

Abstract
Two new cyclic depsipeptides, companeramides A (1) and B (2), have been isolated from the phylogenetically characterized cyanobacterial collection that yielded the previously reported cancer cell toxin coibamide A (collected from Coiba Island, Panama). The planar structures of the companeramides, which contain 3-amino-2-methyl-7-octynoic acid (Amoya), hydroxy isovaleric acid (Hiva), and eight α-amino acid units, were established by NMR spectroscopy and mass spectrometry. The absolute configuration of each companeramide was assigned using a combination of Marfey’s methodology and chiral-phase HPLC analysis of complete and partial hydrolysis products compared to commercial and synthesized standards. Companeramides A (1) and B (2) showed high nanomolar in vitro antiplasmodial activity but were not overtly cytotoxic to four human cancer cell lines at the doses tested.
Cyanobacteria are well known as sources of cytotoxins and have yielded medicinally relevant metabolites with antiproliferative and neurologically active properties.1 Many cyanobacterial cytotoxins are nonribosomally encoded, structurally complex depsipeptides occurring in suites of structurally related analogues. Considerable variation in the level of biological activity among structural analogues is related to differences in substituents and/or absolute configuration, as exemplified by the apratoxins2 and grassypeptolides.3 Accurate determination of the absolute configuration is thus critical, but is made challenging by the biosynthetic incorporation of diverse polyketide-derived segments and nonproteinogenic α- and β- as well as d amino and hydroxy acids, in cyanobacterial depsipeptides, which stem from the convergence of multiple biosynthetic pathways.4 A more recent pharmacological theme emerging for marine cyanobacterial peptides is antiplasmodial activity, as reviewed by Peach and Linington5 as well as others, and is of particular interest when accompanied by low cytotoxicity to mammalian cells. Such differential activity of the natural products to parasite versus host cells has been reported for alkynoic linear lipopeptides related to the dragonamides,6 the cyclic hexapeptide venturamides,4 and gallinamide7 (which is identical to symplostatin 48,9) from Panamanian cyanobacteria. These and other Panamanian marine cyanobacteria have been investigated for their natural products chemistry as part of the International Cooperative Biodiversity Groups (ICBG) project focused on Panamanian biodiversity as a source of tropical disease treatments and antiproliferative agents.
The chemical investigation of previously unexplored marine cyanobacteria in Coiba National Park, Panama, is ongoing and to date has yielded the selective HDAC inhibitory santacruzamates from a cf. Symploca species,10 the polyketide δ-lactone coibacins from a cf. Oscillatoria species,11 which exhibit selective antileishmanial activity, and the potent cancer cell toxin coibamide A.12 The latter antiproliferative depsipeptide was reported previously from a field collection of filamentous cyanobacteria identified morphologically as comprising primarily a Leptolyngbya species,12 although the major organism identified by phylogenetic analysis (16S rDNA) in the field collection was more similar to the Symploca type strains (PAC-10-3, GenBank KC207936)13 and has been proposed to belong to a new genus.14 Collections of the same cyanobacterial assemblage have consistently provided relatively high yields of the two new cyclic depsipeptides, companeramides A (1) and B (2), so named to convey their repeated isolation as companion (Spanish “compañera”) compounds to coibamide, while avoiding their association with a cyanobacterial genus name that may be subject to change. Companeramides A (1) and B (2) show differential activity against chloroquine-sensitive and -insensitive strains of the malarial parasite Plasmodium falciparum with little cytotoxicity to human cancer cell lines.

Results and Discussion
A marine cyanobacterial assemblage comprising a small filament Leptolyngbya species, based on morphological analysis, was collected in 2004 by hand using scuba from a reef pinnacle in Coiba National Park, Panama. The alcohol-preserved tissue was extracted with CH2Cl2–MeOH, and the extract fractionated by normal-phase silica gel vacuum liquid chromatography (NP-VLC). In preliminary biological activity profiling, the 100% EtOAc fraction was cytotoxic to NCI-H460 human lung tumor cells with an IC50 of 300 ng/mL and also showed preliminary activity in the ICBG panel of antiparasitic assays: malaria (IC50 6 μg/mL), American trypanosomiasis (IC50 35 μg/mL), and leishmaniasis (IC50 > 50 μg/mL). RP18 solid-phase extraction (SPE), HPLC, and further cytotoxicity testing of this fraction resulted in the discovery of the potent antiproliferative metabolite coibamide A,12 to which was attributed the preliminary antiparasitic activities observed for the parent fraction. However, 1H NMR analysis of the RP18-SPE subfractions indicated the presence of additional, but unrelated, peptidic metabolites in the 70% MeOH–H2O subfraction preceding that containing coibamide A (90% MeOH–H2O). HPLC purification of this more polar SPE fraction has afforded two new cyclic depsipeptides, companeramides A (1) and B (2), subsequently found to be responsible for the observed antiparasitic activity of the parent fraction.
Laboratory culture of the 2010 field-collected cyanobacterial assemblage yielded predominantly companeramides A (1) and B (2), with a relatively low yield of coibamide A during the first six months of culture. Microscopic examination of these cultures also revealed that a small-filament cyanobacterium was the dominant organism in culture, over its companion large-filament cyanobacterium. Beyond six months in laboratory culture, this assemblage no longer produced coibamide A, but continued to produce the companeramides, and microscopic examination revealed the presence of almost exclusively small filaments, with only sporadic large filaments in the cultured material (Figure S21, Supporting Information). Subsequently a monoculture of this small-filament cyanobacterium was achieved that could be associated with companeramide production (PAC-10-3 csf1, GenBank KM882611). Phylogenetically, this organism is most closely related to a taxonomically unassigned filamentous cyanobacterium associated with Black Band Disease (FLK9, GenBank EU196364, see Supporting Information Figures S19 and S20).
