

Abstract
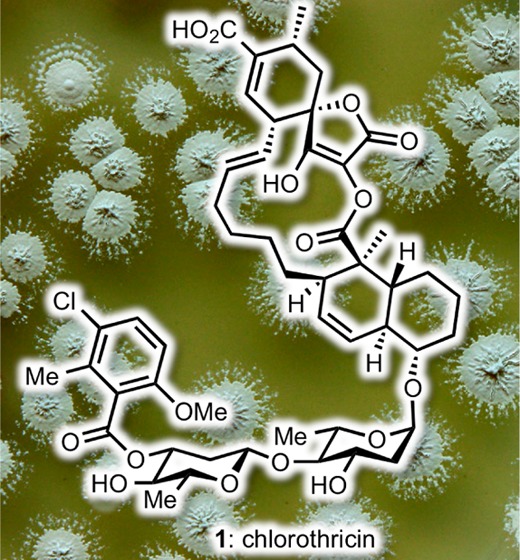
The discovery of chlorothricin (1) defined a new family of microbial metabolites with potent antitumor antibiotic properties collectively referred to as spirotetronate polyketides. These microbial metabolites are structurally distinguished by the presence of a spirotetronate motif embedded within a macrocyclic core. Glycosylation at the periphery of this core contributes to the structural complexity and bioactivity of this motif. The spirotetronate family displays impressive chemical structures, potent bioactivities, and significant pharmacological potential. This review groups the family members based on structural and biosynthetic considerations and summarizes synthetic and biological studies that aim to elucidate their mode of action and explore their pharmacological potential.
Introduction
Since the beginning of mankind, organisms producing natural products have provided a reservoir of therapeutic remedies and medicines for various diseases.1 A subset of these drugs has been classified as antitumor antibiotics based on their ability to “block cell growth by interfering with DNA, the genetic material in cells”.2 Key general features of an antitumor antibiotic include interference with DNA synthesis, membrane transport, and production of reactive oxygen species.3 One of the most notable examples of an antitumor antibiotic is mitomycin C, a microbial metabolite that is used currently for the treatment of breast and bladder cancer.4 Among other antitumor antibiotics, daunorubicin and its semisynthetic derivative doxorubicin represent chemotherapeutic leukemia agents in clinical settings.5
The search for new antitumor antibiotics led to the discovery of chlorothricin (1), a complex polyketide produced by various Streptomyces strains.6 Its intriguing chemical structure and bioactivity defined a new family of microbial metabolites, commonly referred to as spirotetronate polyketides. This family is identified by the presence of a cyclohexene ring spiro-linked to a tetronic acid moiety (Figure 1, fragment A) that is embedded in a macrocycle (Figure 1, fragment B). In several cases, the structure also contains a trans-decalin ring (Figure 1, fragment C) and is decorated by various deoxy oligosaccharides (Figure 1, fragment D). In terms of biological profile, spirotetronate polyketides exhibit potent antibacterial and antitumor activities and a documented value as tools in the elucidation of new biological pathways. As such, they represent highly promising leads in drug discovery. To appreciate their untapped potential, in this review we group the known spirotetronates based on common structural elements and biosynthetic considerations. We then discuss the biological profiles and highlight synthetic efforts toward each group.
Figure 1.
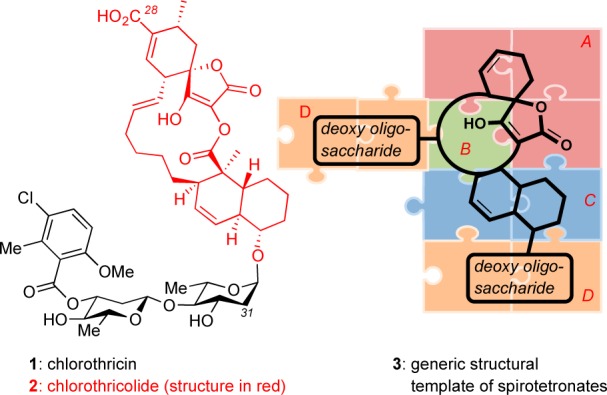
Structure of chlorothricin and general structure of spirotetronate polyketides.
Classification
Recently Süssmuth and co-workers proposed a classification of tetronates based on two main categories: the linear tetronates and the spirotetronates.7 On the basis of biosynthetic considerations, the latter subgroup can be divided into two classes: class I (generic structure 4), which contains the spirotetronate moiety within a varying size macrocycle, and class II (generic structure 5), which additionally contains a decalin moiety (Figure 2). Representative members of the class I spirotetronates in order of increasing macrocyclic length are abyssomicin C (6)8 (containing a C11 macrocycle), okilactomycin D (7)9 (containing a C13 macrocycle), and spirohexenolide A/B (8/9)10 (containing a C15 macrocycle). Representative members of the class II spirotetronates include maklamicin (11)11 (containing a C11 macrocycle), tetronolide (12)12 (the aglycon of tetrocarcin A containing a C13 macrocycle), and chlorothricolide (2)6 (the aglycon of chlorothricin containing a C13 macrolactone). In this class is also included versipelostatin aglycone (13), which contains the largest C17 macrocyclic motif isolated to date.13 Quartromicins 10, unusual spirotetronate polyketides containing four spirotetronate subunits, lie outside these two classes due to their peculiar structure14 and unique biosynthesis.15 The above classification stems from a common biosynthetic pathway that accounts for construction of these compounds.
Figure 2.

Spirotetronate polyketides: general structures and representative members.
Biosynthesis of Spirotetronates
In general, the biosynthesis of spirotetronates occurs through condensation of acetic acid units via the type I polyketide synthase pathway (Scheme 1).16 As shown in the biosynthesis of abyssomicin C17 and okilactomycin,18 construction of the class I spirotetronates proceeds by elongating their carbon chain via incorporation of propanoyl and/or acetyl units (14/15) to the acyl carrier protein (ACP). This iterative operation forms polyketide chain 16. Incorporation of a glyceryl unit, via CoA intermediate 17,19 forms tetronate 18 likely via a Claisen condensation followed by lactonization. The precise mechanism for the elimination of the C-5 hydroxy group was recently elucidated by the Sun and Leadlay groups and shown to proceed via acetylation and subsequent elimination, thereby forming dienophile 19.20 An intramolecular Diels–Alder (IMDA) reaction then generates the characteristic spirotetronate moiety. The resulting substrates subsequently undergo peripheral oxidations to produce the final structures of the natural products.17a
Scheme 1. Proposed Biosynthesis of Class I and Class II Spirotetronate Polyketides.
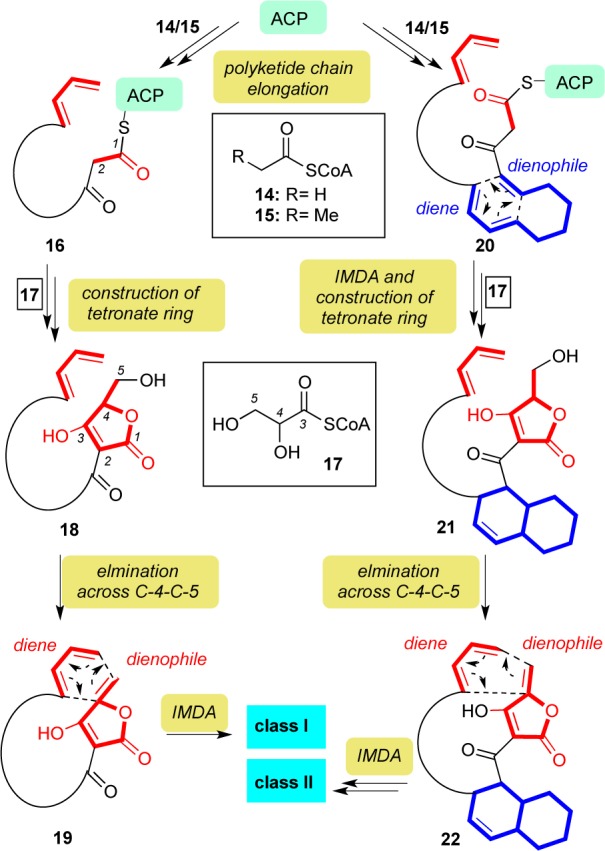
The biosynthetic pathways of the class II polyketide spirotetronates have been elucidated for chlorothricin,21 tetrocarcin A,22 kijanimicin,23 and versipelostatin.24 Following chain elongation, the diene and dienophile groups of 20 undergo an IMDA to construct the characteristic decalin moiety of 21. Glyceryl CoA (17)19 is then inserted to generate tetronate 22, which following a second IMDA gives rise to the aglycones of the class II spirotetronates (Figure 2).
Oxidations and/or glycosylations at the periphery of the aglycone lead to various natural products of the class II spirotetronates. For instance, chlorothricolide (2), the aglycone of chlorothricin, contains an acyl-oxy tetronic acid moiety. This functionality (i.e., oxygenation at the C-2 position) is proposed to result from a Baeyer–Villiger oxidation that takes place after formation of the spirotetronate motif.21b,21d,21e,25 A similar biosynthetic scenario can be proposed for the construction of PA-46101 A and B (see structures 57/58).26 Another interesting example of post-translational modification is found in the structure of tetronolide (12), the aglycone of tetrocarcin A (see structure 47). Compound 12 is highlighted by an enal functionality at C-22–C-23–C-32. This functionality was proposed to result from oxidation at C-32 to the corresponding aldehyde followed by double-bond migration to C-22–C-23 and further allylic oxidation at C-21.22a
Several class II spirotetronates are subject to glycosylation mostly with 2-deoxycarbohydrates such as d-tetronitrose (26, d-kijanose), amicetose (27), and digitoxose (28). These carbohydrates are proposed to arise from thymidine diphosphate (TDP)-6-deoxy-4-keto-d-glucose (24), which, in turn, is available from d-glucose-1-phosphate (23) (Scheme 2). Biosynthesis of the uncommon tetronitrose is proposed to occur from 25 via aminotransferase and methylation, while the precise mechanism for the carbamate biosynthesis still remains elusive.22a,23,27
Scheme 2. Biosynthesis of Deoxysugars.

