

Abstract
Background
A collection of 175 melon (Cucumis melo L.) accessions (including wild relatives, feral types, landraces, breeding lines and commercial cultivars) from 50 countries was selected to study the phenotypic variability for ripening behavior and sugar accumulation. The variability of single nucleotide polymorphisms (SNPs) at 53 selected candidate genes involved in sugar accumulation and fruit ripening processes was studied, as well as their association with phenotypic variation of related traits.
Results
The collection showed a strong genetic structure, defining seven groups plus a number of accessions that could not be associated to any of the groups (admixture), which fitted well with the botanical classification of melon varieties. The variability in candidate genes for ethylene, cell wall and sugar-related traits was high and similar to SNPs located in reference genes. Variability at ripening candidate genes had an important weight on the genetic stratification of melon germplasm, indicating that traditional farmers might have selected for ripening traits during cultivar diversification. A strong relationship was also found between the genetic structure and phenotypic diversity, which could hamper genetic association studies. Accessions belonging to the ameri group are the most appropriate for association analysis given the high phenotypic and molecular diversity within the group, and lack of genetic structure.
The most remarkable association was found between sugar content and SNPs in LG III, where a hotspot of sugar content QTLs has previously been defined. By studying the differences in allelic variation of SNPs within horticultural groups with specific phenotypic features, we also detected differential variation in sugar-related candidates located in LGIX and LGX, and in ripening-related candidates located in LGII and X, all in regions with previously mapped QTLs for the corresponding traits.
Conclusions
In the current study we have found an important variability at both the phenotypic and candidate gene levels for ripening behavior and sugar accumulation in melon fruit. By combination of differences in allelic diversity and association analysis, we have identified several candidate genes that may be involved in the melon phenotypic diversity.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-015-0183-2) contains supplementary material, which is available to authorized users.
Keywords: Melon, Climacteric ripening, Sugar, Germplasm collection
Background
Melon (Cucumis melo L.) is one of the most important crops within the Cucurbitaceae family, presenting a high variability in fruit traits among different cultivars, ranging from non-sweet fruits that are harvested before maturity and consumed as vegetables, to sweet fruits with high sugar concentrations that are eaten in salads or as dessert. Melon has been proposed to have an African and/or Asian origin [1], and was subject to intense diversification after domestication, with primary centers of diversity in Central Asia and secondary centers in the Mediterranean basin and Far East countries.
C. melo has been divided into two subspecies: ssp. melo, and ssp. agrestis [2]. Recently Pitrat [3] split these subspecies into 15 botanical groups: ssp. melo, which includes cantalupensis, reticulatus, adana, chandalak, ameri, inodorus chate, flexuosus, dudaim and tibish (later reclassified as ssp. agrestis by Esteras et al., [4]), and ssp. agrestis, which includes momordica, conomon, chinensis, makuwa and acidulus. Among these, melon cultivars belonging to the cantalupensis, reticulatus and inodorus groups are economically the most important (e.g. cantaloups, western shippers, galias, ‘Piel de Sapo’ and honeydew).
Melon fruits display a broad range of phenotypic variation. Melon fruit weight varies from a few grams to several kilograms (fruits up to 35 kg have been reported), and the shape may be round, oblate, ovate, elliptical or extremely elongated [5-8]. A huge variability also exists for other characteristics associated with fruit quality, such as flesh color, sugar content and aroma [9]. Different combinations can be found, varying from the non-sweet non-aromatic fruits of cultivars from the flexuosus group to the sweet and aromatic cantalupensis melons [10].
Differences in sugar content among cultivars mainly reflect differences in sucrose accumulation [8]. Sucrose accumulation is controlled by a major gene [9] that explains the main differences between sweet and non-sweet cultivars, although multiple minor quantitative trait loci (QTLs) for sugar accumulation have also been reported [11]. The metabolic pathway of sugar metabolism in melon fruit has been investigated in several studies [9,12]. Melon, like other cucurbits is a symplastic phloem loader that synthesize raffinose and stachyose from sucrose in specialized intermediary cells in source leaves. These two raffinose-family oligosaccharides (RFOs), plus sucrose, are translocated from source leaves to the developing melon fruit. After phloem unloading in sink organs, the RFOs are hydrolyzed by acid and neutral α-galactosidases (AAG, NAG), producing sucrose and galactose. The latter is phosphorylated by galactokinase and then converted to glucose 6-phosphate, which can either be respired or used to synthesize sucrose via sucrose-phosphate synthase (SPS) and sucrose-phosphate phosphatase (SPP). Sucrose unloaded from the phloem can be hydrolyzed in the apoplast by cell wall invertase (CIN). The resulting hexose sugars (glucose and fructose) are imported into cells by monosaccharide transporters, phosphorylated by hexokinase (HXK) and fructokinase (FK) and used for respiration or sucrose resynthesis. Sucrose can also be unloaded symplastically. Within the cell, sucrose can be catabolized in the cytosol by sucrose synthase (SUS) or neutral invertase (NIN), or imported into the vacuole for storage or hydrolysis by vacuolar acid invertase (AIN), with potential regulation of the latter by invertase inhibitor proteins (INH). During early fruit development, sucrose catabolism predominates, as the carbon and energy derived from sucrose are needed for growth-related processes. As the fruit develops, more and more sucrose is stored rather than respired, and this transition from sucrose catabolism to storage is characterized by loss of AIN activity. Vacuolar processing enzymes (VPE) participate in protein maturation in the vacuole and are also implicated as factors in sugar storage. For example, a reduction in the rate of the proteolysis of vacuolar invertases can lead to their accumulation and modify sugar metabolism and accumulation [13]. Some of the genes encoding sugar metabolizing enzymes and VPEs have been mapped in melon using specific biparental populations [11,14-16].
Melon also comprises broad genetic variation for ripening behavior, with climacteric and non-climacteric varieties. Typical climacteric melons are found within the cantalupensis group. These exhibit a distinct peak in respiration and ethylene production at maturity, and generally have a short ripening time and rapidly deteriorate in quality after harvest [17]. In contrast, melons from the inodorus group are unable to produce autocatalytic ethylene [18] and, in general, ripen more slowly and have a longer postharvest shelf life [19]. This diversity in ripening behavior makes melon an ideal subject for investigating the physiological and genetic basis for differences between ethylene-dependent and ethylene-independent fruit ripening. There have been several studies of the inheritance of climacteric ripening behavior in melon. Perin et al. [17], investigated the segregation of the formation of the abscission layer and ethylene production in a climacteric x non-climacteric cross. Both traits were controlled by two independent loci (Al-3 and Al-4) in linkage groups (LG) VIII and IX, and four further QTLs in LGs I, II, III and XI. In a collection of near isogenic lines (NILs) derived from two non-climacteric genotypes [20-22], one NIL showed climacteric behavior [14]. This NIL carried two introgressions in LG III and LG VI, and both of them had QTLs involved in climacteric ripening (ETHQB3.5 and ETHQV6.3, respectively), which interacted epistatically [23]. Fine mapping studies narrowed down the position of ETHQV6.3 to a 4.5-Mbp physical region of the melon genome [23].