Companeramide A (1) yielded a prominent [M + H]+ ion by HRTOFMS for a molecular formula of C57H97N9O11, which was supported by NMR spectroscopic analysis. The 1H NMR spectrum for 1 exhibited signals typical for a relatively hydrophobic cyanobacterial peptide metabolite, including four N-methyl singlets (δH 3.14, 3.06, 2.92, 2.69), four NH doublets (δH 8.86, 6.92, 6.91, 6.75), nine α-H multiplets (δH 4.58–5.26), and numerous overlapped methyl doublets (δH 0.77–1.32, Table 1). The 13C NMR spectrum for 1 displayed nine distinguishable signals for ester/amide-type carbonyls (δC 169.4–174.4), one signal for an oxygenated sp3-hybridized carbon (δC 75.4), and two signals at δC 83.8 and 68.6, diagnostic of a terminal acetylenic moiety (Table 1). The HMBC spectrum displayed prominent three-bond HMBC correlations from the four N-methyl singlets, permitting N-methyls H3-15, H3-25, H3-42, and H3-52 to be associated with corresponding α-carbons C-11, C-20, C-38, and C-50, respectively. The side chain spin systems of these four N-methylated amino acid residues were delineated by multiplicity-edited HSQC and HSQC-TOCSY experiments as two N-methyl valine (N-Me-Val), one N-methyl leucine (N-Me-Leu), and one N-methyl alanine (N-Me-Ala) residue. Further analysis of the COSY, HSQC, HSQC-TOCSY, and HMBC experiments identified the regular amino acids alanine (Ala), proline (Pro), and two isoleucines (Ile), as well as hydroxyisovaleric acid (Hiva) (Table S1, Supporting Information). The molecular formula for 1 dictated a tenth residue comprising C9H13NO. COSY and TOCSY NMR experiments were used to identify a spin system in which a methine doublet of quartets (δH 2.51, H-2) was correlated to both a methyl doublet (δH 1.31, H3-9) and a second downfield methine multiplet (δH 3.97, H-3). This H-3 multiplet was in turn relay-coupled to the signals for three contiguous methylenes (H2-4 to H2-6) and also to an NH doublet at δH 6.75 (NH-3; Figure 1). HMBC correlations from the distal methylene 1H signal (H2-6, δH 2.17) to nonprotonated sp-hybridized C-7 (δC 83.8) and methine C-8 (δC 68.6) signals completed the hydrocarbon chain with a terminal acetylene. Finally, three-bond HMBC correlations from the H3-9 doublet to methine C-3 (δC 51.2) and the C-1 carbonyl signal (δC 174.3) aided in the establishment of this α-methyl-β-amino acid as 3-amino-2-methyl-7-octynoic acid (Amoya).
Table 1. 1H and 13C NMR Data for Companeramides A (1) and B (2) in CDCl3 (700 MHz).
| companeramide
A (1) |
companeramide
B (2) |
||||||
|---|---|---|---|---|---|---|---|
| unit | position | δH, mult. (J, Hz) | δC, mult. | unit | position | δH, mult. (J, Hz) | δC, mult. |
| AMOYA | 1 | 174.3, C | AMOYA | 1 | 175.4, C | ||
| 2 | 2.51, dq (7.2, 7.2) | 46.4, CH | 2 | 2.63, m | 46.0, CH | ||
| 3 | 3.97, m | 51.2, CH | 3 | 3.81, m | 52.4, CH | ||
| 3-NH | 6.75, br d (6.5) | 3-NH | 7.44, d (6.0) | ||||
| 4 | 1.71, obs | 31.3, CH2 | 4 | 1.93, m | 30.2, CH2 | ||
| 1.63, obs | 1.73, m | ||||||
| 5 | 1.54, m | 25.2, CH2 | 5 | 1.53, m | 25.5, CH2 | ||
| 1.48, obs | 1.45, m | ||||||
| 6 | 2.17, obs | 17.9, CH2 | 6 | 2.18, m | 18.0, CH2 | ||
| 7 | 83.8, C | 7 | 83.4, C | ||||
| 8 | 1.89, t (2.6) | 68.6, CH | 8 | 1.91, obs | 69.4, CH | ||
| 9 | 1.31, d (7.5) | 14.2, CH3 | 9 | 1.36, obs | 14.2, CH3 | ||
| N-Me-Val-1 | 10 | 170.1, C | N-Me-Val-1 | 10 | 170.2, C | ||
| 11 | 4.58, d (11.2) | 62.8, CH | 11 | 4.72, d (11.2) | 62.4, CH | ||
| 12 | 2.29, obs | 25.6, CH | 12 | 2.27, m | 26.5, CH | ||
| 13 | 0.95, d (6.4) | 19.4, CH3 | 13 | 0.96, obs | 19.9, CH3 | ||
| 14 | 0.88, d (6.7) | 18.4, CH3 | 14 | 0.81, obs | 18.5, CH3 | ||
| 15 | 3.14, s | 30.2, CH3 | 15 | 3.23, s | 30.9, CH3 | ||
| Ala | 16 | 174.4, C | Val-1 | 16 | 173.4, C | ||
| 17 | 4.72, obs | 46.2, CH | 17 | 4.58, dd (8.3, 7.8) | 55.5, CH | ||
| 17-NH | 8.86, d (6.3) | 17-NH | 8.84, d (7.8) | ||||
| 18 | 1.32, d (7.0) | 16.4, CH3 | 18 | 2.13, m | 30.5, CH | ||
| N-Me-Leu | 19 | 169.4, C | 19 | 0.91, obs | 18.8, CH3 | ||
| 20 | 4.67, dd (9.7, 4.9) | 58.6, CH | 20 | 0.93, obs | 18.7, CH3 | ||
| 21 | 1.83, ddd (14.5, 8.5, 4.9) | 36.7, CH2 | N-Me-Ala-1 | 21 | 169.8, C | ||
| 1.59, m | |||||||
| 22 | 1.41, obs | 24.2, CH | 22 | 4.96, q (6.8) | 56.1, CH | ||