Biology of Spirotetronate Polyketides
The majority of spirotetronates have been subjected to biological assays that aim to define their bioactivity as antibiotic and/or anticancer leads as well as compounds that regulate metabolism. With this in mind, we have grouped these molecules in three major classes that describe the commonality of their bioactivities.
Spirotetronates as Potential Antibiotic Leads
The Abyssomicin Family
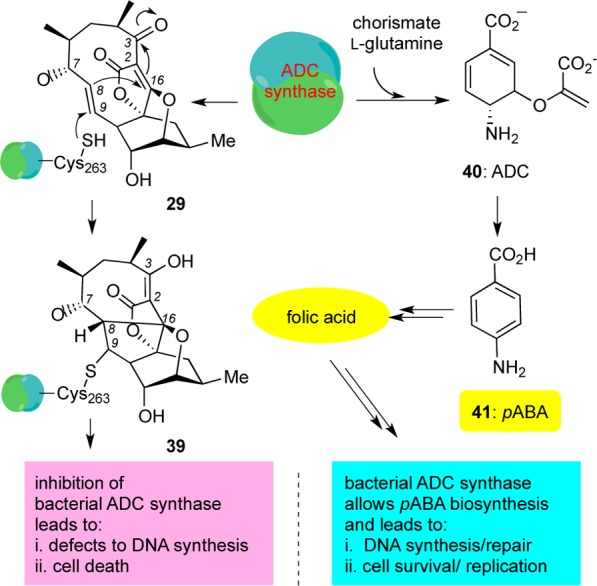
Isolated from a marine Verrucosispora, abyssomicin C (6) and its atropisomer (29) (Figure 3) are the first known natural products to block para-aminobenzoic acid (pABA, 41) biosynthesis.8,28pABA is a biosynthetic precursor of folic acid (vitamin B9), and as such, it is essential for DNA synthesis/repair and cell survival (Scheme 3).29 On the other hand, lack of folic acid is known to induce mutations in DNA resulting in cell death. Importantly, blocking the pABA pathway is detrimental to bacteria but inconsequential to humans since the latter cannot produce folic acid but only absorb it through their diet.30 Studies on the effect of the abyssomicin motif in pABA biosynthesis have been performed with atrop-abyssomicin (29) and are highlighted in Scheme 3. Amino-4-deoxychorismate (ADC) synthase, a heterodimeric protein, catalyzes the biosynthesis of amino-4-deoxychorismate (40), a synthetic precursor of pABA. Compound 29 was found to covalently react with the Cys-263 of the PabB subunit of ADC synthase at the C-9 enone center. The transiently formed C-8 nucleophile then reacts with the spirotetronate subunit at the C-16 center to form compound 39, thus irreversibly binding to ADC synthase.31
Figure 3.

Representative structures of the abyssomicin family of spirotetronates.
Scheme 3. pABA Biosynthesis and Proposed Mode of Action of atrop-Abyssomicin C.
Several natural products of the abyssomicin family have been tested for their ability to inhibit pABA biosynthesis (Scheme 3). Among them, only abyssomicin C (6), atrop-abyssomicin C (29), and abyssomicin J (31) have shown promising bioactivities.8,28,32 Specifically, 6 and 29 potently inhibit proliferation of methicillin-resistant Staphylococcus aureus at MIC values of 5.2 and 3.5 μg/mL, respectively.8,33 Similar cytotoxicities have been reported against various tuberculosis-related mycobacteria.32a,32b On the other hand, abyssomicin D (32) and related analogues lacking the C-7–C-9 enone motif are inactive, attesting to the biological significance of this functionality.28,31,32,32c−32e Moreover, most studies indicate that 29 is more potent than 6. This increased potency has been attributed to an increased conjugation between the C-7 carbonyl group and the C-8–C-9 alkene that renders 29 a stronger Michael acceptor than 6.33 The bioactivity of abyssomicin J (31), a thioether dimer of the abyssomicin scaffold, can be explained by considering that oxidation of the sulfur accelerates a retro-Michael addition, producing the C-7–C-9 enone functionality in situ. In fact, it has been suggested that 31 is a prodrug of 6, and as such, it represents a more attractive drug candidate.32b
Kijanimicin (43) and Related Class II C13 Macrocycles
Isolated from various Micromonospora bacteria, kijanimicin (43)34 and lobophorin B (42)35 are structurally defined by a C13 macrocycle (referred to as kijanolide, 44) in which the C-9 and C-17 hydroxy groups have been glycosylated (Figure 4). Most members of this group show potent activity against Gram-positive bacteria34e,36 as well as cytotoxicity against various cancer cell lines.36a,36b,37 In addition, kijanimicin was shown to exhibit robust anticancer38 and antimalarial activities34e in mouse models. Moreover, Fenical et al. reported promising anti-inflammatory activities of lobophorins in a mouse ear edema model. Interestingly, this is the first report on the untapped potential of spirotetronates as small-molecule leads against inflammation.35
Figure 4.
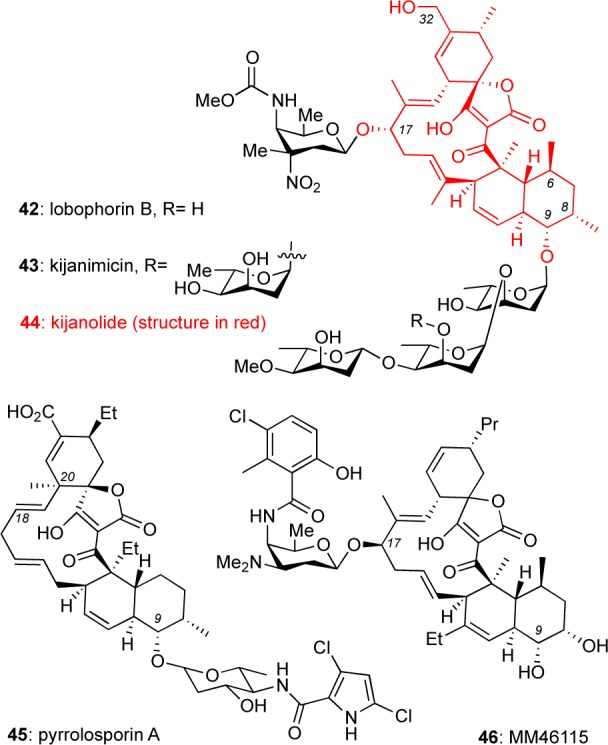
Structures of kijanimicin and related class II C13 macrocyclic spirotetronates.
A similar framework is in the structures of pyrrolosporin A (45, C-9-glycosylation)39 and MM46115 (46, C-17 glycosylation).40 The glycopyranose motif of pyrrolosporin is also found in the structures of decatromicin A/B41 and Nai414-A/B,42 which also exhibit similar antibiotic activity against various strains of Gram-positive bacteria. In addition to its potent antibiotic activities MM46115 was found to exhibit promising antiviral activities.40a Along these lines, the structurally unrelated quartromicins 9(14a,14c) were shown to display potent bioactivity against herpes simplex virus (HSV) and human immunodeficiency virus (HIV) at low μM concentration.14b
Recent studies indicate that kijanimicin binds to the TetR family of transcriptional regulators43 that control expression of various cytoplasmic proteins in prokaryotes. This binding leads to (a) C-9-deglycosylation of kijanimicin, which results in loss of activity, and (b) overexpression of the receptor, thus increasing antibiotic resistance.44 The structurally related saccharocarcins45 are subject to a similar mechanism of deactivation and antibiotic resistance.44,46
Figure 5.
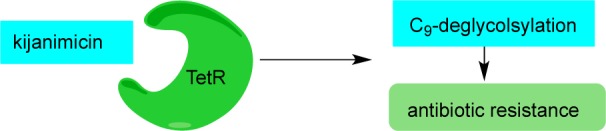
Binding of kijanimicin to TetR leads to C-9-deglycosylation and antibiotic resistance.
Spirotetronates as Potential Anticancer Leads
Spirohexenolides A and B (8, 9)
Burkart et al. reported the isolation of spirohexenolides A (8) and B (9) and their potent cytotoxicities against various cancer cell lines (Figure 2).10 Subsequent immunoaffinity-fluorescent labeling studies indicated that 8 targets human macrophage migration inhibitory factor (hMIF).47 This interaction reduces the phosphorylation levels of PI3K/AKT, ultimately leading to a reduction of tumor cell growth (Figure 6).48 Conjugation of spirohexenolide A with fluorescent tags showed localization in the lysosome of HCT-116 cells, suggesting that spirohexenolides interfere with cellular endocytosis of hMIF.47
Figure 6.

Cancer cellular signaling and mode of action of select spirotetronate polyketides.
Tetronolide-Containing Natural Products
Isolated from various Micromonospora bacteria, tetrocarcin A (47, also known as antlermicin A and AC6D)49 represents the archetype of the tetronolide family of natural products that also includes AC6H (48)50 and arisostatins A (49) and B (50) (Figure 7).51 The antibiotic potential of these spirotetronates against several Gram-positive bacteria has been reported.49a,49e,50−52 Animal studies have shown that 47 is about 4 times more potent than the commonly used antibiotic diminazene. Although 47 has a narrow safety margin, it can be used in combination with diminazene, providing a beneficial synergistic effect.53
Figure 7.
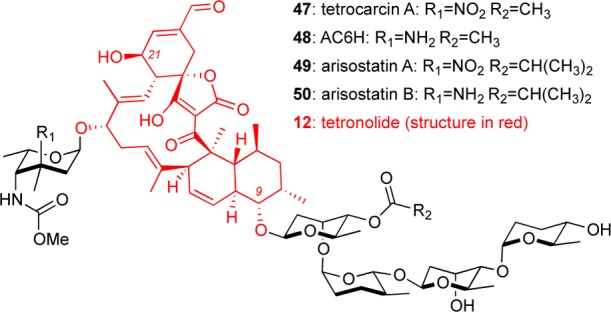
Selected structures of tetronolide-containing natural products.
Various reports on the potential anticancer profile of tetrocarcin A and related family members have been published. Initial studies showed tumor reduction in a mouse sarcoma model upon administration of 10 mg/kg of 47 over a period of 6 days. Similar treatment in a mouse leukemia P388 model led to an increased life expectancy.49b,49e Comparable studies in B16 mouse melanoma showed that the life expectancy more than doubled at a single dose of 27 mg/kg of 47.54 Moreover, AC6H 48 exhibited cytotoxicity against P388 leukemia and B16 melanoma cells at 6.25 and 25 μg/mL, respectively.50 AC6H also showed a moderate increase in the life expectancy of a P388 leukemia mouse model albeit less active than tetrocarcin A.50 Studies in U937 cells indicated that arisostatin A (49) is equipotent to tetrocarcin A, while arisostatin B (50) was 10-fold less active. Arisostatin A was also found to be active in various breast and lung cancer cell lines at low micromolar concentrations.51
Mode-of-action studies in HeLa cells showed that tetrocarcin A (47) potently inhibits Bcl-2, an important antiapoptotic protein that is often overexpressed in cancer cells (Figure 6).55 Although there is no evidence of direct binding to Bcl-2, the phenotypical response induced by 47 is very promising56 and suggests that this compound represents an important and unexplored lead against cancer.57
Studies in lymphoma cells showed that 47 induces a stress response of the endoplasmic reticulum (ER), resulting in upregulation of the heat shock protein HSP70, ultimately triggering cell apoptosis (Figure 6).58 Studies in breast cancer cells have suggested an alternative mechanism of action of 47 that proceeds by inhibiting phosphorylation of the PI3K/Akt signaling cascade.59 Although the main cellular target of tetrocarcin A is still under investigation, preclinical studies have demonstrated its potential as a drug against chemoresistant cancers. In fact, 47 was reported to be more effective than paclitaxel at inducing cell apoptosis in breast cancer cells.58a,60
Arisostatin A (49) was found to induce cell apoptosis by generating reactive oxygen species (ROS), altering mitochondrial transmembrane potential, and releasing cytochrome c (Cyt C) in AMC-HN-4 cells (Figure 6), ultimately leading to activation of caspase-3 and induction of apoptosis. However, Bcl-2 activation was not altered by arisostatin A, indicating a different mode of action from that of 47.61
Screening the potential anticancer and antimicrobial activities of naturally occurring tetrocarcins has produced the main structure–activity relationship (SAR) data for this family. These studies have led to the following observations: (a) the number of carbohydrate units (digitoxose and amicetose) attached at the C-9 center of tetrocarcin A is proportional to its antimicrobial activity;62,49a,49b,49e,63 (b) C-21 acetylation and C-9 glycosylation of tetrocarcin A did not significantly affect Bcl-2 activation.63b The results suggest that the attachments of amicetose (27) and digitoxose (28) at the C-9 position of tetrocarcin A enhance its antibacterial profile but have no significant effect on its anticancer potential.64
Versipelostatins
Versipelostatin A (51) was isolated from a strain of Streptomyces versipellis (Figure 8).13,65 Biological studies showed that 51 is the first known molecule to inhibit gene expression of GRP78. Together with its isoform GRP94, these heat shock proteins are induced by stress responses in the endoplasmic reticulum and are essential for cancer cell survival.13,66 In addition to its role in cancer, ER stress is considered to play a major role in the pathogenesis of various CNS diseases, such as Alzheimer’s and Parkinson’s disease.67
Figure 8.

