116 and SW480 cells were inoculated into 96-well plates and treated with compounds 1–8 for 48 h. The cell viability from each treated sample was assessed in triplicate using a CellTiter-Glo assay kit (Promega) according to the manufacturer’s instructions.
Acknowledgments
Financial support of this study was provided from the National Research Foundation of Korea Grant by the Korean Government (NRF-2010-0020484, 2014R1A2A2A01006793). Other financial support was granted from Kraft Foods and NIH grant R01AT007318. We thank Drs. A. Kochanowska-Karamyan and J. Sims for collecting the sponge samples and the Mississippi Center for Supercomputing Research (MCSR) for providing supercomputer access.
Supporting Information Available
NMR spectra, HRFABMS data, computational details, and Cartesian coordinates of all conformers. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# I. H. Hwang and J. Oh contributed equally.
The authors declare no competing financial interest.
Dedication
Dedicated to Dr. William Fenical of Scripps Institution of Oceanography, University of California–San Diego, for his pioneering work on bioactive natural products.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- National Cancer Institute. http://seer.cancer.gov/ accessed on 4/10/2104.
- Satoh S.; Daigo Y.; Furukawa Y.; Kato T.; Miwa N.; Nishiwaki T.; Kawasoe T.; Ishiguro H.; Fujita M.; Tokino T. Nat. Genet. 2000, 24, 245–250. [DOI] [PubMed] [Google Scholar]
- Lepourcelet M.; Chen Y.-N. P.; France D. S.; Wang H.; Crews P.; Petersen F.; Bruseo C.; Wood A. W.; Shivdasani R. A. Cancer Cell 2004, 5, 91–102. [DOI] [PubMed] [Google Scholar]
- Gwak J.; Oh J.; Cho M.; Bae S. K.; Song I.-S.; Liu K.-H.; Jeong Y.; Kim D.-E.; Chung Y.-H.; Oh S. Mol. Pharmacol. 2011, 79, 1014–1022. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Li L.; Celver J.; Killian C.; Kovoor A.; Seeram N. P. J. Agric. Food Chem. 2009, 58, 3965–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinski T. F.; Dalisay D. S.; Lievens S. L.; Saludes J. P. Nat. Rev. Drug Discovery 2009, 8, 69–85. [DOI] [PubMed] [Google Scholar]
- Lu P.-H.; Chueh S.-C.; Kung F.-L.; Pan S.-L.; Shen Y.-C.; Guh J.-H. Eur. J. Pharmacol. 2007, 556, 45–54. [DOI] [PubMed] [Google Scholar]
- Du L.; Zhou Y.-D.; Nagle D. G. J. Nat. Prod. 2013, 76, 1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Yun E.; Hwang I. H.; Yoon S.; Kim D.-E.; Kim J.; Na M.; Song G.-Y.; Oh S. Mar. Drugs 2014, 12, 3231–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann M. T. Curr. Pharm. Des. 2003, 9, 879–889. [DOI] [PubMed] [Google Scholar]
- Hwang I. H.; Oh J.; Kochanowska-Karamyan A.; Doerksen R. J.; Na M.; Hamann M. T. Tetrahedron Lett. 2013, 54, 3872–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasu S. S.; Yeung B. K.; Hamann M. T.; Scheuer P. J.; Kelly-Borges M.; Goins K. J. Org. Chem. 1995, 60, 7290–7292. [Google Scholar]
- Menna M.; Imperatore C.; D’Aniello F.; Aiello A. Mar. Drugs 2013, 11, 1602–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daletos G.; de Voogd N. J.; Müller W. E.; Wray V.; Lin W.; Feger D.; Kubbutat M.; Aly A. H.; Proksch P. J. Nat. Prod. 2014, 77, 218–226. [DOI] [PubMed] [Google Scholar]
- Rodríguez J.; Quiñoá E.; Riguera R.; Peters B. M.; Abrell L. M.; Crews P. Tetrahedron 1992, 48, 6667–6680. [Google Scholar]
- Salmoun M.; Devijver C.; Daloze D.; Braekman J. C.; Gomez R.; De Kluijver M.; Van Soest R. J. Nat. Prod. 2000, 63, 452–456. [DOI] [PubMed] [Google Scholar]
- Kushlan D. M.; Faulkner D. J.; Parkanyi L.; Clardy J. Tetrahedron 1989, 45, 3307–3312. [Google Scholar]
- Smith S. G.; Goodman J. M. J. Org. Chem. 2009, 74, 4597–4607. [DOI] [PubMed] [Google Scholar]
- Smith S. G.; Goodman J. M. J. Am. Chem. Soc. 2010, 132, 12946–12959. [DOI] [PubMed] [Google Scholar]
- Cho M.; Gwak J.; Park S.; Won J.; Kim D.-E.; Yea S. S.; Cha I.-J.; Kim T. K.; Shin J.-G.; Oh S. FEBS Lett. 2005, 579, 4213–4218. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Oh J.; Wonhwa L.; Kwak S.; Li W.; Chittiboyina A. G.; Ferreira D.; Hamann M. T.; Lee S. H.; Bae J.-S. Biochim. Biophys. Acta, Gen. Subj. 2014, 1840, 2042–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Gwak J.; Cho M.; Song T.; Won J.; Kim D.-E.; Shin J.-G.; Oh S. Mol. Pharmacol. 2006, 70, 960–966. [DOI] [PubMed] [Google Scholar]
- Ryu M.-J.; Cho M.; Song J.-Y.; Yun Y.-S.; Choi I.-W.; Kim D.-E.; Park B.-S.; Oh S. Biochem. Biophys. Res. Commun. 2008, 377, 1304–1308. [DOI] [PubMed] [Google Scholar]
- Dignam J. D.; Lebovitz R. M.; Roeder R. G. Nucleic Acids Res. 1983, 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner S. D.; Radeke H. S.; Tallarico J. A.; Snapper M. L. J. Org. Chem. 1995, 60, 1114–1115. [Google Scholar]
- Carte B.; Rose C. B.; Faulkner D. J. J. Org. Chem. 1985, 50, 2785–2787. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.