| 23 | 0.92, d (6.6) | 22.2, CH3 | 23 | 1.32, d (6.8) | 15.3, CH3 | ||
| 24 | 0.91, d (6.8) | 24.3, CH3 | 24 | 2.74, s | 29.1, CH3 | ||
| 25 | 2.69, s | 28.8, CH3 | Pro | 25 | 171.8, C | ||
| Pro | 26 | 172.4, C | 26 | 4.83, obs | 55.4, CH | ||
| 27 | 4.92, dd (7.4, 6.4) | 55.4, CH | 27 | 2.10, m | 29.8, CH2 | ||
| 2.04, m | |||||||
| 28 | 2.11, m | 29.6, CH2 | 28 | 2.29, m | 25.6, CH2 | ||
| 2.00, m | 1.97, m | ||||||
| 29 | 2.28, obs | 25.6, CH2 | 29 | 3.83, m | 48.0, CH2 | ||
| 1.94, m | 3.72, m | ||||||
| 30 | 3.84, m | 48.0, CH2 | Ile | 30 | 170.2, C | ||
| 3.76, dt (9.9, 7.5) | |||||||
| Ile-1 | 31 | 170.0, C | 31 | 4.76, m | 54.4, CH | ||
| 32 | 4.74, dd (8.9, 6.9) | 54.0, CH | 31-NH | 7.48, d (8.5) | |||
| 32-NH | 6.91, d (8.9) | 32 | 1.70, obs | 39.0, CH | |||
| 33 | 1.69, obs | 38.6, CH | 33 | 0.93, obs | 15.5, CH3 | ||
| 34 | 0.92, d (6.5) | 15.4, CH3 | 34 | 1.39, m | 24.7, CH2 | ||
| 1.06, m | |||||||
| 35 | 1.39, m | 24.3, CH2 | 35 | 0.87, obs | 11.7, CH3 | ||
| 1.06, obs | |||||||
| 36 | 0.83, dd (7.5, 4.8) | 11.0, CH3 | N-Me-Val-2 | 36 | 169.3, C | ||
| N-Me-Val-2 | 37 | 169.5, C | 37 | 4.90, d (11.1) | 62.0, CH | ||
| 38 | 4.71, obs | 61.7, CH | 38 | 2.33, m | 25.8, CH | ||
| 39 | 2.27, obs | 25.7, CH | 39 | 0.98, obs | 19.9, CH3 | ||
| 40 | 0.94, d (6.3) | 19.9, CH3 | 40 | 0.78, d (6.5) | 18.7, CH3 | ||
| 41 | 0.77, d (6.8) | 17.9, CH3 | 41 | 3.06, s | 30.8, CH3 | ||
| 42 | 3.06, s | 30.5, CH3 | Val-2 | 42 | 172.4, C | ||
| Ile-2 | 43 | 172.3, C | 43 | 4.67, dd (9.3, 8.9) | 54.7, CH | ||
| 44 | 4.72, obs | 53.0, C | 43-NH | 6.80, d (9.3) | |||
| 44-NH | 6.92, d (8.8) | 44 | 2.00, m | 31.1, CH | |||
| 45 | 1.74, obs | 37.3, CH | 45 | 0.93, obs | 19.2, CH3 | ||
| 46 | 0.84, d (6.7) | 15.0, CH3 | 46 | 0.91, obs | 18.4, CH3 | ||
| 47 | 1.48, obs | 23.4, CH2 | N-Me-Ala-2 | 47 | 170.3, C | ||
| 1.07, obs | |||||||
| 48 | 0.82, dd (7.4, 5.0) | 11.0, CH3 | 48 | 5.40, q (7.0) | 52.2, CH | ||
| N-Me-Ala | 49 | 170.1, C | 49 | 1.35, obs | 13.8, CH3 | ||
| 50 | 5.26, q (7.3) | 51.7, CH | 50 | 2.89, s | 30.7, CH3 | ||
| 51 | 1.31, d (7.3) | 13.5, CH3 | Hiva | 51 | 170.8, C | ||
| 52 | 2.92, s | 30.5, CH3 | 52 | 4.80, d (7.7) | 75.5, CH | ||
| Hiva | 53 | 171.1, C | 53 | 2.19, m | 30.1, CH | ||
| 54 | 4.80, d (8.3) | 75.4, CH | 54 | 1.08, d (6.9) | 18.2, CH3 | ||
| 55 | 2.19, obs | 30.1, CH | 55 | 1.00, d (6.5) | 18.2, CH3 | ||
| 56 | 1.04, d (6.6) | 18.6, CH3 | |||||
| 57 | 0.93, d (5.7) | 17.8, CH3 | |||||
Figure 1.

Key 2D NMR correlations for (A) companeramide A (1) and (B) companeramide B (2).
Despite several closely overlapping α-proton signals (H-17, H-32, H-38, H-44) correlated to overlapped carbonyl signals (C-10, C-31, and C-49) in the HMBC spectrum for companeramide A (1), all nine amino and one hydroxy acid subunits were sequenced from HMBC and ROESY data collected at 700 MHz. Two obvious fragments consisting of N-Me-Val-1–Ala–N-Me-Leu–Pro (fragment 1) and Ile-1–N-Me-Val-2–Ile-2–N-Me-Ala–Hiva (fragment 2) were assembled by HMBC correlations between N-methyl, amide, and/or α-H and the carbonyl 13C signals for neighboring residues (Figure 1 and Table S1, Supporting Information). At the N-terminus of fragment 2, the ester linkage between Hiva and the Amoya residue was defined by HMBC correlations from the Hiva α-H (H-54, δH 4.80), Amoya α-H (H-2, δH 2.51), and Amoya methyl (H3-9, δH 1.31) to the Amoya carbonyl C-1 signal (δC 174.3). Finally, it remained to connect fragment 1 and the extended fragment 2. ROESY correlations between the Pro H2-30 multiplets (δH 3.84 and 3.76) and the Ile-1 α-H-32 doublet of doublets (δH 4.74) corroborated an HMBC correlation from the Pro α-H-27 doublet of doublets (δH 4.92) to the overlapped Ile carbonyl C-31 signal (δC 170.0). Although HMBC experiments with varying delay times failed to show correlations from the Amoya NH-3 (δH 6.75) or α-H to any carbonyl 13C NMR signals, a ROESY correlation between the NH-3 signal and δH 4.58 (α-H-11) was used to connect the Amoya and N-Me-Val-1 residues to complete the planar macrocyclic structure of companeramide A (1) (Figure 1A). MS/MS data for 1 were consistent with the proposed amino acid sequence (Figure S15, Supporting Information).