Selected members of the versipelostatin family.
Recent studies have shown that versipelostatin A (51) inhibits heat shock proteins and unfolded protein response (UPR) under glucose deprivation conditions.68 As such, it appears to operate via a different mechanism as compared to that of rapamycin, an FDA-approved immunosuppressive drug that activates GRP78 independently of glucose availability.69 Thus, versipelostatin may offer a significant advantage due to its selective effect in hypoglycemic cells.68 Although there is no information for direct binding of 51 to a protein target, its effect on the UPR signaling pathway offers a novel tool to understand ER-induced stress and pharmacologically regulate related illnesses.68a,70
SAR studies on this family have been limited to the bioactivities of naturally occurring versipelostatins.71 The results show that versipelostatins A (51), E (52), and F (53) are the only biologically active compounds, inhibiting GRP78 expression at low micromolar IC50 values.71b Interestingly, 53 was found to be 10 times more potent than 51 in GRP78 expression with an IC50 of 0.3 μM.71a The data attest to the significance of the glycosylation motif to the GRP78 expression and bioactivity.71b In addition to these studies, Takahashi et al. demonstrated the importance of the l-oleandrose sugar for the bioactivity, and changes in the oxidation state of C-7 had no effect on biological activity.72
Okilactomycin (54) and Chrolactomycin (55)
Okilactomycin (54) was isolated from Streptomyces griseoflavus and is noted for its potent antitumor activity against Ehrlich ascites carcinoma in vivo at 2.5 mg/kg with a T/C of 145.7% for mice survival.73 In addition, 54 exhibited in vitro activity against P388 and L1210 leukemia cells, with IC50 values of 89 and 216 nM, respectively.73b Recently, okilactomycin was shown to inhibit rRNA protein synthesis at low μM concentrations,74 suggesting potential applications as an antibacterial agent.75 Although other natural okilactomycins were found to be inactive,9 the related chrolactomycin (55) was reported to exhibit antibacterial and anticancer activity at a low μM concentration.76 It has been reported that 55 inhibits telomerase activity, thus blocking the ability of cancer DNA to replicate.77 The most recently isolated 6-hydroxy chrolactomycin was less active than 55 against Gram-positive bacteria.78
Figure 9.

Structure of okilactomycin and analogues.
PA-46101A/B (57/58), Maklamicin (11), and Nomimicin (59)
The potent antibiotic properties of PA-46101A (57) and B (58) have been reported.26 Recent efforts by Igarashi and co-workers led to the isolation of maklamicin (11)11 and nomimicin (59),79 which contain the smallest macrocyclic ring of the class II spirotetronates. Both compounds display potent activity against many Gram-positive bacteria, while 11 also exhibits moderate antitumor activity against HeLa and MCF7 breast cancer cells.11,79
Figure 10.

Structures of PA46101 A/B, maklamicin, and nomimicin.
Spirotetronates as Potential Leads in Metabolism and Digestion
Chlorothricin (1)/A88696C/F (60/61)/Tetronothiodin (62)
Chlorothricin (1) was shown to inhibit the activity of pyruvate carboxylase,80 a key enzyme that converts pyruvate to oxaloacetate, thus allowing consumption of glucose through the Krebs cycle (Figure 11). Inhibition of pyruvate carboxylase leads to an increase of pyruvate concentration in liver, which through gluconeogenesis accounts for accumulation of glucose, ultimately leading to diabetes.81 Moreover, an inhibitory effect of 1 on malate dehydrogenase, an enzyme that oxidizes malate to oxaloacetate in the Krebs cycle, has also been reported.82 It should be noted, however, that the direct cellular target of 1 is under debate and may involve interaction with components in the cell membrane that may account for the observed downstream effects.83
Figure 11.
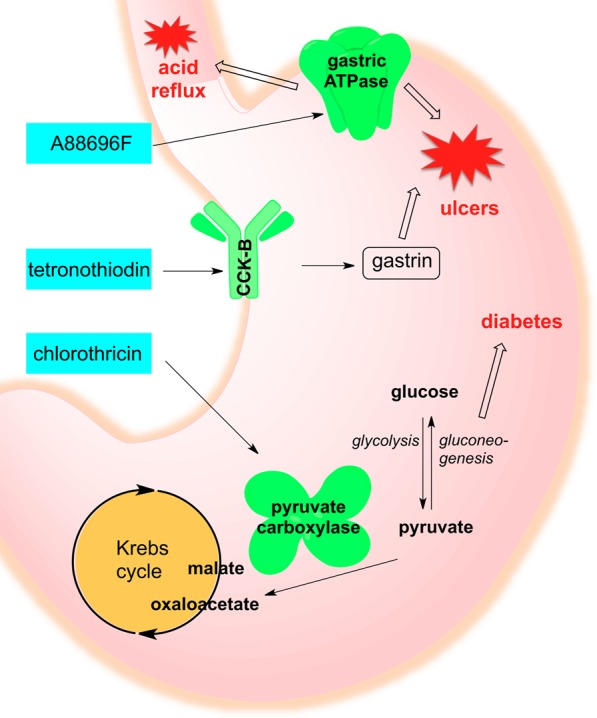
Effects of spirotetronate polyketides on metabolic pathways and digestion.
Although the potential anticancer properties of chlorothricin (1) have not been investigated, C-31 hydroxychlorothricin (Figure 1) was shown to exhibit antitumor activity at 40 mg/kg against implanted Ehrlich carcinoma cells in mice with an LD50 of 295 mg/kg.84 C-28 methyl ester of chlorothricolide (2),85 the aglycone of 1 (Figure 1), also inhibits pyruvate carboxylase albeit at higher concentrations than 1, suggesting that glycosylation enhances biological activity.80a
Efforts to discover new gastric ATP-ase inhibitors86 led to the isolation of A88696F (61) and its dehydroxylated counterpart A88696C (60).87 Hydroxylation at C-3 was found to enhance the biological activities since 61 was the most active, with an IC50 at 0.5 μM, while 60 was considered inactive.87
Figure 12.

Structures of A88696C/F and tetronothiodin.
Isolated from a Streptomyces species, tetronothiodin (62) was shown to inhibit brain-type cholecystokinin (CCK)-B receptor in rat cerebral cortex with an IC50 value of 3.6 nM.88 It is worth noting that CCK receptors are structurally similar to gastrin and are used throughout the central nervous system (CNS) and gastric tract.89 Interestingly, 62 has 27 000 times higher affinity for CCK-B over CCK-A in rat models.89a Thus, in addition to its pharmaceutical promise, 62 could be used as a tool to study the CCK-B/CCK-A signaling pathway.90
Synthetic Approaches toward Spirotetronate Polyketides
In this part of the review, we highlight the key steps toward the synthesis of selected spirotetronates. When possible, we compare the various strategies in terms of overall efficiency.
Class I, C11 Spirotetronates: Abyssomicins (6 and 29)
Abyssomicin C (6) and its atropisomer 29 contain a rigid oxabicyclo [2.2.2] octane substructure that encapsulates the spirotetronate moiety. To date, three chemical syntheses of 6 and 29 have been reported. The key transformations are highlighted in Scheme 4. Sorensen’s group used a biomimetic IMDA to construct spirotetronate moiety 65 from diene 63. C-11–C-12 epoxidation of 65 followed by C-16 intramolecular enol epoxide opening produced a 1:1 mixture of abyssomicin C (6) and atrop-abyssomicin (29).91 A similar strategy has been implemented by the Snider92 and Couladouros93 groups.
Scheme 4. Highlights of Abyssomicin C Syntheses.

The Nicolaou group’s synthesis of abyssomicin C is highlighted by an intermolecular Diels–Alder cycloaddition that furnishes cyclohexene 67 with the desired stereochemistry (Scheme 4).33 A ring-closing metathesis (RCM) was used to generate the macrocyclic skeleton of 6 from diene 68. Interestingly, the authors showed that treatment of 29 with lithium selectride led to formation of abyssomicin D (32). Interestingly, this finding supports the notion that abyssomicin C (6) is a biosynthetic progenitor of 32 and further validates the proposed mechanism of abyssomicin C deactivation as presented in Scheme 3.33
More recently, the groups of Bihelovic and Saicic reported a synthesis of 29.94 Key to their approach was a Tsuji–Trost cyclization that constructed cyclohexene 70. The C11 macrocycle of 29 was subsequently formed using an intramolecular Nozaki–Hiyama–Kishi coupling. Interestingly, this strategy produces exclusively atrop-abyssomycin C.94 It is likely that the restricted rotation around the C-2 and C-3 centers, due to the sp2 hybridization, affects the formation of the two isomers. In support of this hypothesis, the Nicolaou group has shown that 29 can be converted to 6 by protonating the C-16 oxygen under mild acidic conditions.33 Other studies toward the abyssomicin scaffold have been reported in addition to the mentioned total syntheses.95
Class I, C13 Spirotetronates: Okilactomycins (54 and 7)
Smith et al. reported the first total synthesis of okilactomycin 54 in 29 steps.96 Key to the strategy was a Petasis–Ferrier union/rearrangement of 72(97) that yielded the 2,6-cis-tetrahydropyranone ring 73. Ring-closing metathesis of 77 using Hoveyda–Grubbs second-generation catalyst was used to construct the 13-membered macrocycle of 54.96
More recently, the Scheidt group also reported a synthesis of okilactomycin. Key to this approach was a Prins-type fragment assembly98 between cyclohexene 75 and β-keto-ester 74 that formed the 2,6-cis-tetrahydropyranone ring of 76. Similarly to the Smith approach, an intramolecular ring-closing metathesis using Grubbs second-generation catalyst constructed the macrocycle.99 Additional synthetic studies toward okilactomycin have been reported by the Yoshii100 and Paquette groups.101
Scheme 5. Highlights of Okilactomycin Syntheses.

Hoye et al. reported the first total synthesis of (±)/(−)-okilactomycin D (7). Key to this strategy was an IMDA cycloaddition that formed spirotetronate 80 from precursor 79. The overall synthesis proceeds in 13 linear steps (17 total steps) and 17% yield. Remarkably, demethylation of tetronate 80 was efficiently conducted on a 3 g scale.102
Scheme 6. Highlights of Okilactomycin D (7) Synthesis.

Class I, C15 Spirotetronates: Spirohexenolides A and B
The Burkart group reported a strategy toward spirohexenolides based on an intermolecular Diels–Alder cycloaddition (Scheme 7).103 A ring-closing Julia–Kocienski coupling was applied for the synthesis of macrocycle 82. Although the projected intramolecular hemiacetalation to 84 failed due to an oxidative rearrangement of 82 to 83, the overall strategy has successfully installed the major skeletal features of spirohexenolides.104
Scheme 7. Highlights of Synthetic Efforts toward Spirohexenolides.