Ethylene affects the expression of many ripening related genes, in both climacteric and non-climacteric fruits, but expression of other genes is ethylene independent even in climacteric fruits [24-26]. Most of the research based on the regulation of ripening has been focused on the climacteric tomato fruit. The deciphering of the ethylene biosynthetic pathway, including the isolation of the two key enzymes, 1-aminocyclopropane-1-carboxylate (ACC) synthase and ACC oxidase (ACS and ACO) [25], represented substantial advances in our understanding of the role of this hormone in tomato ripening. Further insights came from identification of components of the ethylene perception and signal transduction pathways. These includes ERS (ETHYLENE RESPONSE SENSOR) and ETR (ETHYLENE RESPONSE), which encode membrane proteins involved in signal reception, RTE1 (REVERSION TO ETHYLENE SENSITIVITY 1) that might be involved in negative feedback of ethylene responses, and CTR1 (CONSTITUTIVE TRIPLE RESPONSE 1), which encode a Raf-like kinase that negatively regulates the downstream ethylene response pathway. Also transcription factors such as ERF (ETHYLENE RESPONSIVE FACTOR), EIN (ETHYLENE INSENSITIVE), EIL (ETHYLENE-INSENSITIVE LIKE), and EBF (EIN3-BINDING F-BOX) are involved in ethylene responses [26-28].
In tomato, mapping of mutants showing defects in fruit ripening, such as ripening inhibitor (rin; also called MADS-RIN), non-ripening (nor, also called NAC-NOR), colorless non-ripening (Cnr, also called SPL-CNR) and NR (never ripe) revealed that all the underlying lesions were in transcription factor genes [29-31]. Other transcription factors shown to be involved in ripening include SlHB-1, ETO1, E8 and E4/E8BP. SlHB-1 is a HD-zip homeodomain protein that interacts with ACO1, decreases ethylene synthesis and delays ripening [32]. ETO1 (ETHYLENE OVERPRODUCER 1) is a negative regulator of ethylene ACS type2 [33]. E8 is induced in mature fruits in response to ethylene, although its precise function is still not well defined [34]. E4/E8 binding protein is a protein that interacts with E8 promoter sequences, acting as a positive regulator during fruit ripening [35]. Despite the discovery of the key factors in fruit ripening [36] and interactions between them [37], much remains to be learned before we have a complete understanding of this complex process.
An array of genomic and genetic tools has become available in the last few years for melon research, including genetic maps [11], microarrays [38], TILLING and EcoTILLING platforms [39,40], new mapping populations such as NILs [41] and double haploid lines (DHLs) [42], deep transcriptomic sequencing data [43,44], and a complete genome sequence [45]. These tools are now being deployed to investigate the physiological and genetic basis for agronomically important traits in melon, including fruit ripening and sugar content.
The huge genetic diversity of the species has been studied with different molecular markers [46]. However, despite the availability of massive collections of SNPs, these are still underexploited. Blanca et al. [44] created the most complete version of the melon transcriptome to date, using a combination of expressed sequence tags (ESTs) from Sanger sequencing and next generation sequencing methods, e.g. 454 (Roche) and SOLID (Life Technologies Inc). The resulting database contains thousands of in silico identified SNPs, representing the largest collection existing for melon (www.melogene.net).
In an attempt to study the variation of genes involved in sugar metabolism and the ripening process, we searched the Melogene database for SNPs located in a set of candidate genes involved in these processes. We used this set of SNPs, along with reference SNPs evenly distributed in the genome, to genotype a set of 175 melon accessions, including commercial varieties, landraces, and wild or feral melons from over 50 countries, representing the wide diversity within the species. We report the variability of a set of genes involved in ripening behavior and sugar accumulation in melon fruit, providing a framework for studying the putative role of those genes in the diversification of the species, and to associate allelic variants with the phenotypic differences within the germplasm.
Results
Germplasm population structure
The genetic diversity of the whole germplasm collection (Additional file 1) based on SNP variability (Additional file 2) was analyzed using principal component analysis (PCA) and STRUCTURE. The PCA approach showed a clear differentiation between the two subspecies, melo and agrestis (Figure 1a), so in order to investigate more subtle genetic structure, we performed the PCA for each subspecies separately (Figure 1b and c). Within ssp melo (Figure 1b), the PC1 axis separated inodorus from cantalupensis and reticulatus cultivars. Taking together the first two PC dimensions (11.8% and 7.9% of the total variance for PC1 and PC2 respectively), a group of Spanish inodorus landraces (located in the upper-right part of the plot) is clearly differentiated from a group that includes other Spanish landraces and inodorus and ameri cultivars from Eastern Europe, Asia and North Africa (located in the center/lower-right part of the plot). The cantalupensis types could also be split into two groups: modern ‘Charentais’ and reticulatus cultivars in the upper-left area, and older cantaloup landraces in the central part of the plot. The non-sweet flexuosus types were grouped together, but clearly separate from all the sweet melons.
Figure 1.
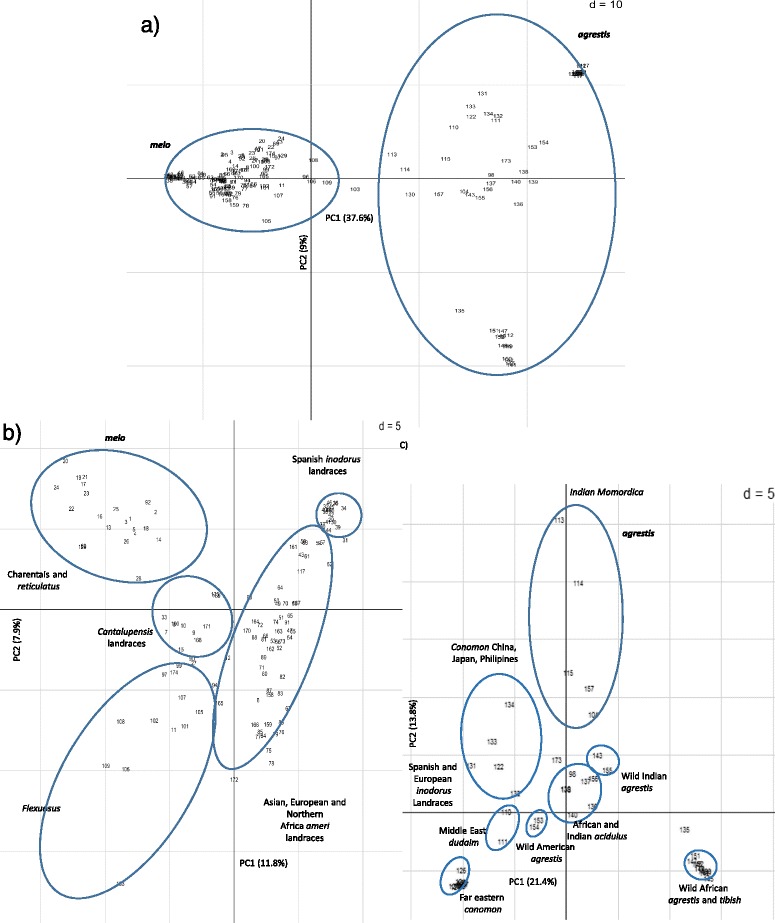
Principal Component Analysis (PCA) based on 210 SNP markers distributed through the melon genome and in candidate genes for quality traits. a) the PCA analysis for all the germplasm collection. b and c) C. melo ssp. melo and agrestis considered separately.