Companeramide B (2) gave a prominent HRESIMS peak at m/z 1056.7013, which was 28 mass units less than the major [M + H]+ ion for companeramide A (1) and indicated a molecular formula of C55H93N9O11 for 2. The 1H NMR spectrum for 2 displayed four N-methyl amide singlets (δH 3.23, 3.06, 2.89, 2.74), four amide proton doublets (δH 8.84, 7.48, 7.44, 6.80), and midfield multiplets integrating to 10 α-H (δH 3.81–5.40). In the 13C NMR spectrum for 2 there were two signals indicative of a terminal acetylene (δC 83.4 and 69.4), as for 1, and eight distinct signals for ester/amide carbonyls, one of which was noticeably broad and of relatively higher intensity. Analysis of the 2D NMR data for companeramide B (2), including the COSY, HSQC, HSQC-TOCSY, and HMBC spectra, revealed a similar structural composition to 1, with the presence of Amoya, two N-Me-Val, Pro, Ile, and Hiva residues (Tables 1 and S2, Supporting Information). The HSQC-TOCSY spectrum for 2 additionally defined two Val spin systems, each comprising an amide (NH-17 or NH-43), two coupled methines, and two methyl 1H NMR signals. Two remaining sets of coupled α-methine and methyl doublet 1H NMR signals could be attributed to two N-Me-Ala residues on the basis of HMBC correlations from the N-methyl singlets at δH 2.74 (H3-24) and 2.89 (H3-50) to their respective coupled α-methine signals (δH 4.96, H-22; δ 5.40, H-48).
As in the case of 1, the sequential assembly of amino and hydroxy acids in 2 was complicated due to a number of closely overlapping α-H chemical shifts (H-11 and H-31, H-11 and H-43, H-31 and H-52) correlated to closely overlapped carbonyl signals (C-10, C-30, and C-47) in the HMBC spectrum. In this case, HMBC and ROESY correlations supported a fragment 1 sequence of N-Me-Val-1–Val-1–N-Me-Ala-1–Pro (Figure 1B). Although the order of Val-1 and N-Me-Ala-1 in 2 was originally in question, when compared to the corresponding second and third residues (Ala and N-Me-Leu, respectively) in 1, key HMBC correlations were present from the N-CH3-15 singlet (δH 3.23, N-Me-Val-1) to the C-16 carbonyl signal (δC 173.4, Val-1), from the α-H-17 triplet (δH 4.58, Val-1) to carbonyl C-21 (δC 169.8 N-Me-Ala-1), and from the N-CH3-24 singlet (δH 2.74, N-Me-Ala-1) to the C-25 carbonyl signal (δC 171.8, Pro). In addition, MS/MS fragments of m/z 183.11 (b2), 282.18 (b3), and 395.26 (b4) (Figure S16, Supporting Information) supported the assigned fragment 1 sequence for 2. This consistent pattern of N-methylation for 1 and 2 is biosynthetically logical. HMBC and ROESY correlations supported an extended fragment 2 for 2, in which the Ile present in 1 was substituted for a Val, providing a sequence of Ile-1–N-Me-Val-2–Val-2–N-Me-Ala–Hiva. Again, the two fragments could be connected by a ROESY correlation between the Pro methylene (H2-29) signal and Ile α-H-31 signal in combination with a HMBC correlation from the Pro α-H-26 signal (δH 4.83) to the overlapped Ile carbonyl C-30 signal (δC 170.2). Finally, the macrocycle could be closed by a ROESY correlation between the signals at δH 7.44 (Amoya NH-3) and 4.72 (α-H-11) together with an HMBC correlation from the NH-3 doublet to the C-10 carbonyl signal (δC 170.2) of N-Me-Val-1. The resulting planar structure of companeramide B (2, Figure 1B) was again supported by MS/MS data (Figure S16, Supporting Information).
The absolute configurations of the amino and hydroxy acid units in companeramides A (1) and B (2) were determined by both Marfey’s methodology and direct chiral-phase HPLC analyses of acid hydrolysates (0.5 mg, 6 N HCl, 110 °C, overnight). For each natural product, a portion (ca. 0.25 mg) of the acid hydrolysate was derivatized with N-α-(2,4-dinitro-5-fluorophenyl)-l-alaninamide (l-FDAA, Marfey’s reagent) and analyzed by comparative RP18 HPLC with FDAA-derivatized d- and l-amino acid standards. The additional 0.25 mg of each natural product acid hydrolysate was reconstituted with H2O, analyzed by chiral-phase HPLC, and compared with the retention times of authentic R- and S-Hiva standards. For companeramide A (1), an l-configuration for Ala, N-Me-Ala, Pro, and both Ile, N-Me-Leu, and N-Me-Val residues, as well as S-Hiva, was established. The hydrolysate from companeramide B (2) contained l-Pro, two l-N-Me-Val, two l-Val, l-Ile, and both d- and l-N-Me-Ala, as well as S-Hiva.
In order to correctly assign the relative position of the d- and l-N-Me-Ala residues in companeramide B (2), a partial hydrolysis and purification of resulting fragments containing N-Me-Ala was attempted, for subsequent complete hydrolysis and Marfey’s analyses. To this end, 2 (4 mg) was partially hydrolyzed with 3 N HCl, and the product mixture analyzed by LC-MS to identify fragments containing a single N-Me-Ala residue. A tetrapeptide fragment (m/z 413.9) identified as N-Me-Val–Val–N-Me-Ala–Pro from MS/MS data (Figure S17, Supporting Information) was purified from the product mixture and subjected to complete hydrolysis and derivatization with N-α-(2,4-dinitro-5-fluorophenyl)-l-leucinamide (l-FDLA, advanced Marfey’s reagent) for analysis. The retention times for the derivatized amino acids from the latter (tetrapeptide fragment 1 of 2) corresponded to FDLA-derivatized standards for N-Me-l-Val, l-Val, N-Me-d-Ala, and l-Pro. No other partial hydrolysis fragments could be isolated in reasonable amount for Marfey’s analysis; however, assignment of the N-Me-d-Ala in the northern hemisphere (fragment 1) of 2 led N-Me-l-Ala to be assigned in the southern hemisphere (fragment 2) of 2.