Class I, C17 Spirotetronates: Tetronothiodin (62)
Structurally tetronothiodin is highlighted by an α-acyl tetronic acid moiety and tetrahydrothiophene moiety. Page et al. have reported a synthesis of the spirotetronate subunit isomer 87 using a Diels–Alder reaction with propenal and the hydroxyl diene 85 to install the desired stereochemistry of 86 (Scheme 8). Further functional modifications led to the synthesis of spirotetronate 87.105
Scheme 8. Synthetic Studies toward Tetronothiodin.

Class II, C13 Spirotetronates: Tetronolide (12)/Kijanolide (44) and Chlorothricolide (2)
To date there are no reported total syntheses of any class II, C13 spirotetronates. Several strategies have been employed for the synthesis of tetronolide (12), the aglycone of tetrocarcin A (47), kijanolide (44), the aglycone of kijanimicin (42), and chlorothricolide (2), the aglycone of chlorothricin (1). Tetronolide has been synthesized by Yoshii106 and Boeckman,107 while an improved formal synthesis has also been reported by Roush.108 In general, these strategies rely upon independently constructing the spirotetronate and decalin moieties and then connecting them to form the C13 macrocycle. A remarkable synthesis of chlorothricolide (2) was reported by the Roush group.109
The Yoshii and Roush syntheses of the decalin moiety 92, common to both tetronolide and kijanolide, are summarized in Scheme 9. In Yoshii’s approach a Horner–Wadsworth–Emmons (HWE) olefination between 88 and 89 was used to construct polyene 90, which underwent an IMDA reaction to produce decalin 92.106,110 The Roush group implemented a Suzuki coupling between 93 and 94 to form polyene 91, which, following further functionalizations, gave rise to decalin 92 via an IMDA cycloaddition.108,111 A similar approach toward decalin 92 has been reported by the Marshall group.112
Scheme 9. Highlights of Tetronolide Syntheses.
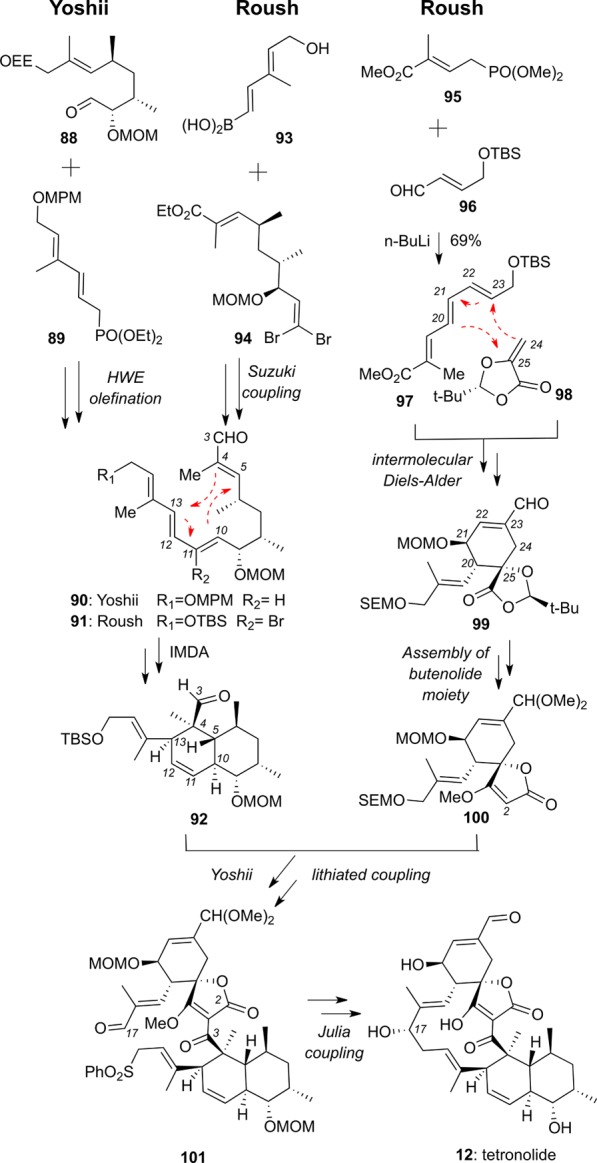
A synthetic approach toward spirotetronate 100 has been reported by Yoshii113 and subsequently optimized by Roush.108,114 This approach is based on constructing triene 97 via a HWE olefination between 95 and 96. An intermolecular Diels–Alder of diene 97 and chiral dienophile 98, followed by oxidative functionalization and double-bond migration, yielded enal 99. Coupling of lithiated spirotetronate 100 with aldehyde 92 followed by subsequent functionalizations yielded sulfone 101, which, under Julia coupling conditions, gave rise to the 13-membered macrocycle of 12.106
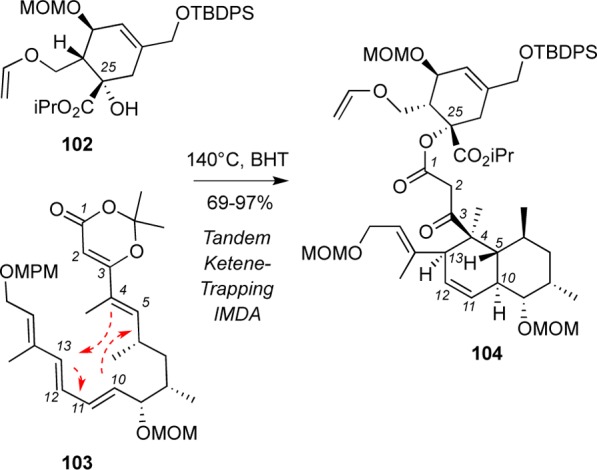
Boeckman’s group synthesis of 12 is highlighted by a tandem ketene-trapping [4+2] cycloaddition of diene 103 and alcohol 102 to form spirotetronate subunit 104. Conversion of 104 to tetronolide 12 was accomplished under Julia conditions. Overall, this approach significantly reduces the number of steps required for completion of the tetronolide synthesis.107,115
Various synthetic studies toward kijanolide (44) have been reported by the groups of Marshall,116 Yoshii,117 and Roush.118 These strategies rely on intermolecular Diels–Alder reactions and ketene-trapping strategy to form the desired macrocycle.119 Application of a Julia coupling to the synthesis of 28,29-bisnor-(+)-kijanolide has been reported by the Yoshii group.120
Scheme 10. Highlights of the Boeckman Strategy toward Tetronolide.
A tandem intra/intermolecular Diels–Alder reaction between polyene 105 and chiral dienophile 98 was implemented for the synthesis of chlorothricolide (2) (Scheme 11). The reaction gave the desired cycloadduct in 40% yield together with partially reacted decalin 107. Upon treatment with dienophile 98, 107 was converted to the desired product 106 in 58% yield.109 Construction of the spirotetronate unit followed by coupling with the allyl ester completed the synthesis of 2.
Scheme 11. Highlights of the Roush Strategy toward Chlorothricolide.

A late-stage IMDA reaction was used by Yoshii’s group for the synthesis of (±)-24-O-methylchlorothricolide (Scheme 12). Although the selectivity of the IMDA reaction was moderate, the overall strategy represents a noteworthy bioinspired approach toward these compounds.121 The groups of Marshall,112c,112d,122 Ireland,123 Snider,124 Schmidt,125 and Meyers126 have also reported studies toward the synthesis of 2.
Scheme 12. Highlights of the Yoshii Strategy toward (±)-24-O-Methylchlorothricolide.

Class II, C17 Spirotetronates: Versipelostatin (51)
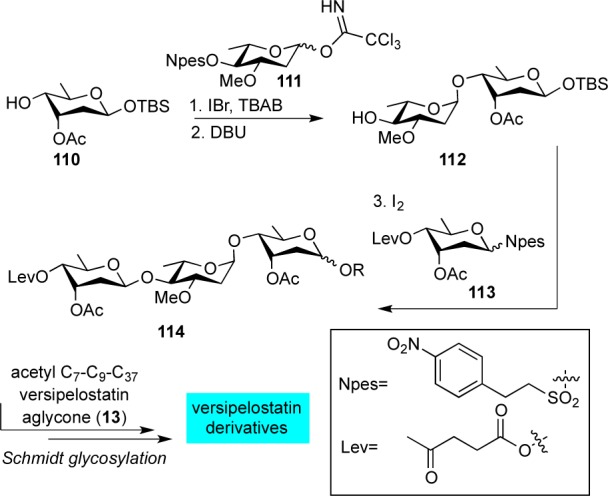
Numerous synthetic studies have been reported toward the total synthesis of versipelostatin A (51), but to date its total synthesis has not been completed. Kirschning’s127 and Takahashi’s72 groups provided synthetic strategies to the trisaccharide moiety. A synthesis of the versipelostatin (51) trisaccharide 114 is shown in Scheme 13.72 Key to the synthesis is a Schmidt glycosylation of 110 with trichloroacetimidate 111. The resulting disaccharide 112 was deprotected and coupled with l-oleandrosyl imidate 113 to produce 114 (Scheme 13). Further functionalization of glycosyl 114 and Schmidt glycosylation with acetyl C-7–C-9–C-37 versipelostatin aglycone 13 (Figure 2) yielded a versipelostatin derivative used for biological studies. On the basis of NMR and biological consideration, the oleandrose sugar was structurally reassigned from d to l. An alternate strategy used was adding each sugar individually to the versipelostatin aglycone, thus elongating the glycosyl chain.72 Various approaches toward the spirotetronate unit of the versipelostatin have been reported.128
Scheme 13. Synthetic Studies toward the Glycosyl Moiety of Versipelostatin A.
Quartromicins (10)
A stereocontrolled Diels–Alder reaction has been implemented by the Roush group for the synthesis of the quartromicin spirotetronate unit.103a,129 In addition, this group reported a strategy of connecting subunits 115 and 116 together using lithium halogen exchange and CeCl3 coupling.130 Bedel’s group offered an alternative strategy of constructing the spirotetronate subunits using RCM, but to date no total syntheses of quartromicins have been completed.131
Scheme 14. Connection of Spirotetronates for Quartromicins.