For ssp. agrestis (Figure 1c), two groups could be distinguished along the PC1 axis (21.4% of the total variance): African wild agrestis accessions with tibish varieties, and conomon types that originate from the Far East. The other accessions of this subspecies, including Indian momordica, wild Indian agrestis, Indian and African acidulus, Middle East dudaim and additional Far East conomon were distributed between the two extreme populations. Some wild American melons, likely representing African or Asian introductions, were also found between those populations. Along the PC2 axis (13.8% of the total variance), the Indian momordica accessions could be distinguished from the rest. This distribution of the genetic variability in the PCA space supports previous observations, indicating that Far East conomon varieties represent one extreme within the overall genetic distribution of melons [5,47], and that the African and Indian wild melons are genetically distinct [4].
Analysis with STRUCTURE following the Evanno ∆K approach [48] to determine the number of populations gave a maximum value when K = 2, with lesser maxima for K = 5 and K = 7 (Additional file 3). The most strongly supported division into two subpopulations (K = 2) reflects the classification into two subspecies, agrestis and melo (Figure 2a), which is supported by most previous molecular studies [4] and the initial PCA of our complete dataset. Further resolution into seven sub-groups (K = 7) was consistent with groupings based on geographical origin and fruit characteristics (Figure 2b). Five of these groups belong to ssp. melo and two to ssp. agrestis, with a small number of accessions in a mixed group that was not clearly resolved. Within subspecies melo, the cantalupensis varieties split into two groups. The first includes mostly French ‘Charentais’ varieties (population 1, dark blue line in Figure 2b), such as ‘Vedrantais’ and ‘Nantais Oblong’. The second includes reticulatus melons (population 2, dark purple line in Figure 2b), with both commercial cultivars and breeding lines, most of which have an American origin (e.g. ‘Top Mark’, ‘Dulce’, ‘PMR 45’). Some other commercial American reticulatus cultivars (e.g. ‘Golden Honey’, ‘Golden Champlain’) seem to be a mixture of Charentais and reticulatus populations. Some other cultivars from different origins were also included within one of these groups. Thus, the Japanese ‘Yamato Purinsu’ and the Chinese ‘China 151’, both considered makuwa types, were included with the French group although they showed some commonality with the conomon population (population 6, light purple line in Figure 2b). These two cultivars have been used in breeding commercial melons due to their high fruit quality and resistance to viruses [49].
Figure 2.
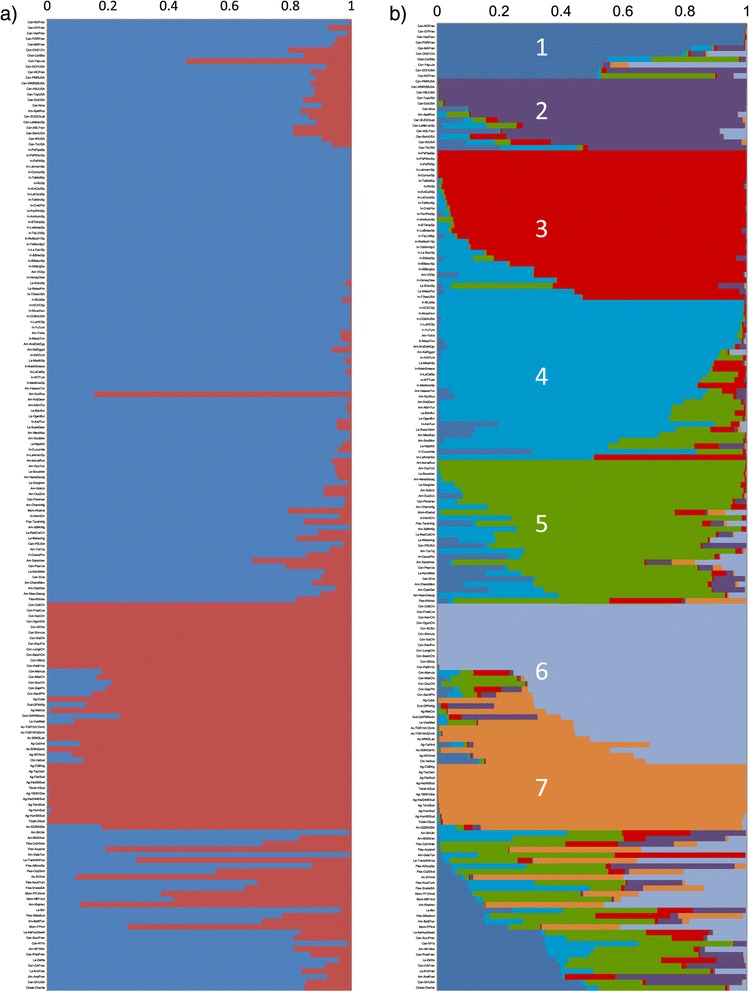
Inferred population structure of the collection using STRUCTURE [ 75 ]. Each accession is represented by a line that is partitioned into coloured segments in proportion to the estimated membership in the corresponding populations. a) Best K choice based on the ΔK method K = 2; blue line represents melo subspecies and red line agrestis. b) second best choice K = 7; Dark blue line represents ‘Charentains’ group (1), purple line reticulatus (2), red line Spanish Inodorus accessions (3), light blue line a mixture of inodorus and ameri (4), green line mostly ameri (5), light purple line conomon (6) and orange line African agrestis (7). Abbreviations: Can = cantalupensis, In = inodorus, Am = ameri, Flex = flexuosus, Cha = chate, Dud = dudaim, Con = conomon, Mom = momordica, Chi = chito, Tibish = tibish, Ag = agrestis. La = landraces. Last three letters indicate the country of origin.
Population 3 (red line in Figure 2b) contains mostly inodorus varieties, especially Spanish and Portuguese casaba melons, commercial cultivars and landraces of the market classes ‘Piel de sapo’ (e.g. ‘Pipa de oro’, ‘Piñoncillo’, ‘Piñonet’ , ‘Verde Pinto’), ‘Amarillo’ (e.g. ‘Amarillo Oro’ , ‘Caña Dulce’), ‘Tendral’ (e.g. ‘Mollerusa’ , ‘Negro de Invierno’), ‘Rochet’ (e.g. ‘Mochuelo’), and ‘Blanco’ (e.g. ‘Crabranco’, ‘Tempranillo’). Other singular Spanish landraces that show some traits of climacteric ripening including aroma and flesh softening (e.g.‘Hilo carrete’ , ‘Madura Amarilla’ , ‘Amarillo Manchado’ , ‘Calamonte’ , and some ‘Blanco’ types), are not classified in the common market classes. These formed a separate population (Population 4 light blue in Figure 2b) along with inodorus varieties from Northern Africa, Eastern Europe and Western Asia (e.g. ‘Muscatello’ , ‘Maazoon’ , ‘Cassaba golden’ , ‘Kirkagac’ , ‘Yuva’). This population also included some Turkish, Russian, Israeli and Egyptian varieties that belong to the highly variable ameri group (e.g. ‘Hassanbey’, ‘Kuvinska’ , ‘Ananas Yokneam’ , ‘Ananas Dokki’).