The β-amino acid Amoya present in companeramides A (1) and B (2) was first identified in the molluscan metabolite onchidin,15 but has since been reported as a component of several marine cyanobacterial metabolites including ulongapeptin,16 guineamide C,17 and malevamide C.18 While the absolute configurations of the Amoya residues in guineamide C and malevamide C have not been determined, those in ulongapeptin were assigned as 2S, 3S by Marfey’s analysis using comparison with a synthetic mixture of C-2 diastereomers (2S, 3R and 2R, 3R) derivatized with both d- and l-FDLA enantiomers. The (2S,3S)-configuration was also assigned to the Amoya unit in onchidin based on NOE studies and 1H NMR coupling constant analysis; however, the total synthesis of onchidin suggested that a revision of the structure was necessary.19 Therefore, it was planned to use Marfey’s analysis for assignment of the Amoya unit in companeramides A and B, facilitated by the availability of the synthetic 3(R)-amino-2(R,S)-methyloctanoate (Amo) standards.20
Two portions of the synthetic material were derivatized separately with d- or l-FDLA to provide HPLC retention times for all four possible Amo diastereomers, with retention times of l-FDLA-(2R,3S)-Amo and l-FDLA-(2S,3S)-Amo being inferred from the retention times of the enantiomeric d-FDLA-(2S,3R)-Amo and d-FDLA-(2R,3R)-Amo standards, respectively. Hydrogenation of each companeramide to reduce the terminal alkyne was followed by acid hydrolysis to release Amo for separate derivatization with d- and l-FDLA. Reported retention times for the four Amo diastereomers using similar HPLC conditions were used to assign the order of elution of the pairs of standards generated in our protocol. For each companeramide, the d-FDLA-Amo product coeluted with the d-FDLA-derivatized (2S,3R)-Amo standard, and similarly the l-FDLA-Amo product coeluted with the l-FDLA-derivatized (2S,3R)-Amo, supporting a 2S,3R-Amoya unit in companeramides A (1) and B (2).
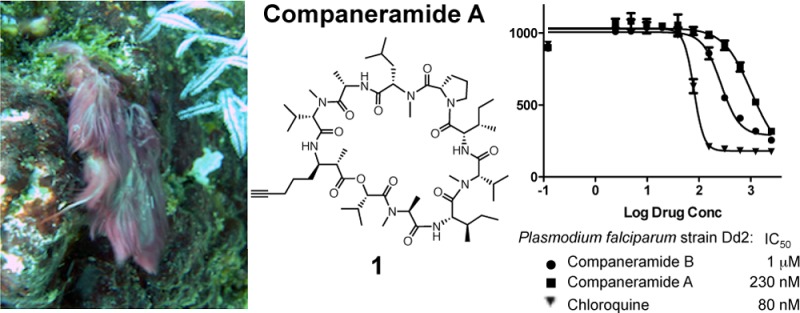
Companeramides A (1) and B (2) showed no significant cytotoxicity at 1 μM against four human cancer cell lines (NCI-H460 non-small-cell lung carcinoma, MDA-MB-231 breast adenocarcinoma, SF-295 glioblastoma, and SK-OV3 ovarian carcinoma cells). Instead preliminary antiparasitic activity of the parent fractions led to testing of the two pure compounds against three strains of the malaria parasite Plasmodium falciparum in a fluorescence-based assay. Neither compound was as active as the chloroquine control against the chloroquine-sensitive D6 or chloroquine-insensitive Dd2 and 7G8 strains (Table 2, Figure S18, Supporting Information). However, the differential activity between the two companeramides across all strains is noteworthy considering their structural similarity. The chloroquine-sensitive D6 strain was approximately twice as sensitive to companeramide A (1) as the chloroquine-resistant Dd2 and 7G8 strains. In contrast, companeramide B (2) showed comparable activity against the chloroquine-sensitive D6 and chloroquine-resistant strain Dd2, but was about three times less active against the chloroquine-resistant 7G8 strain.
Table 2. Antiplasmodial Activities for 1 and 2 Compared to Chloroquine against Chloroquine-Sensitive (D6) and -Resistant (Dd2 and 7G8) Strains of Plasmodium falciparum.
| IC50 (nM) against plasmodial strain |
|||
|---|---|---|---|
| test compound | D6 | Dd2 | 7G8 |
| companeramide A (1) | 570 | 1000 | 1100 |
| companeramide B (2) | 220 | 230 | 700 |
| chloroquine | 5 | 80 | 71 |
The companeramides add to the growing repertoire of cyanobacterial depsipeptides that show some degree of antiplasmodial activity, including dolastatin 10,21 the venturamides,4 carmabins and dragonamides,6 and symplostatin 49 (first reported as gallinamide7). While some of these compounds (e.g., dolastatin 10) are potently toxic to mammalian cells as well as to the malarial parasite, others such as the venturamides and carmabin A show relatively little toxicity to mammalian cells. At the concentrations tested, it is not possible to distinguish any selective antiplasmodial activity for companeramide A (1), although the results for companeramide B (2) suggest potential antiplasmodial selectivity. The requisite testing of 2 for mammalian cell toxicity at higher micromolar concentrations was not pursued in the interest of saving material for other assays in which the target activity may be more pronounced than the moderate antiplasmodial activity presented here. It is interesting to speculate that related large cyclic alkynoic depsipeptides that are reported to be nontoxic, such as the malevamides,18 may possess antiparasitic activity. Noteworthy also is that despite the structural relationship between the companeramides and ulongapeptin, only the heptameric ulongapeptin displays nanomolar cytotoxicity.16
Experimental Section
General Experimental Procedures
Optical rotations were measured on a JASCO P-1010 polarimeter. UV and IR data were obtained on a Hitachi U-2000 spectrophotometer and a Nicolet IR100 FT-IR instrument, respectively. NMR spectra were acquired on Bruker Avance 700 MHz and Bruker Avance DRX 600 MHz spectrometers with the residual CHCl3 solvent used as an internal standard (δC 77.23, δH 7.26). LR FABMS and HRTOFMS (ES+) mass spectra were recorded on ABSciex 3200 QTRAP and Waters Micromass mass spectrometers. The isolation of compounds 1 and 2 was performed using a Waters HPLC system consisting of two Waters 515 pumps, a Rheodyne 7725i injector, and a Waters 996 photodiode array detector. Marfey’s and chiral-phase HPLC analyses were conducted on a Shimadzu HPLC system equipped with two LC-20AD pumps and an SPD-M20A photodiode array detector. General reagents were from Sigma-Aldrich Corp and VWR International.