Conclusions
The discovery of penicillin revolutionized pharmaceutical research by demonstrating, for the first time, that microorganisms can produce secondary metabolites of value to medicine. Since then, cultured microorganisms have been recognized as prolific producers of secondary metabolites that are used either directly as drugs or have inspired the design of drugs.66a,132 On the other hand, the intricate structures of these compounds represent exceptional tools to explore new biological pathways and unknown mechanisms of action. These qualities, although scattered, are observed in the family of spirotetronate polyketides and provide evidence for their significant but still untapped pharmacological value.
More than 40 years after the discovery of chlorothricin, the spirotetronate family has grown to include over 70 macrocycles of various sizes that, in certain cases, are decorated with carbohydrate side chains. In addition to their potent antitumor and antibiotic activities, certain spirotetronates were characterized as “the first” tools to elucidate a biological effect.8a,55a,66a For example, versipelostatin was found to induce potent and selective cytotoxicity in glucose-deprived tumor cells.68 Moreover, abyssomicin C was found to be the first natural product to block pABA biosynthesis, a pathway essential to bacteria but insignificant to humans.8a Impressive synthetic and chemical biology efforts were combined to decipher the mode-of-action of abyssomicins at the molecular level.31,33c This underscores the enormous significance of the spirotetronate polyketide family to biology in addition to their pharmacological potential.
Several studies have documented the significance of the carbohydrate chains for the observed antibiotic activity of spirotetronates.44,63b However, with the exception of abyssomicins, there is no clear understanding of the biological significance of the spirotetronate aglycone core. At present, chemical strategies developed toward the synthesis of spirotetronates have uncovered the value of certain key reactions, such as Diels–Alder cycloaddition, ring -closing metathesis, and Julia olefination. Nonetheless, the vast majority of these strategies have not yielded sufficient amounts of compound for a methodical structure–activity relationship study, thereby hampering rational drug design. It is evident that a methodical fragment-based approach to this structure, in combination with chemical biology studies, will be highly beneficial, as it could reveal the role of the spirotetronate motif, the effect of the macrocyclic size, and the role of the decalin system. In turn, this effort would allow a detailed evaluation and optimization of the spirotetronate pharmacophore. In addition to a dearly needed scalable synthesis,133 advances in microbial biosynthesis134 should offer a potential solution to large-scale production or semisynthesis of a lead candidate. A combination of these efforts should unveil the pharmacological value of spirotetronates and would have significant impact in current efforts toward personalized medicine.
Acknowledgments
Financial support from the National Institutes of Health (CA 133002) is gratefully acknowledged. We also thank the UCSD Academic Senate and the U.S. Department of Education for a Fellowship to M.H.L. through GAANN grant P200A120223. We thank the National Science Foundation for instrumentation grants CHE9709183 and CHE0741968.
The authors declare no competing financial interest.
Dedication
Dedicated to Dr. William Fenical of Scripps Institution of Oceanography, University of California–San Diego, for his pioneering work on bioactive natural products.
Funding Statement
National Institutes of Health, United States
References
- a Hughes C. C.; Fenical W. Chem.—Eur. J. 2010, 16, 12512–12525. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gerwick W. H.; Moore B. S. Chem. Biol. 2012, 19, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Molinski T. F.; Dalisay D. S.; Lievens S. L.; Saludes J. P. Nat. Rev. Drug Discovery 2009, 8, 69–85. [DOI] [PubMed] [Google Scholar]; d Nicolaou K. C.; Chen J. S.; Dalby S. M. Biorg. Med. Chem. 2009, 17, 2290–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Remers W. A.The Chemistry of Antitumor Antibiotics; Wiley-Interscience: New York, 1988; Vol. 1. [Google Scholar]; b Remers W. A.The Chemistry of Antitumor Antibiotics; Wiley-Interscience: New York, 1988; Vol. 2. [Google Scholar]; c http://www.cancer.gov/dictionary?cdrid=44488.
- http://www.pharmacology2000.com/Anticancer/classes4.htm.
- Galm U.; Hager M. H.; Van Lanen S. G.; Ju J. H.; Thorson J. S.; Shen B. Chem. Rev. 2005, 105, 739–758. [DOI] [PubMed] [Google Scholar]
- a Tan C.; Tasaka H.; Yu K. P.; Murphy M. L.; Karnofsk D. Cancer 1967, 20, 333–353. [DOI] [PubMed] [Google Scholar]; b Tacar O.; Sriamornsak P.; Dass C. R. J. Pharm. Pharmacol. 2013, 65, 157–170. [DOI] [PubMed] [Google Scholar]
- Keller-Schierlein W.; Muntwyler R.; Pache W.; Zahner H. Helv. Chim. Acta 1969, 52, 127–142. [Google Scholar]
- Vieweg L.; Reichau S.; Schobert R.; Leadlay P. F.; Sussmuth R. D. Nat. Prod. Rep. 2014, 31, 1554–1584. [DOI] [PubMed] [Google Scholar]
- a Bister B.; Bischoff D.; Strobele M.; Riedlinger J.; Reicke A.; Wolter F.; Bull A. T.; Zahner H.; Fiedler H. P.; Sussmuth R. D. Angew. Chem., Int. Ed. 2004, 43, 2574–2576. [DOI] [PubMed] [Google Scholar]; b Riedlinger J.; Reicke A.; Zahner H.; Krismer B.; Bull A. T.; Maldonado L. A.; Ward A. C.; Goodfellow M.; Bister B.; Bischoff D.; Sussmuth R. D.; Fiedler H. P. J. Antibiot. 2004, 57, 271–279. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Ondeyka J. G.; Zink D. L.; Basilio A.; Vicente F.; Salazar O.; Genilloud O.; Dorso K.; Motyl M.; Byrne K.; Singh S. B. J. Antibiot. 2009, 62, 55–61. [DOI] [PubMed] [Google Scholar]
- Kang M. J.; Jones B. D.; Mandel A. L.; Hammons J. C.; DiPasquale A. G.; Rheingold A. L.; La Clair J. J.; Burkart M. D. J. Org. Chem. 2009, 74, 9054–9061. [DOI] [PubMed] [Google Scholar]
- Igarashi Y.; Ogura H.; Furihata K.; Oku N.; Indananda C.; Thamchaipenet A. J. Nat. Prod. 2011, 74, 670–674. [DOI] [PubMed] [Google Scholar]
- Hirayama N.; Kasai M.; Shirahata K.; Ohashi Y.; Sasada Y. Tetrahedron Lett. 1980, 21, 2559–2560. [Google Scholar]
- Park H. R.; Furihata K.; Hayakawa Y.; Shin-ya K. Tetrahedron Lett. 2002, 43, 6941–6945. [Google Scholar]
- a Roush W. R.; Barda D. A. Org. Lett. 2002, 4, 1539–1542. [DOI] [PubMed] [Google Scholar]; b Tsunakawa M.; Tenmyo O.; Tomita K.; Naruse N.; Kotake C.; Miyaki T.; Konishi M.; Oki T. J. Antibiot. 1992, 45, 180–188. [DOI] [PubMed] [Google Scholar]; c Kusumi T.; Ichikawa A.; Kakisawa H.; Tsunakawa M.; Konishi M.; Oki T. J. Am. Chem. Soc. 1991, 113, 8947–8948. [Google Scholar]
- a Wu L. F.; He H. Y.; Pan H. X.; Han L.; Wang R. X.; Tang G. L. Org. Lett. 2014, 16, 1578–1581. [DOI] [PubMed] [Google Scholar]; b Montgomery L. J.; Challis G. L. Synlett 2008, 2164–2168. [Google Scholar]; c He H. Y.; Pan H. X.; Wu L. F.; Zhang B. B.; Chai H. B.; Liu W.; Tang G. L. Chem. Biol. 2012, 19, 1313–1323. [DOI] [PubMed] [Google Scholar]
- a Dewick P. M.Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; John Wiley & Sons Ltd, 2009. [Google Scholar]; b Shen B. Curr. Opin. Chem. Biol. 2003, 7, 285–295. [DOI] [PubMed] [Google Scholar]; c Staunton J.; Weissman K. J. Nat. Prod. Rep. 2001, 18, 380–416. [DOI] [PubMed] [Google Scholar]
- a Gottardi E. M.; Krawczyk J. M.; von Suchodoletz H.; Schadt S.; Muhlenweg A.; Uguru G. C.; Pelzer S.; Fiedler H. P.; Bibb M. J.; Stach J. E. M.; Sussmuth R. D. ChemBioChem 2011, 12, 1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Roh H.; Uguru G. C.; Ko H. J.; Kim S.; Kim B. Y.; Goodfellow M.; Bull A. T.; Kim K. H.; Bibb M. J.; Choi I. G.; Stach J. E. M. J. Bacteriol. 2011, 193, 3391–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H.; Nakagawa A.; Omura S. J. Antibiot. 1989, 42, 1321–1323. [DOI] [PubMed] [Google Scholar]
- Sun Y. H.; Hong H.; Gillies F.; Spencer J. B.; Leadlay P. F. ChemBioChem 2008, 9, 150–156. [DOI] [PubMed] [Google Scholar]
- Kanchanabanca C.; Tao W. X.; Hong H.; Liu Y. J.; Hahn F.; Samborskyy M.; Deng Z. X.; Sun Y. H.; Leadlay P. F. Angew. Chem., Int. Ed. 2013, 52, 5785–5788. [DOI] [PubMed] [Google Scholar]