A group of accessions from Iran, Uzbekistan and Russia (e.g. ‘Korca’ , ‘Souski’ , ‘Ouzbeque’ , ‘Gorgab’ , ‘Persia’), which mostly belong to the ameri pool, formed the last population of subspecies melo (population 5 green line in Figure 2b). This population showed the highest genetic variability (gene diversity = 0.22), whereas population 3 showed the lowest (gene diversity = 0.09) (Additional file 4). Most of the landraces from Eastern and Central Asia or the Middle East, show high levels of admixture with one or more ssp. melo populations. There seems to be a continuous degree of overlap between population 4 and 5 found in most ameri landraces, such as ‘Koljonitza’ and ‘Mucha Nesvi’ from Georgia, ‘Altinbas’ from Turkey, ‘Chandalak’ from Rusia and Mongolia, ‘Tokash’ from Tajikistan or ‘Mestnaia’ from Kazajistan.
Within the agrestis subspecies, two clear populations could be distinguished, in agreement with the PCA. Accessions belonging to different types of the conomon group (makuwa, chinensis and conomon) from Far Eastern countries (China, Japan, Korea, and Philippines) were grouped in population 6 (light purple in Figure 2b). Wild African agrestis from Ghana, Nigeria and Sudan, as well as the cultivated tibish from Sudan, which is considered to represent a first step of domestication in Africa, are clearly separated from conomon melons (population 7 orange color in Figure 2b). Most agrestis accessions from India and America showed a clear mix between these two populations, supporting a common ancestral origin.
None of the remaining accessions belonged to any specific population, but exhibited a high degree of admixture between populations of both subspecies. For example, the flexuosus varieties from Mediterranean and Middle East countries, traditionally included in the subspecies melo, show mixed agrestis-melo patterns. A similar situation is observed in the dudaim (mix of conomon and reticulatus) and momordica varieties (mix of the two agrestis populations with ameri and cantalupensis). Momordica has been traditionally assigned to the agrestis subspecies, but our results reflect the proximity of these agrestis varieties to the melo group. The subgroups demarcated by the PCA coincide with the sub-populations identified by STRUCTURE analysis, corroborating division of the overall population into the designated groups.
Variability in fruit traits and ripening behavior
All accessions have been characterized for different fruits traits: fruit weight, flesh color, sugar and malate content, and also for traits related to climacteric behavior, such as abscission layer formation, fruit detachment, and flesh firmness (Additional file 5). In general, the traits showed a continuous distribution, fitting or approximating a normal distribution. Variability within and between sub-populations was observed for most traits. All cantalupensis and reticulatus cultivars of the structure populations 1 and 2 had fruits with medium size (average fruit weight ± sd = 816 ± 348 g), mostly with orange flesh, with medium to high sugar content (°Brix = 9.5 ± 1.5 and 9.1 ± 1.2 in VCO and COMAV trials respectively, and average sucrose content = 97 ± 33 μg/g fresh weight). These accessions also clearly show strong climacteric behavior, most with a fully formed abscission layer and fruit detachment, whereas the Spanish group of inodorus landraces (population 3) had bigger fruits (fruit weight = 1,027 ± 315 g), with green, white or cream flesh, with higher sugar content (°Brix = 11.1 ± 1.1 and 10 ± 1.7 and average sucrose = 134 ± 44 μg/g fresh weight), and were mostly non-climacteric with no abscission layer or fruit detachment.
A higher variability in most traits was observed among accessions assigned to sub-populations 4 and 5. These developed medium to large sized fruits (fruit weight = 1,017 ± 425 and 975 ± 554 g, for populations 4 and 5 respectively), with green, white, yellow, cream or light orange flesh, and variable sugar content (°Brix = 9.1 ± 2.0 -7.1 ± 2.3 in the VCO trial and 8.0 ± 1.8-7.0 ± 1.9 at COMAV, and sucrose content = 114 ± 42 and 60 ± 46 μg/g fresh weight, respectively in both populations). The ripening behavior was also variable, including some Spanish landraces with certain climacteric behavior, typical non-climacteric inodorus and some ameri cultivars with different degrees of climacteric behavior (from no to full fruit slip).
Other ssp. melo accessions (cantalupensis, reticulatus, ameri, and inodorus, and other landraces) that show admixture of two or more of these sub-populations (populations 1 to 5) were also variable for fruit size and flesh color. Most had medium to high sugar content, but exhibited different degrees of climacteric behavior, ranging from clearly non-climacteric cultivars to some fully climacteric cantaloups and ameri accessions, with a wide range of intermediate behaviors (Additional file 5).
Within the subspecies agrestis, sub-population 6, which includes conomon, makuwa and chinensis types, showed small fruits (fruit weight = 464 ± 266 g), mostly with green or white flesh, and wide variation in sugar levels (°Brix = 7.5 ± 3.1-7.2 ± 2.1 in VCO and COMAV, and sucrose content = 78 ± 43 μg/g fresh weight). They also showed different ripening behaviors, ranging from non-climacteric to weakly climacteric. Similar variation in ripening behavior was also observed in some wild African agrestis melons (population 7) that turn yellow during ripening and show signs of forming an abscission layer. However, this population was quite uniform for fruit size, flesh color and sugar content, developing very small, green-fleshed, non-sweet fruits (fruit weight = 31 ± 30 g, °Brix = 6.3 ± 3.8-6.9 ± 2.6 in VCO and COMAV, and sucrose content = 37 ± 53 μg/g fresh weight). Accessions included in the admixture melo-agrestis group, consisting of momordica, dudaim, flexuosus and chate cultivar types, generally had little or no sugar, and showed weak to strong climacteric behavior (Additional file 5).
Variability in candidate genes
Out of a total of 251 SNPs assayed, 210 were polymorphic in the population. Of these, 37 were located in ethylene metabolism or cell wall related genes and 27 in sugar metabolism candidates (Additional file 2). Variability at ethylene and cell wall related SNPs (gene diversity = 0.37) is slightly higher than at sugar related SNPs (gene diversity = 0.31), and similar to reference SNPs (gene diversity = 0.41) (Additional file 6). SNP variability among the ethylene and cell wall related SNPs had a higher weighting than sugar related SNPs in the first component of the PCA (PC1 in Figure 1a), which separates the two subspecies (Additional file 7). This indicated that variability in the ethylene and cell wall related genes made a greater contribution to the sub specific genetic structure of the collection than the sugar-related genes. However, SNPs among all three groups of genes appeared to make similar contributions to the separation in PC2, which mainly distinguishes African agrestis from the others. Interestingly, the candidate genes for ethylene metabolism and cell wall have a larger contribution in the population differentiation found with STRUCTURE than candidate genes for sugar content (Additional file 8).