Collection and Identification
The marine cyanobacterial assemblage (McPhail laboratory voucher number PAC-6/25/04-2) was first collected in June 2004 by hand using scuba from a reef pinnacle off the west coast of Coiba National Park. The material collected for chemical extraction (1 L) was stored in 50% EtOH for transport and then stored at −20 °C until extraction. For phylogenetic analyses, subsamples of the field collection (0.5 mL of cyanobacteria in 5 mL of RNA Later solution) were stored at room temperature (rt) and then 4 °C before DNA extraction, and live cultures were also initiated. Additional field collections were made subsequently in 2010 (voucher number PAC-10-03) and 2012 (voucher number PAC-7/10/12-1), for chemical and DNA extraction, as well as laboratory culture. DNA extraction of the 2010 RNA Later samples and comparison of the genomic 16S rDNA (rDNA) sequence with the NCBI BLAST database led to the identification of the cf. Symploca strain,13 which will be assigned to a new genus, “Hyalidium”.14 Monoculture of the small-filament cyanobacterium (PAC-10-3 csf1) associated with companeramide production in the 2010 collection provided material for genomic DNA isolation (GenBank accession number KM882611), and this organism was found to be 99% identical to that of filamentous cyanobacteria associated with Black Band Disease (see Supporting Information, S19 and S20).
Extraction and Isolation
Approximately 95.6 g dry weight of the 2004 cyanobacterial collection was extracted repeatedly with CH2Cl2–MeOH (2:1) to produce 5.75 g of an organic extract. The extract was fractionated by normal-phase VLC on Si gel with a stepwise gradient solvent system from hexanes to EtOAc to MeOH, yielding nine fractions. The fraction eluting with 100% EtOAc was cytotoxic (IC50 300 ng/mL) to NCI-H460 lung cancer cells and further chromatographed by RP18 SPE using a stepwise solvent gradient (MeOH–H2O, 6:4, 7:3, 8:2, 9:1, 100% MeOH and CH2Cl2). The noncytotoxic SPE fraction eluting with 70% MeOH possessed interesting peptide-like 1H NMR spectroscopic parameters and was purified further using RP18 HPLC (85:15 MeOH–H2O, Phenomenex Synergi Fusion 4 μm, 10 × 250 mm, 2 mL/min, UV detection at 216 nm) to yield companeramide A (1, 2.3 mg, tR 30.5 min) and companeramide B (2, 5.3 mg, tR 21.4 min).
Companeramide A (1):
colorless oil; [α]23D −140 (c 0.6, MeOH); UV (MeOH) λmax (log ε) 215 (3.45) nm; IR νmax (neat) 3311, 2963, 2930, 1720, 1628, 1517, 1462, 1231, 1098 cm–1; 1H and 13C NMR data, see Tables 1 and S1 (Supporting Information); HRTOFMS (ES+) m/z 1084.7395 [M + H]+ (calcd for C57H98N9O11, 1084.7386).
Companeramide B (2):
colorless oil; [α]23D −78 (c 0.5, MeOH); UV (MeOH) λmax (log ε) 225 (4.06) nm; IR νmax (neat) 3311, 2963, 2930, 1720, 1628, 1517, 1462, 1231, 1098 cm–1; 1H and 13C NMR data, see Tables 1 and S2 (Supporting Information); HRTOFMS (ES+) m/z 1056.7115 [M + H]+ (calcd for C55H94N9O11, 1056.7073), m/z 1078.6865 [M + Na]+ (calcd for C55H93N9O11Na, 1078.6892).
Absolute Configuration of Companeramides A (1) and B (2)
The absolute configurations of the amino acid residues in companeramides A (1) and B (2) were determined by Marfey’s methodology. Approximately 0.5 mg of each of companeramides A and B were separately hydrolyzed (6 N HCl at 110 °C for 16 h), evaporated to dryness, and reconstituted in H2O (50 μL). FDAA solutions in acetone (0.1%, 100 μL) and 1 N NaHCO3 (50 μL) were added to each hydrolysate and heated to 37 °C for 1 h. The solutions were allowed to cool to rt, neutralized with 2 N HCl (25 μL), and evaporated to dryness. The residues were resuspended in DMSO–H2O (1:1, 100 μL) and analyzed by RP18 HPLC (Waters Symmetry Shield C18 column, 3.9 × 150 mm, 1 mL/min, UV detection at 340 nm) using a linear gradient of 9:1 40 mM ammonium acetate buffer (pH 5.2)–CH3CN to 1:1 40 mM ammonium acetate buffer–CH3CN over 60 min. The absolute configurations of the amino acids in the hydrolysates of 1 and 2 were determined by direct comparison with the retention times (tR, min) for Marfey’s derivatives of authentic standards.
The retention times (min) of the FDAA-derivatized α-amino acids in the hydrolysate of 1 matched those of l-Pro (12.8; d-Pro, 13.5), N-Me-l-Val (23.2; d-N-Me-Val, 27.5), l-allo-Ile (21.4; l-Ile, 21.2, d-Ile, 28.3, d-allo-Ile, 28.8), l-Ala (12.3; d-Ala, 16.9), N-Me-l-Ala (14.3; N-Me-d-Ala, 16.6), and N-Me-l-Leu (26.6; N-Me-d-Leu, 30.3). The retention times (min) of the derivatized amino acids in the hydrolysate of 2 matched l-Pro (12.8; d-Pro, 13.5), N-Me-l-Val (23.2; N-Me-d-Val, 27.5), l-Val (17.3; d-Val, 24.0), l-allo-Ile (21.4; l-Ile, 21.2, d-Ile, 28.3 and d-allo-Ile, 28.8), and both N-Me-l-Ala (14.3) and N-Me-d-Ala (16.6).
Chiral-phase HPLC analysis was used to determine the absolute configurations of the Hiva residues in the two depsipeptides. A portion of the acid hydrolysate of 1 (0.25 mg) and 2 (0.25 mg) was reconstituted in H2O prior to chiral-phase HPLC analysis (85:15 2 mM CuSO4–CH3CN; column Phenomenex Chirex 3126 (D), 4.6 × 250 mm, flow 1.0 mL/min, UV detection at 254 nm). The Hiva residue in the hydrolysates of 1 and 2 eluted with the same retention time (tR, min) as the S-Hiva standard (40.0) but not that of R-Hiva (61.7).