- a Mascaretti O.; Hook D.; Kreutzer E. F.; Chang C. J.; Floss H. G. Planta Med. 1978, 33, 919–924. [DOI] [PubMed] [Google Scholar]; b Holzbach R.; Pape H.; Hook D.; Kreutzer E. F.; Chang C.; Floss H. G. Biochemistry 1978, 17, 556–560. [DOI] [PubMed] [Google Scholar]; c He Q. L.; Jia X. Y.; Tang M. C.; Tian Z. H.; Tang G. L.; Liu W. ChemBioChem 2009, 10, 813–819. [DOI] [PubMed] [Google Scholar]; d Lee J. J.; Lee J. P.; Keller P. J.; Cottrell C. E.; Chang C. J.; Zahner H.; Floss H. G. J. Antibiot. 1986, 39, 1123–1134. [DOI] [PubMed] [Google Scholar]; e Jia X. Y.; Tian Z. H.; Shao L.; Qu X. D.; Zhao Q. F.; Tang J.; Tang G. L.; Liu W. Chem. Biol. 2006, 13, 575–585. [DOI] [PubMed] [Google Scholar]; f Mascaretti O.; Chang C. J.; Floss H. G. J. Nat. Prod. 1979, 42, 455–462. [Google Scholar]; g Mascaretti O. A.; Chang C. J.; Hook D.; Otsuka H.; Kreutzer E. F.; Floss H. G. Biochemistry 1981, 20, 919–924. [DOI] [PubMed] [Google Scholar]; h Kawashima A.; Nakamura Y.; Ohta Y.; Akama T.; Yamagishi M.; Hanada K. J. Antibiot. 1992, 45, 207–212. [DOI] [PubMed] [Google Scholar]; i Shao L.; Qu X. D.; Jia X. Y.; Zhao Q. F.; Tian Z. H.; Wang M.; Tang G. L.; Liu W. Biochem. Biophys. Res. Commun. 2006, 345, 133–139. [DOI] [PubMed] [Google Scholar]
- a Fang J.; Zhang Y. P.; Huang L. J.; Jia X. Y.; Zhang Q.; Zhang X.; Tang G. L.; Liu W. J. Bacteriol. 2008, 190, 6014–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tamaoki T.; Tomita F. J. Antibiot. 1983, 36, 595–598. [DOI] [PubMed] [Google Scholar]; c Tamaoki T.; Tomita F. Agric. Biol. Chem. 1982, 46, 1021–1026. [Google Scholar]
- a Zhang H.; White-Phillip J. A.; Melancon C. E.; Kwon H. J.; Yu W. L.; Liu H. W. J. Am. Chem. Soc. 2007, 129, 14670–14683. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bruender N. A.; Thoden J. B.; Holden H. M. Biochemistry 2010, 49, 3517–3524. [DOI] [PubMed] [Google Scholar]
- Chijiwa S.; Park H. R.; Furihata K.; Ogata M.; Endo T.; Kuzuyama T.; Hayakawa Y.; Shin-ya K. Tetrahedron Lett. 2003, 44, 5897–5900. [Google Scholar]
- Mascaretti O.; Otsuka H.; Chang C. J.; Floss H. G. J. Nat. Prod. 1979, 42, 679–679. [Google Scholar]
- Matsumoto M.; Kawamura Y.; Yoshimura Y.; Terui Y.; Nakai H.; Yoshida T.; Shoji J. i. J. Antibiot. 1990, 43, 739–747. [DOI] [PubMed] [Google Scholar]
- a Bruender N. A.; Holden H. M. Protein Sci. 2012, 21, 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li S. M.; Xiao J.; Zhu Y. G.; Zhang G. T.; Yang C. F.; Zhang H. B.; Ma L.; Zhang C. S. Org. Lett. 2013, 15, 1374–1377. [DOI] [PubMed] [Google Scholar]; c Thoden J. B.; Branch M. C.; Zimmer A. L.; Bruender N. A.; Holden H. M. Biochemistry 2013, 52, 3191–3193. [DOI] [PubMed] [Google Scholar]; d Xiao J.; Zhang Q. B.; Zhu Y. G.; Li S. M.; Zhang G. T.; Zhang H. B.; Saurav K.; Zhang C. S. Appl. Microbiol. Biotechnol. 2013, 97, 9043–9053. [DOI] [PubMed] [Google Scholar]
- Keller S.; Nicholson G.; Drahl C.; Sorensen E.; Fiedler H. P.; Sussmuth R. D. J. Antibiot. 2007, 60, 391–394. [DOI] [PubMed] [Google Scholar]
- Wegkamp A.; van Oorschot W.; de Vos W. M.; Smid E. J. Appl. Environ. Microbiol. 2007, 73, 2673–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts F.; Roberts C. W.; Johnson J. J.; Kyle D. E.; Krell T.; Coggins J. R.; Coombs G. H.; Milhous W. K.; Tzipori S.; Ferguson D. J. P.; Chakrabarti D.; McLeod R. Nature 1998, 393, 801–805. [DOI] [PubMed] [Google Scholar]
- Keller S.; Schadt H. S.; Ortel I.; Sussmuth R. D. Angew. Chem., Int. Ed. 2007, 46, 8284–8286. [DOI] [PubMed] [Google Scholar]
- a Freundlich J. S.; Lalgondar M.; Wei J. R.; Swanson S.; Sorensen E. J.; Rubin E. J.; Sacchettini J. C. Tuberculosis 2010, 90, 298–300. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang Q.; Song F.; Xiao X.; Huang P.; Li L.; Monte A.; Abdel-Mageed W. M.; Wang J.; Guo H.; He W.; Xie F.; Dai H.; Liu M.; Chen C.; Xu H.; Liu M.; Piggott A. M.; Liu X.; Capon R. J.; Zhang L. Angew. Chem., Int. Ed. 2013, 52, 1231–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Niu X. M.; Li S. H.; Gorls H.; Schollmeyer D.; Hilliger M.; Grabley S.; Sattler I. Org. Lett. 2007, 9, 2437–2440. [DOI] [PubMed] [Google Scholar]; d Igarashi Y.; Yu L. K.; Miyanaga S.; Fukuda T.; Saitoh N.; Sakurai H.; Saiki I.; Alonso-Vega P.; Trujillo M. E. J. Nat. Prod. 2010, 73, 1943–1946. [DOI] [PubMed] [Google Scholar]; e Abdalla M. A.; Yadav P. P.; Dittrich B.; Schuffler A.; Laatsch H. Org. Lett. 2011, 13, 2156–2159. [DOI] [PubMed] [Google Scholar]
- a Nicolaou K. C.; Harrison S. T. Angew. Chem., Int. Ed. 2006, 45, 3256–3260. [DOI] [PubMed] [Google Scholar]; b Nicolaou K. C.; Harrison S. T. J. Am. Chem. Soc. 2007, 129, 429–440. [DOI] [PubMed] [Google Scholar]; c Nicolaou K. C.; Harrison S. T.; Chen J. S. Synthesis-Stuttgart 2009, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mallams A. K.; Puar M. S.; Rossman R. R. J. Am. Chem. Soc. 1981, 103, 3938–3940. [Google Scholar]; b Mallams A. K.; Puar M. S.; Rossman R. R.; McPhail A. T.; Macfarlane R. D. J. Am. Chem. Soc. 1981, 103, 3940–3943. [Google Scholar]; c Mallams A. K.; Puar M. S.; Rossman R. R.; McPhail A. T.; Macfarlane R. D.; Stephens R. L. J. Chem. Soc., Perkin Trans. 1 1983, 1497–1534. [Google Scholar]; d Kopper S. Liebigs Ann. Chem. 1994, 581–592. [Google Scholar]; e Waitz J. A.; Horan A. C.; Kalyanpur M.; Lee B. K.; Loebenberg D.; Marquez J. A.; Miller G.; Patel M. G. J. Antibiot. 1981, 34, 1101–1106. [DOI] [PubMed] [Google Scholar]
- Jiang Z. D.; Jensen P. R.; Fenical W. Bioorg. Med. Chem. Lett. 1999, 9, 2003–2006. [DOI] [PubMed] [Google Scholar]
- a Pan H. Q.; Zhang S. Y.; Wang N.; Li Z. L.; Hua H. M.; Hu J. C.; Wang S. J. Mar. Drugs 2013, 11, 3891–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Niu S.; Li S.; Chen Y.; Tian X.; Zhang H.; Zhang G.; Zhang W.; Yang X.; Zhang S.; Ju J.; Zhang C. J. Antibiot. 2011, 64, 711–716. [DOI] [PubMed] [Google Scholar]; c Lin Z. J.; Koch M.; Pond C. D.; Mabeza G.; Seronay R. A.; Concepcion G. P.; Barrows L. R.; Olivera B. M.; Schmidt E. W. J. Antibiot. 2014, 67, 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R. B.; Xi T.; Li J.; Wang P.; Li F. C.; Lin Y. C.; Qin S. Mar. Drugs 2011, 9, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner W. T.; Claridge C. A.; Huftalen J. B. J. Antibiot. 1983, 36, 1078–1079. [DOI] [PubMed] [Google Scholar]
- a Lam K. S.; Hesler G. A.; Gustavson D. R.; Berry R. L.; Tomita K.; MacBeth J. L.; Ross J.; Miller D.; Forenza S. J. Antibiot. 1996, 49, 860–864. [DOI] [PubMed] [Google Scholar]; b Schroeder D. R.; Colson K. L.; Klohr S. E.; Lee M. S.; Matson J. A.; Brinen L. S.; Clardy J. J. Antibiot. 1996, 49, 865–872. [DOI] [PubMed] [Google Scholar]
- a Ashton R. J.; Kenig M. D.; Luk K.; Planterose D. N.; Scottwood G. J. Antibiot. 1990, 43, 1387–1393. [DOI] [PubMed] [Google Scholar]; b Luk K.; Readshaw S. A. J. Chem. Soc., Perkin Trans. 1 1991, 1641–1644. [Google Scholar]
- a Momose I.; Iinuma H.; Kinoshita N.; Momose Y.; Kunimoto S.; Hamada M.; Takeuchi T. J. Antibiot. 1999, 52, 781–786. [DOI] [PubMed] [Google Scholar]; b Momose I.; Hirosawa S.; Nakamura H.; Naganawa H.; Iinuma H.; Ikeda D.; Takeuchi T. J. Antibiot. 1999, 52, 787–796. [DOI] [PubMed] [Google Scholar]
- Mazzetti C.; Ornaghi M.; Gaspari E.; Parapin S.; Maffioli S.; Sosio M.; Donadio S. J. Nat. Prod. 2012, 75, 1044–1050. [DOI] [PubMed] [Google Scholar]
- Ramos J. L.; Martinez-Bueno M.; Molina-Henares A. J.; Teran W.; Watanabe K.; Zhang X. D.; Gallegos M. T.; Brennan R.; Tobes R. Microbiol. Mol. Biol. Rev. 2005, 69, 326–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbertson L.; Ahn S. K.; Nodwell J. R. Chem. Biol. 2013, 20, 232–240. [DOI] [PubMed] [Google Scholar]
- a Hegde V. R.; Patel M. G.; Das P. R.; Pramanik B.; Puar M. S. J. Antibiot. 1997, 50, 126–134. [DOI] [PubMed] [Google Scholar]; b Horan A. C.; Shearer M. C.; Hegde V.; Beyazova M. L.; Brodsky B. C.; King A.; Berrie R.; Cardaci K.; Nimeck M. J. Antibiot. 1997, 50, 119–125. [DOI] [PubMed] [Google Scholar]; c Langner M.; Laschat S.; Grunenberg A. Eur. J. Org. Chem. 2003, 1494–1499. [Google Scholar]
- Kren V.; Rezanka T. FEMS Microbiol. Rev. 2008, 32, 858–889. [DOI] [PubMed] [Google Scholar]
- Yu W.-L.; Jones B. D.; Kang M.; Hammons J. C.; Clair J. J. L.; Burkart M. D. J. Nat. Prod. 2013, 76, 817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lindsley C. W. Curr. Top. Med. Chem. 2010, 10, 458–477. [DOI] [PubMed] [Google Scholar]; b Conroy H.; Mawhinney L.; Donnelly S. C. Q. J. Med. 2010, 103, 831–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tomita F.; Tamaoki T. J. Antibiot. 1980, 33, 940–945. [DOI] [PubMed] [Google Scholar]; b Tamaoki T.; Kasai M.; Shirahata K.; Ohkubo S.; Morimoto M.; Mineura K.; Ishii S.; Tomita F. J. Antibiot. 1980, 33, 946–950. [DOI] [PubMed] [Google Scholar]; c Kobinata K.; Uramoto M.; Mizuno T.; Isono K. J. Antibiot. 1980, 33, 244–246. [DOI] [PubMed] [Google Scholar]; d Kamada H.; Izaki K.; Isono K.; Takahashi H. Agric. Biol. Chem. 1984, 2, 419–424. [Google Scholar]; e Morimoto M.; Fukui M.; Ohkubo S.; Tamaoki T.; Tomita F. J. Antibiot. 1982, 35, 1033–1037. [DOI] [PubMed] [Google Scholar]