Relationship between candidate gene variation and cultivar classification
Some SNPs in sugar-related genes have an allele specific for one of the groups defined by STRUCTURE (Additional file 9). For example, the C/T SNP in CmINH1, which causes a non-tolerated (according to SIFT) A126V amino acid substitution in invertase inhibitor 1 (CmINH1.1), appears only in the French ‘Vedrantais’ cultivar and some closely related ‘Charentais’ melons, and in the makuwa ‘Yamato Purinsu’ and ‘China51’ genotypes that are known to have been used in cantaloup breeding [49]. All these cultivars belong to the STRUCTURE-defined sub-population 1 (Figure 2). They are sweet climacteric varieties that show a decline in sucrose content upon harvest. Invertase inhibitors potentially play a role in reducing invertase activity, thereby allowing sucrose to accumulate in the developing fruits. CmINH1 is the mostly highly expressed of the three invertase inhibitors at the onset of sucrose accumulation in the reticulatus ‘Dulce’ genotype [12]. Another SNP in the 3´-UTR region of the same gene, CmINH1.4, is almost exclusively present in wild agrestis types and tibish from Sudan. Similar SNPs in other invertase inhibitors, such as a C/T mutation that causes a tolerated P31S substitution in CmINHLIKE2.1, have a slightly less restricted distribution, occurring in other non-sweet, wild African agrestis types as well as agrestis and tibish from Sudan (all in STRUCTURE sub-population 7) (Additional file 9). The same allelic distribution is found in a 3´-UTR mutation in the fructokinase gene (CmFK3), in a non-tolerated C/T (F9L) mutation in the vacuolar processing enzyme CmVPELIKE2.3, and in a synonymous C/T (Q721Q) change in CmSUS3 encoding one isoform of sucrose synthase (CmSUS3.1). Similarly, the non-tolerated C/A (L267I) mutation in CmSPP1 (CmSPP1.1) is present only in a few African wild types, tibish cultivars and in some African acidulus. CmSUS3 is the mostly highly expressed SUS gene during the sucrose accumulating period in reticulatus melons, and CmSPP1 increases its expression during ripening [12]. Most of these genes make a major contribution to PC2 in Figure 1a (Additional file 7), which mainly separates African agrestis from other melons. Some of these genes also make a substantial contribution to the genetic differentiation among groups (high Fst and R2 from AMOVA) (Additional file 8).
Other SNPs in sugar-related candidates had more balanced frequencies for both alleles, and allelic variation was found within specific populations inferred by STRUCTURE and within the admixture group. Some of these SNPs showed an interesting pattern among sucrose accumulating and non-accumulating accessions. For example, most non-sweet or low-sugar genotypes in several populations (African, Indian and American agrestis, tibish, acidulus, flexuosus-chate, dudaim, conomon and momordica) had a non-tolerated T/C (S173P) mutation in CmAIN2 (CmAIN2.3), which also appears in some exotic medium sugar ameri and cantaloups (e.g. ‘Chandalack’, ‘Pearl’, ‘Earl favourite’, ‘Seminole’, ‘Persian’), but is absent in the other sweet genotypes. Most cantaloups and all inodorus had the alternative allele (Additional file 9). CmAIN2 encodes an acid invertase that is expressed in young fruits but not at maturity [12,50]. If invertase activity reflects the change in expression at the transcript level, the enzyme is likely to contribute to sucrose hydrolysis in young fruit but then decrease in activity as the fruit develops, allowing sucrose to accumulate in mature fruit.
There are SNPs in the 3´-UTRs of two invertase inhibitor genes (CmINH1.3 and CmINH3.1), and in each case, one allele tends to occur more frequently in sweet genotypes (inodorus, cantalupensis and ameri), although also present in a few low sugar momordica, dudaim and chate types. Also the CmINHLIKE2.4 SNP (C/T giving rise to a tolerated S137A substitution), shows a variation pattern similar to that of CmAIN2, with most of the non-sweet agrestis and melo genotypes (agrestis, tibish, acidulus, momordica, conomon, dudaim, flexuosus, chate) sharing the same allele as a few cantaloups. One synonymous mutation in the coding region of CmVPELIKE3 (CmVPELIKE3.2) and one 3´-UTR change in CmAAG2 (CmAAG2.1) could also be related with sugar content. The expression studies by Dai et al [12] suggested that the acid α-galactosidase encoded by the AAG2 gene plays a role in hexose production only in the early stages of fruit development in the ‘Dulce’ reticulatus genotype. This genotype has the C allele, more common in the sweet genotypes. Most of these mutations are located in the sugar candidate genes with the highest contributions to the PC1 in Figure 1a (Additional file 7), which separate both subspecies. QTLs for sugar accumulation have been reported previously in the genomic regions linked to some of the discussed genes, especially CmAIN2 and CmVPE-LIKE3 (LG IX), and CmAAG2 (LG X) [11].
The ripening-related gene candidates that contribute most strongly to differentiation between the subspecies (such as the CNR, AtEIN3, CmACO3 andCmERF3 genes linked to ethylene metabolism, and the cell wall related gene CmEXP3) (Additional file 7) have different alleles in ssp. melo (sub-populations 1-5) versus ssp. agrestis, (sub-populations 6 and 7), and are almost fixed within the respective subspecies (Additional file 9). This echoes the pattern seen for sugar related genes, where some of the STRUCTURE sub-populations carry a specific allele for some of the SNPs. For example, the rare alleles of two SNPs, CmEIN3LIKEex2 (a G/T change that causes a tolerated L36V substitution) and AtEIN3ex2 (a C/T synonymous substitution of I499I), both located in the same melon gene MELO3C015633 (an EIL3 transcription factor involved in ethylene signaling [51]), were fixed in the population of inodorus (sub-population 3), mostly composed of clearly non-climacteric genotypes. The cantalupensis and reticulatus populations, which are both highly climacteric, have the alternative allele, which is also more frequent in all of the remaining populations composed of genotypes with different degrees of climacteric behavior.
Other SNPs are also differentially distributed between highly climacteric cantalupensis and reticulatus (sub-populations 1 and 2) and non-climateric inodorus (sub-population 3). However, in this case the allele associated with climacteric behavior is also fixed in the conomon population (sub-population 6), while the allele that is more commonly associated with non-climacteric behavior appears in populations 4, 5 and 7. The latter three populations show variable levels of climacteric behavior. These SNPs include a tolerated C/G mutation (H121D) in the coding region of the cell wall related gene CmXTH5, a SNP in the 3´-UTR of CmACO3, and a synonymous T/G mutation (G171G) in CmERF2. All these SNPs contributed to the genetic differentiation of STRUCTURE populations (Additional file 7).
Linkage disequilibrium
Linkage disequilibrium (LD) was studied in order to assess the possible degree of linkage between SNPs associated with the studied traits (see below) and real causal SNPs. Wild melon accessions were excluded because the fruits from these accessions differed in too many respects from fruits of cultivated accessions, making direct comparisons difficult if not impossible. Intra-chromosomal LD showed a rapid decay within a physical distance of less than 0.5 kbp, but increases from 0.5 to 1 kbp and then decreases rapidly with larger distances (Figure 3). This is similar to the results of Esteras et al. [4], who found that the LD extension decayed at less than 3 kbp. Thus, in the current data set, causal SNPs are expected to be very closely linked to the SNPs that are significantly associated with trait variation.
Figure 3.
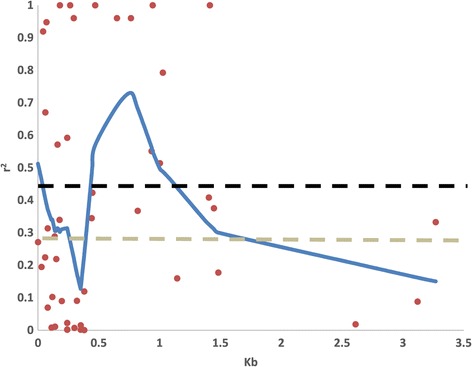
Linkage disequilibrium (r 2 ) versus physical distance (kb) in the accessions considered for the association linkage analysis. LD extension is shown for a subpopulation of melons, excluding African agrestis, and was used in the association analysis for fruit quality traits. The false discovery rates p < 0.05 and p < 0.01 are indicated with black and grey dashed lines respectively. Curves were fitted by second degree LOESS.