To assign the position of N-Me-l-Ala versus N-Me-d-Ala in companeramide B (2), 4 mg of 2 was partially hydrolyzed in 3 N HCl (2 mL, constant stirring, 100 °C); the reaction was monitored by LC-MS at 30 min intervals and was halted after 3 h by cooling to −20 °C, after which the acid was removed under high vacuum. The residual hydrolysate was suspended in 75% CH3CN–H2O (100 μL) for LC-MS/MS analysis (ES+) to identify fragments containing single N-Me-Ala residues. Tetrapeptide N-Me-Val-Val-N-Me-Ala-Pro (m/z 413.9, 0.1 mg) was purified from the total hydrolysate by RP18 HLC using a linear gradient of 5–100% CH3CN–H2O/0.1% TFA over 60 min (Phenomenex Synergi Hydro column, 10 × 250 mm, 2.5 mL/min, 340 nm). Subsequent complete hydrolysis of the tetrapeptide (6 N HCl, microwave irradiation, 1000 W, 50 s) was followed by Marfey’s derivatization of the resulting hydrolysate (with l-FDLA) and commercial amino acid standards (with l-FDLA and d-FDLA). In each case, 1 M NaHCO3 (20 μL) was added to a 50 mM amino acid solution in H2O, followed by FDLA (1% w/v in acetone, 44 μL), and the reaction mixture heated to 40 °C (1 h). The reactions were quenched by cooling to rt and acidification (20 μL of 1 N HCl), before drying under high vacuum and resuspension in 100 μL of DMSO for LC-MS analysis (Thermo Aquasil C18 column, 3 × 150 mm, 25–55% linear gradient of CH3CN–H2O/0.1% TFA over 45 min, 1 mL/min, 340 nm, ES+). Column eluent was split ca. 1:500 prior to entrance into the ESI source to optimize ionization. Retention times (m/z [M + Na]+, tR min) for derivatized residues of the tetrapeptide corresponded to standards for N-Me-l-Val (426.2, 23.55; N-Me-d-Val, 28.00), l-Val (412.2, 19.06; d-Val, 28.40), N-Me-d-Ala (398.2, 18.31; N-Me-l-Ala, 17.84), and l-Pro (ion not identified, 16.34, d-Pro, 19.50).
Assignment of the Amoya residues relied on hydrogenation of the Amoya unit in 1 and 2 to 3-amino-2-methyloctanoic acid (Amo), peptide hydrolysis, Marfey’s derivatization with FDLA, and comparison with FDLA-derivatized synthetic Amo standards. In each case, approximately 0.5 mg of cyclic depsipeptide was dissolved in dry MeOH and stirred over 5% Pd/C under an atmosphere of hydrogen (rt, 18 h). After filtration over Celite, washing with EtOH, and concentration in vacuo, the products (m/z 1088, tetrahydro-1, and 1060, tetrahydro-2) were resuspended in 6 N HCl and stirred overnight at 100 °C. The acid hydrolysate of tetrahydro-1 was derivatized with l-FDLA, as described above, while that of tetrahydro-2 was divided into two portions and derivatized with d- and l-FDLA. Similarly, synthetic 3(R)-amino-2(R,S)-methyloctanoate (0.3 mg, in 1:1 MeOH–H2O) was derivatized with each of d- and l-FDLA for RP18 HPLC-MS analysis (50% CH3CN–H2O; Phenomenex Synergi Hydro column, 4.6 × 250 mm, flow 0.2 mL/min, UV detection at 340 nm). Observed elution times of the standards were 9.4 min (d-FDLA-2S,3R-Amo = l-FDLA-2R,3S-Amo), 10.0 min (d-FDLA-2R,3R-Amo = l-FDLA-2S,3S-Amo), 17.4 min (l-FDLA-2R,3R-Amo), and 20.9 min (l-FDLA-2S,3R-Amo), as assigned by comparison to reported elution times.16 The l-FDLA-hydrolysate of tetrahydro-1 provided a small peak at 20.9 min for l-FDLA-2S,3R-Amo, which was supported by retention times of 20.9 min (l-FDLA-2S,3R-Amo) and 9.4 min (d-FDLA-2S,3R-Amo) for the two derivatized portions of tetrahydro-2.
In Vitro Antimalarial Activity Assay
Plasmodium falciparum strains D6, Dd2, and 7G8 were cultured in human erythrocytes at 2% hematocrit in RPMI 1640 containing 0.5% Albumax, 45 μg/L hypoxanthine, and 50 μg/L gentamicin, as previously described.22 In vitro antimalarial activity was determined by the malaria SYBR Green I-based fluorescence (MSF) assay described previously23 with slight modification.22 Stock solutions of each test sample were prepared in sterile distilled H2O at a concentration of 10 mM. The sample solutions were serially diluted with culture medium and distributed to asynchronous parasite cultures on 96-well plates in quadruplicate in a total volume of 100 μL to achieve 0.2% parasitemia with a 2% hematocrit. Automated pipetting and dilution were carried out with a programmable Precision 2000 robotic station (Bio-Tek). The plates were then incubated for 72 h at 37 °C. After incubation, 100 μL of lysis buffer with 0.2 μL/mL SYBR Green I was added to each well. The plates were incubated at rt for 1 h in the dark and then placed in a 96-well fluorescence plate reader (Spectramax Gemini-EM; Molecular Diagnostics) with excitation and emission wavelengths at 497 and 520 nm, respectively, for measurement of fluorescence. The 50% inhibitory concentration (IC50) was determined by nonlinear regression analysis of logistic dose–response curves (GraphPad Prism software).
Cancer Cell Cytotoxicity
Cell viability was assessed with a standard MTT assay as described previously with the viability of vehicle-treated cells defined as 100% and coibamide A (30 nM) included as a control cancer cell toxin.24 All compounds were reconstituted in 100% DMSO and stored at −20 °C until the day of treatment; final concentrations of DMSO never exceeded 0.1%. Human H460 lung and SF-295 glioblastoma cells were from the National Cancer Institute cell line repository, and MDA-MB-231 breast and SK-OV3 ovarian carcinoma cells were from the American Type Culture Collection (ATCC). All cells were maintained under standard growth conditions and seeded into 96-well plates at a density of 3000 cells per well the day before treatment.