- Shimotohno K. W.; Endo T.; Furihata K. J. Antibiot. 1993, 46, 682–684. [DOI] [PubMed] [Google Scholar]
- a Furumai T.; Takagi K.; Igarashi Y.; Saito N.; Oki T. J. Antibiot. 2000, 53, 227–232. [DOI] [PubMed] [Google Scholar]; b Igarashi Y.; Takagi K.; Kan Y.; Fujii K.; Harada K.; Furumai T.; Oki T. J. Antibiot. 2000, 53, 233–240. [DOI] [PubMed] [Google Scholar]
- a Tamaoki T.; Tomita F.; Kawamura F.; Saito H. Agric. Biol. Chem. 1983, 47, 59–65. [Google Scholar]; b Ohtomo M.; Yamazaki K.; Ito S.; Shimura K.; Shimizu S.; Minami T.; Fujinaga T.; Shimada K. Jpn. J. Vet. Sci. 1985, 47, 581–587. [DOI] [PubMed] [Google Scholar]
- a Namikawa K.; Sakuma Y.; Sunaga F.; Kanno Y. Jpn. J. Vet. Sci. 1988, 50, 968–970. [DOI] [PubMed] [Google Scholar]; b Namikawa K.; Sakuma Y.; Sunaga F.; Kanno Y. Jpn. J. Vet. Sci. 1988, 50, 605–612. [DOI] [PubMed] [Google Scholar]
- a http://dtp.nci.nih.gov/dtpstandard/servlet/InvivoScreen?testshortname=Tumor+B1+%28ip%29+in+1A&searchlist=333856&searchtype=NSC.; b http://dtp.nci.nih.gov/dtpstandard/servlet/InvivoScreen?testshortname=Tumor+B1+%28ip%29+in+02&searchlist=333856&searchtype=NSC.
- a Nakashima T.; Miura M.; Hara W. Cancer Res. 2000, 60, 1229–1235. [PubMed] [Google Scholar]; b Yang J.; Liu X. S.; Bhalla K.; Kim C. N.; Ibrado A. M.; Cai J. Y.; Peng T. I.; Jones D. P.; Wang X. D. Science 1997, 275, 1129–1132. [DOI] [PubMed] [Google Scholar]; c Cory S.; Huang D. C. S.; Adams J. M. Oncogene 2003, 22, 8590–8607. [DOI] [PubMed] [Google Scholar]
- Hara T.; Omura-Minamisawa M.; Chao C.; Nakagami Y.; Ito M.; Inoue T. Int. J. Radiat. Oncol. 2005, 61, 517–528. [DOI] [PubMed] [Google Scholar]
- a Azmi A. S.; Mohammad R. M. J. Cell. Physiol. 2009, 218, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rutledge S. E.; Chin J. W.; Schepartz A. Curr. Opin. Chem. Biol. 2002, 6, 479–485. [DOI] [PubMed] [Google Scholar]; c Fischer U.; Schulze-Osthoff K. Cell Death Differ. 2005, 12, 942–961. [DOI] [PubMed] [Google Scholar]; d Huang Z. W. Oncogene 2000, 19, 6627–6631. [DOI] [PubMed] [Google Scholar]
- a Anether G.; Tinhofer I.; Senfter M.; Greil R. Blood 2003, 101, 4561–4568. [DOI] [PubMed] [Google Scholar]; b Mayer M. P.; Bukau B. Cell. Mol. Life Sci. 2005, 62, 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nakajima H.; Sakaguchi K.; Fujiwara I.; Mizuta M.; Tsuruga M.; Magae J.; Mizuta N. Biochem. Biophys. Res. Commun. 2007, 356, 260–265. [DOI] [PubMed] [Google Scholar]; b Osaki M.; Oshimura M.; Ito H. Apoptosis 2004, 9, 667–676. [DOI] [PubMed] [Google Scholar]; c Luo J.; Manning B. D.; Cantley L. C. Cancer Cell 2003, 4, 257–262. [DOI] [PubMed] [Google Scholar]
- a Tinhofer I.; Anether G.; Senfter M.; Pfaller K.; Bernhard D.; Hara M.; Greil R. FASEB J. 2002, 16, 1295–1321. [DOI] [PubMed] [Google Scholar]; b Tinhofer I.; Anether G.; Senfter M.; Greil R. Blood 2001, 98, 224B–224B. [DOI] [PubMed] [Google Scholar]
- Kim Y. H.; Shin H. C.; Song D. W.; Lee S. H.; Furumai T.; Park J. W.; Kwon T. K. Biochem. Biophys. Res. Commun. 2003, 309, 449–456. [DOI] [PubMed] [Google Scholar]
- Tomita F.; Tamaoki T.; Shirahata K.; Kasai M.; Morimoto M.; Ohkubo S.; Mineura K.; Ishii S. J. Antibiot. 1980, 33, 668–670. [DOI] [PubMed] [Google Scholar]
- a Tamaoki T.; Kasai M.; Shirahata K.; Tomita F. J. Antibiot. 1982, 35, 979–984. [DOI] [PubMed] [Google Scholar]; b Kaneko M.; Nakashima T.; Uosaki Y.; Hara M.; Ikeda S.; Kanda Y. Bioorg. Med. Chem. Lett. 2001, 11, 887–890. [DOI] [PubMed] [Google Scholar]
- Kren V.; Martinkova L. Curr. Med. Chem. 2001, 8, 1303–1328. [DOI] [PubMed] [Google Scholar]
- Park H. R.; Chijiwa S.; Furihata K.; Hayakawa Y.; Shin-Ya K. Org. Lett. 2007, 9, 1457–1460. [DOI] [PubMed] [Google Scholar]
- a Shin-Ya K. Biosci., Biotechnol., Biochem. 2005, 69, 867–872. [DOI] [PubMed] [Google Scholar]; b Li J. Z.; Lee A. S. Curr. Mol. Med. 2006, 6, 45–54. [DOI] [PubMed] [Google Scholar]
- Yu Z. F.; Luo H.; Fu W. M.; Mattson M. P. Exp. Neurol. 1999, 155, 302–314. [DOI] [PubMed] [Google Scholar]
- a Matsuo J.; Tsukumo Y.; Sakurai J.; Tsukahara S.; Park H. R.; Shin-ya K.; Watanabe T.; Tsuruo T.; Tomida A. Cancer Sci. 2009, 100, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Park H. R.; Tomida A.; Sato S.; Tsukumo Y.; Yun J.; Yamori T.; Hayakawa Y.; Tsuruo T.; Shin-Ya K. J. Natl. Cancer Inst. 2004, 96, 1300–1310. [DOI] [PubMed] [Google Scholar]
- Arsham A. M.; Howell J. J.; Simon M. C. J. Biol. Chem. 2003, 278, 29655–29660. [DOI] [PubMed] [Google Scholar]
- Sausville E. A. J. Natl. Cancer Inst. 2004, 96, 1266–1267. [DOI] [PubMed] [Google Scholar]
- a Ueda J. Y.; Chijiwa S.; Takagi M.; Shin-Ya K. J. Antibiot. 2008, 61, 752–755. [DOI] [PubMed] [Google Scholar]; b Zhao P.; Ueda J.; Kozone I.; Chijiwa S.; Takagi M.; Kudo F.; Nishiyama M.; Shin-Ya K.; Kuzuyama T. Org. Biomol. Chem. 2009, 7, 1454–1460. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Yoshizawa A.; Chijiwa S.; Ueda J.; Takagi M.; Shin-ya K.; Takahashi T. Chem.—Asian J. 2009, 4, 1114–1125. [DOI] [PubMed] [Google Scholar]
- a Imai H.; Kaniwa H.; Tokunaga T.; Fujita S.; Furuya T.; Matsumoto H.; Shimizu M. J. Antibiot. 1987, 40, 1483–1489. [DOI] [PubMed] [Google Scholar]; b Imai H.; Suzuki K. I.; Morioka M.; Numasaki Y.; Kadota S.; Nagai K.; Sato T.; Iwanami M.; Saito T. J. Antibiot. 1987, 40, 1475–1482. [DOI] [PubMed] [Google Scholar]
- Iwatsuki M.; Otoguro K.; Ishiyama A.; Namatame M.; Nishihara-Tukashima A.; Hashida J.; Nakashima T.; Masuma R.; Takahashi Y.; Yamada H.; Omura S. J. Antibiot. 2010, 63, 619–622. [DOI] [PubMed] [Google Scholar]
- Poehlsgaard J.; Douthwaite S. Nat. Rev. Microbiol. 2005, 3, 870–881. [DOI] [PubMed] [Google Scholar]
- Nakai R.; Kakita S.; Asai A.; Chiba S.; Akinaga S.; Mizukami T.; Yamashita Y. J. Antibiot. 2001, 54, 836–839. [DOI] [PubMed] [Google Scholar]
- a Kim N. W.; Piatyszek M. A.; Prowse K. R.; Harley C. B.; West M. D.; Ho P. L. C.; Coviello G. M.; Wright W. E.; Weinrich S. L.; Shay J. W. Science 1994, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]; b Nakai R.; Ishida H.; Asai A.; Ogawa H.; Yamamoto Y.; Kawasaki H.; Akinaga S.; Mizukami T.; Yamashita Y. Chem. Biol. 2006, 13, 183–190. [DOI] [PubMed] [Google Scholar]
- Iorio M.; Maffioli S. I.; Gaspari E.; Rossi R.; Mauri P.; Sosio M.; Dionadio S. J. Nat. Prod. 2012, 75, 1991–1993. [DOI] [PubMed] [Google Scholar]
- Igarashi Y.; Iida T.; Oku N.; Watanabe H.; Furihata K.; Miyanouchi K. J. Antibiot. 2012, 65, 355–359. [DOI] [PubMed] [Google Scholar]
- a Schindler P. W.; Scrutton M. C. Eur. J. Biochem. 1975, 55, 543–553. [DOI] [PubMed] [Google Scholar]; b Schindler P. W.; Zahner H. Eur. J. Biochem. 1973, 39, 591–600. [DOI] [PubMed] [Google Scholar]
- Zeczycki T. N.; Maurice M. S.; Attwood P. V. Open Enzym. Inhib. J. 2010, 3, 8–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler P. W. Eur. J. Biochem. 1975, 51, 579–585. [DOI] [PubMed] [Google Scholar]
- Pache W.; Chapman D. Biochim. Biophys. Acta 1972, 255, 348–357. [DOI] [PubMed] [Google Scholar]
- Yamamoto I.; Nakagawa M.; Hayakawa Y.; Adadchi K.; Kobayashi E. J. Antibiot. 1987, 40, 1452–1454. [DOI] [PubMed] [Google Scholar]
- Muntwyler R.; Keller-Schierlein W. Helv. Chim. Acta 1972, 55, 2071–2094. [DOI] [PubMed] [Google Scholar]
- Shin J. M.; Munson K.; Vagin O.; Sachs G. Pflug. Arch. Eur. J. Physiol. 2009, 457, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bonjouklian R. Tetrahedron Lett. 1993, 34, 7861–7864. [Google Scholar]; b Bonjouklian R.; Mynderse J. S.; Hunt A. H.; Deeter J. B. Tetrahedron Lett. 1993, 34, 7857–7860. [Google Scholar]
- a Ohtsuka T.; Kotaki H.; Nakayama N.; Itezono Y.; Shimma N.; Kudoh T.; Kuwahara T.; Arisawa M.; Yokose K. J. Antibiot. 1992, 46, 11–17. [DOI] [PubMed] [Google Scholar]; b Ohtsuka T.; Kudoh T.; Shimma N.; Kotaki H.; Nakayama N.; Itezono Y.; Fujisaki N.; Watanabe J.; Yokose K.; Seto H. J. Antibiot. 1992, 45, 140–143. [DOI] [PubMed] [Google Scholar]; c Ohtsuka T.; Nakayama N.; Itezono Y.; Shimma N.; Kuwahara T.; Yokose K.; Seto H. J. Antibiot. 1993, 46, 18–24. [DOI] [PubMed] [Google Scholar]