Association analysis for sugar and organic acid content
Association analysis was investigated using either a general linear model (GLM) or mixed linear model (MLM) approach, with the latter method being used to correct for the effect of genetic structure. GLM analysis gave a high number of significant associations (Additional file 10), including a consistent association of SNPs in sugar candidates (such as CmINHLIKE2.1 and CmAIN2.3) with Brix and sucrose content, whereas MLM analysis provided only a few significant associations (Table 1). The larger number of associations obtained by GLM is attributable to the strong genetic structure in the current germplasm sample, so we focussed on the MLM results, which are likely to be more robust.
Table 1.
Markers associated with melon fruit traits of interest
| Marker | Transcription model | Chr | locus_position | R 2 | p value | Nucleotide position | SNP | AA change | |
|---|---|---|---|---|---|---|---|---|---|
| Climacteric behavior | |||||||||
| ABCISSION LAYER | MLO65044.1 | MELO3C005044 | 12 | 3625217 | 0.111 | 5.475E-05 | 1481 | C/T | L454L |
| Firmness | |||||||||
| FLESHFIRMESSVCO | PSI_41-B07 | MELO3C021426 | 11 | 22820373 | 0.134 | 1.996E-05 | 593 | C/T | F167F |
| Sugar | |||||||||
| BRIXVCO | SlERF1 | MELO3C011287 | 3 | 22138473 | 0.133 | 2.598E-05 | 208 | C/T | N62N |
| BRIXCOMAV | CMPSNP711 | MELO3C021106 | 1 | 26308138 | 0.103 | 0.0001498 | 2382 | C/T | 3´UTR |
| BRIXCOMAV | SlERF1 | MELO3C011287 | 3 | 22138473 | 0.131 | 1.839E-05 | 208 | C/T | N62N |
| Sucrose | SlERF1 | MELO3C011287 | 3 | 22138473 | 0.112 | 7.56E-05 | 208 | C/T | N62N |
| Sucrose | SlERF3 | MELO3C002624 | 12 | 20075012 | 0.132 | 3.144E-05 | 481 | A/G | Q138Q |
| Organic acid | |||||||||
| malic acid | CmINHLIKE2.2 | MELO3C017187 | 2 | 22908768 | 0.101 | 0.0001873 | 245 | C/T | S60S |
| malic acid | AtEIN3ex4.2 | MELO3C019931 | 3 | 17934192 | 0.084 | 0.0003247 | 1464 | C/T | F375F |
| malic acid | CMPSNP677 | MELO3C009586 | 4 | 26788563 | 0.088 | 0.0002695 | 2976 | C/G | 3´UTR |
| Fruit color | |||||||||
| C_FLESH | CmXTH5 | MELO3C012004 | 10 | 3358353 | 0.167 | 1.339E-06 | 583 | C/G | H121D |
| C_FLESH | CmERF2ex2 | MELO3C012242 | 10 | 1742964 | 0,149 | 7.931E-06 | 741 | G/T | G171G |
| C_FLESH | MLO625760.1 | MELO3C025760 | 11 | 22071980 | 0,095 | 0.0002283 | 384 | A/G | Q128Q |
Results are shown for markers that are significantly associated with various fruit ripening and sugar content traits, based on a mixed linear model (MLM) analysis of association using TASSEL [77]. Bonferroni’s correction was applied and the R2 and p-values for each association are indicated.
Three SNPs located in different genomic regions, CMPSNP711, SlERF1 and SlERF3 (Additional file 2) were associated with sugar content (Table 1). These were also significant with GLM analysis (Additional file 10). Although initially selected as a reference marker [4], CMPSNP711 in LG I was found to be associated with soluble solids content (°Brix) in the COMAV trial. This SNP is a 3´-UTR mutation in the MELO3C021106 gene, which putatively encodes xyloglucan glycosyltransferase 6. The C allele is more common in accessions producing fruits with low sugar content. It is fixed in sub-populations 6 and 7, comprised of low sugar conomon and non-sweet African agrestis accessions. The alternative allele is fixed in populations 1, 2 and 3 (sweet cantalupensis and Spanish inodorus), except in the Apelsinaja cultivar from Russia, the accession with the lowest brix degree in sub-population 2. Sub-populations 4 and 5 and the admixture group, with variable sugar content, contained both alleles, with the C allele occurring more frequently in the less sweet genotypes (Additional file 9). CMPSNP711 is located in a more distant part of chromosome I than other sugar content QTLs that have been mapped to the same linkage group [11].
Two melon orthologs of tomato ethylene responsive factors genes, SlERF3 and SlERF1 [52], carrying synonymous A/G (Q138Q) and C/T (N62N) mutations, respectively, were also found to be significantly associated with sugar content (Table 1). The A allele of the SlERF3 SNP was fixed in the non-sweet African agrestis (sub-population 7), and frequent in the low sugar genotypes of the admixture group. The alternative G allele was fixed in the remaining populations, as defined by the STRUCTURE analysis, and correlates with higher °Brix content in the admixture group. This SNP is located in LG XII in a region in which QTLs for sugar content have been reported previously [11]. Interestingly, mutation of SlERF1, located in LG III was consistently associated with soluble solids content in both trials and to sucrose content. The C allele is more frequent in conomon, cantalupensis groups (both Charentais and reticulatus) and Spanish inodorus, while European and Asiatic inodorus and ameri share the alternative allele with African agrestis and most of the low sugar types of the admixture group (acidulus, flexuosus, chate and momordica) (Additional file 9). A hotspot of QTLs involved in sugar accumulation has been described previously on this region of LG III [11].
Fruit flavor is also affected by acidity. Malic acid content was significantly associated with CmINHLIKE2.2, CMPSNP677 and AtEIN3ex4 (Table 1, Additional file 9). The SNP in the invertase inhibitor gene CmINHLIKE2 corresponds to a synonymous T/C (S60S) mutation. The C allele found in all conomon was also present in flexuosus, chate, dudaim, momordica, acidulus and some wild American and Indian agrestis, all accessions with acid pulp rich in malic acid. The 3´-UTR mutation in CMPSNP677 (MELO3C009586 encoding a melon orthologue of the Arabidopsis ubiquitin carboxyl-terminal hydrolase 12-like protein) was initially selected as a reference SNP, but was also found to be related to malic acid content. The allelic distribution was similar to that of CmINH-LIKE2.2, except that the conomon allele is shared not only with flexuosus, dudaim, momordica, acidulus and wild American and Indian agrestis, but also with African agrestis. QTLs for glucose, fructose and soluble solids have previously been located in the chromosomal region of LG IV where this gene is located [11]. Finally, a synonymous C/T mutation (AtEIN3ex4.2 F375F) in AtEIN3 was also associated with malic acid content. For this SNP, the conomon allele is shared with acidulus and Indian and American wild types. In Arabidopsis, the AtEIN3 transcription factor plays a role in ethylene signaling downstream of the ETR1 ethylene receptor, and is involved in fine tuning of the ethylene response [28]. The association observed in the present work suggests a potential role for this transcription factor in regulating organic acid content in fruit.
Association analysis for fruit ripening
Similarly to what occurred with sugar related gene candidates, the GLM analysis showed associations with some ripening candidates. For example, a non-synonymous, tolerated mutation (H121D) in CmXTH5 located in LG X, was associated with formation of an abscission layer and fruit detachment (Additional file 10). The alternative alleles of this gene are fixed in the highly climacteric and the non-climacteric cantalupensis and inodorus, as described above (Additional file 9). Furthermore CmXTH5 was mapped in the same region as a QTL for fruit firmness on LG X [14].