Acknowledgments
Financial support from the NIH Fogarty International Center (ICBG Grant TW006634) is gratefully acknowledged. We thank the Smithsonian Tropical Research Institute, the crew of R/VUrraca, T. Capson, C. Guevara, D. Sherman, and R. Thacker for collection of the cyanobacterium, Autoridad Nacional del Ambiente (ANAM), Panama, for permission to make this collection, and J. Morré and the OSU EIHS Center for MS data acquisition (NIEHS P30 ES00210). We thank Drs. A. Fenner and M. Balunas for generously providing a sample of synthetic 3(R)-amino-2(R,S)-methyloctanoate.
Supporting Information Available
1D and 2D NMR spectra for companeramides A (1) and B (2). MS/MS spectra for 1 and 2 and tetrapeptide hydrolysis product. Concentration–response profiles for 1 and 2 against three strains of Plasmodium falciparum. Experimental details for DNA extraction and amplification of cyanobacterial 16S rRNA, phylogenetic analysis, and phylogenetic alignment tree. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Dedication
Dedicated to Dr. William Fenical of Scripps Institution of Oceanography, University of California–San Diego, for his pioneering work on bioactive natural products.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nunnery J. K.; Mevers E.; Gerwick W. H. Curr. Opin. Biotechnol. 2010, 21, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg C. C.; Cowley E. S.; Sikorska J.; Shaala L. A.; Ishmael J. E.; Youssef D. T. A.; McPhail K. L. J. Nat. Prod. 2013, 76, 1781–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg C. C.; Thimmaiah M.; Shaala L. A.; Hau A. M.; Malmo J. M.; Ishmael J. E.; Youssef D. T.; McPhail K. L. J. Nat. Prod. 2011, 74, 1677–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linington R. G.; Gonzáles J.; Ureña L. D.; Romero L. I.; Ortega-Barría E.; Gerwick W. H. J. Nat. Prod. 2007, 70, 397–401. [DOI] [PubMed] [Google Scholar]
- Peach K. C.; Linington R. G. Future Med. Chem. 2009, 1, 593–617. [DOI] [PubMed] [Google Scholar]
- McPhail K. L.; Correa J.; Linington R. G.; Gonzalez J.; Ortega-Barria E.; Capson T. L.; Gerwick W. H. J. Nat. Prod. 2007, 70, 984–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linington R. G.; Clark B. R.; Trimble E. E.; Almanza A.; Urena L. D.; Kyle D. E.; Gerwick W. H. J. Nat. Prod. 2009, 72, 14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taori K.; Liu Y.; Paul V. J.; Luesch H. ChemBioChem 2009, 10, 1634–1639. [DOI] [PubMed] [Google Scholar]
- Stolze S. C.; Deu E.; Kaschani F.; Li N.; Florea B. I.; Richau K. H.; Colby T.; van der Hoorn R. A. L.; Overkleeft H. S.; Bogyo M.; Kaiser M. Chem. Biol. 2012, 19, 1546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlik C. M.; Wong C. Y. B.; Ononye S.; Lopez D. D.; Engene N.; McPhail K. L.; Gerwick W. H.; Balunas M. J. J. Nat. Prod. 2013, 76, 2026–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balunas M. J.; Grosso M. F.; Villa F. A.; Engene N.; McPhail K. L.; Tidgewell K.; Pineda L. M.; Gerwick L.; Spadafora C.; Kyle D. E.; Gerwick W. H. Org. Lett. 2012, 14, 3878–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R. A.; Goeger D. E.; Hills P.; Mooberry S. L.; Huang N.; Romero L. I.; Ortega-Barria E.; Gerwick W. H.; McPhail K. L. J. Am. Chem. Soc. 2008, 130, 6324–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engene N.; Gunasekera S. P.; Gerwick W. H.; Paul V. J. Appl. Environ. Microbiol. 2013, 79, 1882–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In a manuscript in preparation Niclas Engene has assigned this organism to the new genus “Hyalidium” (personal communication to K.L.M.).
- Fernández R.; Rodríguez J.; Quíñoá E.; Riguera R.; Debitus C.; Bouchet P. Tetrahedron Lett. 1994, 35, 9239–9242. [Google Scholar]
- Williams P. G.; Yoshida W. Y.; Quon M. K.; Moore R. E.; Paul V. J. J. Nat. Prod. 2003, 66, 651–654. [DOI] [PubMed] [Google Scholar]
- Tan L. T.; Sitachitta N.; Gerwick W. H. J. Nat. Prod. 2003, 66, 764–771. [DOI] [PubMed] [Google Scholar]
- Horgen F. D.; Yoshida W. Y.; Scheuer P. J. J. Nat. Prod. 2000, 63, 461–467. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Kobayashi J.; Yazaki R.; Ueno M. Chem.—Asian J. 2007, 2, 135–144. [DOI] [PubMed] [Google Scholar]
- Balunas M. J.; Grosso M. F.; Villa F. A.; Engene N.; McPhail K. L.; Tidgewell K.; Pineda L. M.; Gerwick L.; Spadafora C.; Kyle D. E.; Gerwick W. H. Org. Lett. 2012, 14, 3878–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennell B. J.; Carolan S.; Pettit G. R.; Bell A. J. Antimicrob. Chemother. 2003, 51, 833–841. [DOI] [PubMed] [Google Scholar]
- Kelly J. X.; Smilkstein M. J.; Cooper R. A.; Lane K. D.; Johnson R. A.; Janowsky A.; Dodean R. A.; Hinrichs D. J.; Winter R.; Riscoe M. Antimicrob. Agents Chemother. 2007, 51, 4133–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilkstein M.; Sriwilaijaroen N.; Kelly J. X.; Wilairat P.; Riscoe M. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hau A. M.; Greenwood J. A.; Lohr C. V.; Serrill J. D.; Proteau P. J.; Ganley I. G.; McPhail K. L.; Ishmael J. E. PloS One 2013, 8, e65250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.