- a Jensen R. T. Yale J. Biol. Med. 1996, 69, 245–259. [PMC free article] [PubMed] [Google Scholar]; b Reubi J. C.; Schaer J. C.; Waser B. Cancer Res. 1997, 57, 1377–1386. [PubMed] [Google Scholar]
- a Kuwahara T.; Kudoh T.; Nagase H.; Takamiya M.; Nakano A.; Ohtsuka T.; Yoshizaki H.; Arisawa M. Eur. J. Pharmacol. 1992, 221, 99–105. [DOI] [PubMed] [Google Scholar]; b Watanabe J.; Fujisaki N.; Fujimori K.; Anzai Y.; Oshima S.; Sano T.; Ohtsuka T.; Watanabe K.; Okuda T. J. Antibiot. 1993, 46, 1–10. [DOI] [PubMed] [Google Scholar]
- Zapf C. W.; Harrison B. A.; Drahl C.; Sorensen E. J. Angew. Chem., Int. Ed. 2005, 44, 6533–6537. [DOI] [PubMed] [Google Scholar]
- Snider B. B.; Zou Y. F. Org. Lett. 2005, 7, 4939–4941. [DOI] [PubMed] [Google Scholar]
- Couladouros E. A.; Bouzas E. A.; Magos A. D. Tetrahedron 2006, 62, 5272–5279. [Google Scholar]
- a Bihelovic F.; Karadzic I.; Matovic R.; Saicic R. N. Org. Biomol. Chem. 2013, 11, 5413–5424. [DOI] [PubMed] [Google Scholar]; b Bihelovic F.; Saicic R. N. Angew. Chem., Int. Ed. 2012, 51, 5687–5691. [DOI] [PubMed] [Google Scholar]
- a Rath J. P.; Kinast S.; Maier M. E. Org. Lett. 2005, 7, 3089–3092. [DOI] [PubMed] [Google Scholar]; b Rath J. P.; Eipert M.; Kinast S.; Maier M. E. Synlett 2005, 314–318. [Google Scholar]; c Zografos A. L.; Yiotakis A.; Georgiadis D. Org. Lett. 2005, 7, 4515–4518. [DOI] [PubMed] [Google Scholar]
- a Smith A. B.; Bosanac T.; Basu K. J. Am. Chem. Soc. 2009, 131, 2348–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smith A. B.; Basu K.; Bosanac T. J. Am. Chem. Soc. 2007, 129, 14872–14874. [DOI] [PubMed] [Google Scholar]
- Smith A. B.; Fox R. J.; Razler T. M. Acc. Chem. Res. 2008, 41, 675–687. [DOI] [PubMed] [Google Scholar]
- Tenenbaum J. M.; Morris W. J.; Custar D. W.; Scheidt K. A.. Exploring Prins Strategies for the Synthesis of Okilactomycin; San Diego, 2013; Vol. 9. [Google Scholar]
- Tenenbaum J. M.; Morris W. J.; Custar D. W.; Scheidt K. A. Angew. Chem., Int. Ed. 2011, 50, 5892–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K.; Shimotani A.; Yoshii E.; Yamaguchi K. Heterocycles 1992, 34, 2259–2261. [Google Scholar]
- a Paquette L. A.; Boulet S. L. Synthesis-Stuttgart 2002, 888–894. [Google Scholar]; b Boulet S. L.; Paquette L. A. Synthesis-Stuttgart 2002, 895–900. [Google Scholar]
- Niu D.; Hoye T. R. Org. Lett. 2012, 14, 828–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Roush W. R.; Limberakis C.; Kunz R. K.; Barda D. A. Org. Lett. 2002, 4, 1543–1546. [DOI] [PubMed] [Google Scholar]; b Roush W. R.; Barda D. A. J. Am. Chem. Soc. 1997, 119, 7402–7403. [Google Scholar]
- Jones B. D.; Clair J. J. L.; Moore C. E.; Rheingold A. L.; Burkart M. D. Org. Lett. 2010, 12, 4516–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page P. C. B.; Vahedi H.; Batchelor K. J.; Hindley S. J.; Edgar M.; Beswick P. Synlett 2003, 1022–1024. [Google Scholar]
- Takeda K.; Kawanishi E.; Nakamura H.; Yoshii E. Tetrahedron Lett. 1991, 32, 4925–4928. [Google Scholar]
- Boeckman R. K.; Shao P. C.; Wrobleski S. T.; Boehmler D. J.; Heintzelman G. R.; Barbosa A. J. J. Am. Chem. Soc. 2006, 128, 10572–10588. [DOI] [PubMed] [Google Scholar]
- Roush W. R.; Reilly M. L.; Koyama K.; Brown B. B. J. Org. Chem. 1997, 62, 8708–8721. [Google Scholar]
- a Roush W. R.; Sciotti R. J. J. Am. Chem. Soc. 1994, 116, 6457–6458. [Google Scholar]; b Roush W. R.; Sciotti R. J. J. Am. Chem. Soc. 1998, 120, 7411–7419. [Google Scholar]
- Takeda K.; Kobayashi T.; Saito K.; Yoshii E. J. Org. Chem. 1988, 53, 1092–1095. [Google Scholar]
- Roush W. R.; Brown B. B.; Drozda S. E. Tetrahedron Lett. 1988, 29, 3541–3544. [Google Scholar]
- a Marshall J. A.; Salovich J. M.; Shearer B. G. J. Org. Chem. 1990, 55, 2398–2403. [Google Scholar]; b Marshall J. A.; Grote J.; Shearer B. J. Org. Chem. 1986, 51, 1633–1635. [Google Scholar]; c Marshall J. A.; Grote J.; Audia J. E. J. Am. Chem. Soc. 1987, 109, 1186–1194. [Google Scholar]; d Marshall J. A.; Audia J. E.; Grote J.; Shearer B. G. Tetrahedron 1986, 42, 2893–2902. [Google Scholar]
- a Matsuda K.; Nomura K.; Yoshii E. J. Chem. Soc., Chem. Commun. 1989, 221–223. [Google Scholar]; b Takeda K.; Kato H.; Sasahara H.; Yoshii E. J. Chem. Soc., Chem. Commun. 1986, 1197–1198. [Google Scholar]; c Takeda K.; Shibata Y.; Sagawa Y.; Urahata M.; Funaki K.; Hori K.; Sasahara H.; Yoshii E. J. Org. Chem. 1985, 50, 4673–4681. [Google Scholar]
- Roush W. R.; Koyama K. Tetrahedron Lett. 1992, 33, 6227–6230. [Google Scholar]
- a Boeckman R. K.; Barta T. E.; Nelson S. G. Tetrahedron Lett. 1991, 32, 4091–4094. [Google Scholar]; b Boeckman R. K.; Estep K. G.; Nelson S. G.; Walters M. A. Tetrahedron Lett. 1991, 32, 4095–4098. [Google Scholar]; c Boeckman R. K.; Wrobleski S. T. J. Org. Chem. 1996, 61, 7238–7239. [DOI] [PubMed] [Google Scholar]
- Marshall J. A.; Xie S. P. J. Org. Chem. 1992, 57, 2987–2989. [Google Scholar]
- Takeda K.; Yano S.; Sato M.; Yoshii E. J. Org. Chem. 1987, 52, 4135–4137. [Google Scholar]
- a Roush W. R.; Brown B. B. J. Org. Chem. 1993, 58, 2151–2161. [Google Scholar]; b Roush W. R.; Brown B. B. Tetrahedron Lett. 1989, 30, 7309–7312. [Google Scholar]
- Roush W. R.; Brown B. B. J. Org. Chem. 1993, 58, 2162–2172. [Google Scholar]
- Takeda K.; Yano S.; Yoshii E. Tetrahedron Lett. 1988, 29, 6951–6954. [Google Scholar]
- Takeda K.; Igarashi Y.; Okazaki K.; Yoshii E.; Yamaguchi K. J. Org. Chem. 1990, 55, 3431–3434. [Google Scholar]
- Marshall J. A.; Audia J. E.; Grote J. J. Org. Chem. 1984, 49, 5277–5279. [Google Scholar]
- a Ireland R. E.; Thompson W. J.; Srouji G. H.; Etter R. J. Org. Chem. 1981, 46, 4863–4873. [Google Scholar]; b Ireland R. E.; Thompson W. J. J. Org. Chem. 1979, 44, 3041–3052. [Google Scholar]
- Snider B. B.; Burbaum B. W. J. Org. Chem. 1983, 48, 4370–4374. [Google Scholar]
- a Hirsenkorn R.; Haagzeino B.; Schmidt R. R. Tetrahedron Lett. 1990, 31, 4433–4436. [Google Scholar]; b Hirsenkorn R.; Schmidt R. R. Liebigs Ann. Chem. 1990, 883–899. [Google Scholar]; c Schmidt R. R.; Hirsenkorn R. Tetrahedron Lett. 1984, 25, 4357–4360. [Google Scholar]
- Roth G. P.; Rithner C. D.; Meyers A. I. Tetrahedron 1989, 45, 6949–6962. [Google Scholar]
- Kunst E.; Kirschning A. Synthesis-Stuttgart 2006, 2397–2403. [Google Scholar]
- a Katsuta R.; Arai K.; Yajima A.; Nukada T. Synlett 2012, 397–400. [Google Scholar]; b Sasaki S.; Samejima S.; Uruga T.; Anzai K.; Nishi N.; Kawakita E.; Takao K.; Tadano K. J. Antibiot. 2013, 66, 147–154. [DOI] [PubMed] [Google Scholar]
- a Roush W. R.; Barda D. A. Tetrahedron Lett. 1997, 38, 8781–8784. [Google Scholar]; b Roush W. R.; Barda D. A. Tetrahedron Lett. 1997, 38, 8785–8788. [Google Scholar]; c Roush W. R.; Barda D. A.; Limberakis C.; Kunz R. K. Tetrahedron 2002, 58, 6433–6454. [Google Scholar]
- Trullinger T. K.; Qi J.; Roush W. R. J. Org. Chem. 2006, 71, 6915–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedel O.; Francais A.; Haudrechy A. Synlett 2005, 2313–2316. [Google Scholar]
- a Jensen P. R.; Chavarria K. L.; Fenical W.; Moore B. S.; Ziemert N. J. Ind. Microbiol. Biotechnol. 2014, 41, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gulder T. A. M.; Moore B. S. Curr. Opin. Microbiol. 2009, 12, 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gerwick W. H.; Fenner A. M. Microb. Ecol. 2013, 65, 800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Newman D. J.; Cragg G. M. J. Nat. Prod. 2012, 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lane A. L.; Moore B. S. Nat. Prod. Rep. 2011, 28, 411–428. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Dixon N.; Wong L. S.; Geerlings T. H.; Micklefield J. Nat. Prod. Rep. 2007, 24, 1288–1310. [DOI] [PubMed] [Google Scholar]
- a Newhouse T.; Baran P. S.; Hoffmann R. W. Chem. Soc. Rev. 2009, 38, 3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wender P. A.; Verma V. A.; Paxton T. J.; Pillow T. H. Acc. Chem. Res. 2008, 41, 40–49. [DOI] [PubMed] [Google Scholar]; c Kuttruff C. A.; Eastgate M. D.; Baran P. S. Nat. Prod. Rep. 2014, 31, 419–432. [DOI] [PubMed] [Google Scholar]; d Wender P. A.; Miller B. L. Nature 2009, 460, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chang M. C. Y.; Keasling J. D. Nat. Chem. Biol. 2006, 2, 674–681. [DOI] [PubMed] [Google Scholar]; b Khosla C.; Keasling J. D. Nat. Rev. Drug Discovery 2003, 2, 1019–1025. [DOI] [PubMed] [Google Scholar]; c Pfeifer B. A.; Admiraal S. J.; Gramajo H.; Cane D. E.; Khosla C. Science 2001, 291, 1790–1792. [DOI] [PubMed] [Google Scholar]; d Lau J.; Tran C.; Licari P.; Galazzo J. J. Biotechnol. 2004, 110, 95–103. [DOI] [PubMed] [Google Scholar]