MLM showed a significant association with the same trait of MLO65044.1, a synonymous C/T mutation (L454L) in an MLO-like gene [53] located in LG XII (Table 1). The homozygous CC genotype is found exclusively in most inodorus types of Spanish, Turkish or Portuguese origin, which are mostly non-climacteric, whereas most other accessions have a homozygous TT genotype, including both climacteric and non-climacteric types. No QTLs related to climacteric ripening or fruit abscission have been reported before in this chromosome region [11,17].
Fruit firmness is also related to ripening behavior. MLM found the marker PSI_41-B07 SNP (C/T, F167F), on LG XI, significantly associated with the variation in this trait. In this region a QTL for flesh firmness was previously reported [11]. The T allele is present in the great majority of the germplasm, while the C allele is characteristic of conomon, African and Indian wild agrestis and acidulus, which had the highest values for firmness.
Association analysis for flesh color
Fruit flesh color is also related with both fruit quality and ripening behavior, as the flesh of most climacteric genotypes changes from green to orange during ripening. Flesh color is controlled by two major genes, green flesh (gf) and white flesh (wf), which interact epistatically [54] and give rise to orange, white or green flesh depending on the gene combination. The gf gene has been mapped to chromosome VIII [20,47], while wf is located on chromosome IX [17]. Nevertheless the variability in the intensity of flesh color suggests that the phenotypic influence of the two major genes is modulated by the action of other genes, and minor QTLs for color intensity have been mapped to chromosomes III, VI and VII [10,20]. MLM showed several mutations associated with the variation in the color parameter chroma C, the non-synonymous mutation in CmXTH5, the synonymous changes in CmERF2ex2, and MLO625760.1, a gene in LG XI that encodes an MLO-like protein (Table 1). The same region (16-27 cM) contains a SNP in a ZEAXANTHIN EPOXIDASE (ZEP) gene [11]. The results obtained here suggest a contribution from chromosome X and XI that is worth further investigation.
Discussion
PCA and STRUCTURE analyses carried out in the current study, based on SNPs located in reference and candidate genes, provided a germplasm stratification and classification very similar to previous works based on other marker systems or un-selected SNPs [4,5,47]. Therefore, the variability in sugar- and ripening-related genes appears to reflect the known genetic variability of this species. This observation is supported by the fact that gene diversity at candidate genes is just slightly lower than in reference genes.
Our results confirm that the highest genetic variability among cultivated melons occurs in Western and Central Asia and in the Middle East, where a high degree of genetic admixture was found. This admixture is not unexpected in melon, as no major geophysical barriers are found in the geographical distribution of traditional melon varieties, so gene flow among different regions is not impeded. Asian varieties are thought to be the likely ancestors of the cantalupensis and inodorus group [5,55]. In fact, some known current cultivars and landraces from France, Spain, Italy, Israel, Japan, and USA that do not fit to the cantalupensis Charentais, reticulatus or inodorus morphotypes completely, show variable admixture degrees of several sub-populations.
Our results indicate that SNPs in ripening related candidates, both ethylene and cell wall related, have a higher weight than SNPs in sugar candidates on both sub-species and sub-population stratification. This might reflect the effect of human selection in the development of strictly non-climacteric and high climacteric cultivars within ssp. melo. In contrast, selection for strictly climacteric or non-climacteric ripening was probably less intense within the ssp. agrestis cultivars, which more frequently show an intermediate ripening behaviour. A number of consistent QTLs involved in climacteric ripening have been described previously [14,17,18,23]. However, QTLs for sugar content have been found less consistently [16,20,47], indicating a higher heritability of ripening-related QTLs. Traditional farmers might have exerted a stronger selection for ripening traits, selecting alleles underlying ripening phenotypic variability more efficiently and thereby fixing the alleles within a particular cultivar group.
Association of candidate SNPs with phenotypic variability has been investigated by two approaches: (i) by looking for alleles that are fixed or have a higher frequency within horticultural groups with characteristic phenotypic features, and (ii) by association analysis. Sugar content and ripening behavior were clearly related with genetic structure, so the use of MLM models to adjust for genetic structure would also reduce the power to detect associations. MLM analysis has resulted in a much smaller number of SNPs associated with the studied traits. This could be due to lack of linkage or to the association between genetic structure and trait variability, as alleles that are close to being fixed within groups could not be associated significantly after adjusting for population structure in the MLM analysis. Nevertheless, a consistent association of the SlERF1 SNP and sugar content has been found on LG III. This linkage group contains a sugar content QTL hotspot [11], and was also found to be significantly associated with sugar content in a previous study [56], providing good evidence that this region contains genes that influence sugar accumulation.
Allelic differentiation analysis provided information that is complementary to the association results. Some SNPs in sugar-related candidate genes appeared in genotypes with common features in sugar content. These include SNPs that could be related with the mechanisms that prevent sugar accumulation in African melons, such as SNPs in invertase inhibitor genes (CmINH1 and CmINHLIKE2) and in other genes encoding enzymes or proteins involved in sugar metabolism (CmFK3, CmVPELIKE2, CmSUS3 and CmSPP1). Also of interest are mutations with more balanced allele frequencies and those with differential patterns of variation within sugar accumulating and non-accumulating groups, such as the mutation in the acid invertase CmAIN2 and other sugar-related genes (CmVPELIKE3 or CmAAG2). The fact that these three genes co-localize with previously reported QTLs for sugar content in LG IX and X [11] gives support to their role in sugar content variation. It is worth noting that the more permissive GLM analysis provided consistent association of SNPs in these three candidates (CmAIN2.3, CmINHLIKE 2.1 and CmVPELIKE3.2) with °Brix values in both locations and with sucrose content.
Both GLM and MLM gave significant associations of traits related with ripening behavior, such as the formation of the abscission layer and flesh firmness, with reference SNPs in genes located in LG XII and XI, the latter being co-localized with a QTL for flesh firmness [11] (Table 1 and Additional file 10). However, association of ripening-related candidate genes with these traits was only detected by GLM. For example, the mutation in CmXTH5, a xyloglucan endotransglycolase/hydrolase that contributes to xyloglucan depolymerization in ripening fruit, had contrasting genotypes in cantalupensis and inodorus groups and was consistently associated with the formation of abscission layer and fruit detachment. The co-localization of this gene with a QTL for fruit firmness in LG X [14] provides further evidence that CmXTH5 could be a candidate gene for the QTL. Other candidate regions found to be associated to the formation of the abscission layer with GLM mapped in regions of LG II, III, and VI that carry QTLs involved in climacteric ripening [11,23]. In addition, ethylene pathway candidate genes (ACS1, ETR1, AtEIN3), that map to regions in which QTLs for ripening in LG III, V and VII have been previously reported, were found to be associated with fruit firmness. However, further studies are needed to see if these mutations affect gene expression or gene function, as this differential allelic distribution might also be an effect of the population structure.
Our study shows that association and allelic differentiation analysis could be used as complementary approaches in highly structured populations, such as the current one, in order to define candidate genes. A highly variable germplasm collection with low genetic structure would be necessary to increase the power of association studies in melon. The ameri group showed both high phenotypic and genotypic diversity. Therefore, cultivars from this group, along with cultivars from related admixture groups from the Near-Middle East region, would provide a suitable genotype set for association analysis in melon.
Conclusions
The analysis of SNPs in sugar metabolism and ripening-related genes, and also in reference genes, has revealed differences in the amount of genetic diversity among the main groups. Ripening-related genes seem to contribute more to this structure than sugar metabolism related genes. We found specific alleles of candidate genes fixed in cultivar groups concomitant with differences in sugar or ripening behavior. This could be due to ancient selection of these alleles by early farmers, but given the strong genetic structure, it is not currently possible to distinguish between direct selection or genetic drift. Nevertheless, some SNPs were still found to be associated with ripening behavior or sugar accumulation even after taking the genetic structure of the collection into consideration. As LD is extremely low in melon germplasm, these SNPs should be tightly linked to the causal SNPs. Taking into consideration the low LD, the strong genetic structure of melon germplasm, and the relatively high phenotypic and genotypic diversity found among accessions of Near-Middle East origin within the admixture group, it should be feasible to define a panel of accessions derived from this group that has minimal genetic structure but retains sufficient genetic diversity for association analysis after more extensive genotyping.
Methods
Plant material
The panel of melon genotypes used in this study consisted of 175 accessions of diverse origins (Additional file 1), with all of the major types identified by Pitrat [3] within the two subspecies, C. melo ssp. melo and C. melo ssp. agrestis, being represented by multiple accessions. This was based on a previously assembled core germplasm collection [4,57], supplemented by germplasm from the Cucurbit Breeding group at the Institute for the Conservation and Breeding of Agrobiodiversity (COMAV) in Valencia and is currently conserved at the COMAV genebank (www.comav.upv.es). Subspecies melo accessions included the commercially important groups inodorus, cantalupensis and reticulatus. The inodorus group contains commercial cultivars, Spanish landraces and traditional cultivars from Southern and Eastern Europe, North Africa and Asia. The cantalupensis group includes accessions widely grown in Europe, such as ‘Charentais’ types and other French, American, Israeli and Japanese cantaloupes, commercial reticulatus varieties and other cantalupensis types. The rest of the ssp. melo accessions belong to ameri, chandalak, adana, flexuosus, chate and dudaim groups from Southern and Eastern Europe, Northern Africa, Central Asia and India. The agrestis subspecies was represented mostly by accessions of Asian and African origin. Momordica from India, conomon, makuwa and chinensis from Far-East Asia, and Indian wild melons together represented the Asian melon diversity. Tibish cultivars from Sudan, considered as the most primitive melon known, cultivated acidulus and wild melons from Africa were also included.
SNP selection
Three collections of SNPs were selected: (i) reference (148 SNPs), (ii) sugar metabolism related (43 SNPs), and (iii) ripening related (including ethylene related elements as well as genes encoding enzymes for cell wall degradation) (60 SNPs) (Additional file 2). The reference SNPs were chosen to define the genetic structure of the collection, and were extracted from a larger SNP collection genotyped in a set of accessions that collectively represents the full range of variation within the species, most of which were included in the current study [4]. The criteria for reference SNPs selection were: (i) even distribution throughout the genome; and (ii) variation in the germplasm collection representative of the variability within the species (preferably with a major allele frequency (MAF) of less than 80%) [4].
The SNPs of the candidate genes were selected from a SNP collection generated after resequencing eight pools of accessions representing the main melon botanical groups [44], most of which are common with the collection described in [4] and are included in the current study. All are in the SNP collection in the Melogene database (www.melogene.net).
Candidate SNPs were selected from the full SNP collection by using the BLAST algorithm to search the sequences for candidate genes against the Melogene database. The genome position of candidate gene SNPs was defined according to melon genome assembly v3.5, and their position in the genetic map was also established relative to the mapped refrence SNPs, using the anchorage of the genome sequence to the genetic map available in the Melonomics database (www.melonomics.net) [4,45]. The following criteria were also used to select SNPs in candidate genes: (i) location in the coding region (preferably causing non-synonymous substitutions) or in the 5´- or 3´-untranslated regions (UTRs) of candidate genes; (ii) not linked to other SNPs (≥100-bp distance); (iii) not close to an intronic region; and (iv) preferably having a MAF of less than 80%.
Forty-three SNPs were located in nineteen candidate genes related to sugar metabolism. Thirteen of these candidate genes were selected from those reported in previous studies (acid α-galactosidases CmAAG1 and CmAAG2, sucrose synthases CmSUS2 and CmSUS3, sucrose P phosphatases CmSPP1 and CmSPP2, fructokinase CmFK3, soluble acid invertase CmAIN2, cell wall acid invertases CmCIN3 and CmCIN4, and invertase inhibitors CmINH1, CmINH2 and CmINH3). Dai et al. [12] analyzed the expression of these candidate genes during fruit development and ripening in cv. ‘Dulce’, a sweet melon from the reticulatus group, using publicly available data from the ICUGI melon EST database (www.icugi.org). Some of them were mapped by Harel-Beja et al [16] and Diaz et al [11] and colocalize with QTLs associated with sugar content. Additional candidates were identified directly in the melon genome database according to their annotation (www.melonomics.net) (CmAAG3, CmAAG4, CmINHLIKE2, CmVPELIKE 1, 2 and 3 (Additional file 2).
Thirty four candidates involved in ethylene biosynthesis, signal reception and signaling, and cell wall disassembly were selected from the Melonomics database based on their genome annotation and information from previous studies in melon [58], tomato mutants [52], grapevine [59], Arabidopsis thaliana [60], apple [61] and peach [62] (Additional file 2). Ethylene receptors and signal transduction elements such as CmETR1 [11] and the melon orthologs of the following genes were selected: (i) PpERS1 from Prunus persica (ERS1PRUPE); (ii) VvETR2, VvRTE1 and VvCTR1 from Vitis vinifera; (iii) AtCTR1 from Arabidopsis thaliana; and (iv) SlETR3 from Solanum lycopersicum. SlETR3 (NR) was previously identified in the NR (never ripe) tomato mutant [63-66]. Furthermore, genes encoding components of the ethylene signalling pathway downstream of CTR1 were also analyzed. These included the melon orthologue of AtEIN3, other genes annotated as EIN3 or EIL (CmEIN3LIKE, CmEIN3LIKE2 and CmEIL3) in the Melonomics database, and the melon orthologue of the SlEBF gene of tomato [67], which controls EIN3 degradation in the nucleus. Other candidates were regulatory elements that affect ACS activity (CmE8, E4/E8, CmETO1LIKE and the melon orthologue of SlHB1) [32,36], ethylene responsive factors (CmERF1, CmERF2, CmERF3, the melon othologue of the apple MdERF2, two melon orthologs of the tomato genes SlERF1 and SlERF3) [14,52,62], and the melon orthologs of the tomato ripening mutants Cnr and rin [31,68]. Genes co-located with the major QTLs for ripening, or coding for proteins involved in ethylene synthesis and signaling, e.g. SlSBP and CmETHIND were also included in the SNP analysis, using information from melon genetic maps [11,14,15,23]. Finally, genes encoding proteins involved in cell wall degradation were considered, such as expasin3 (CmEXP3), which has been hypothesized to contribute to hemicellulose depolymerization, and CmXTH5, which encodes a xyloglucan endotransglycolase/hydrolase that contributes to xyloglucan depolymerization in ripening fruit [69].
The effect of the variants was analyzed with SIFT (Sorting Intolerant from Tolerant, http://sift.jcvi.org/), which predicts whether an amino acid substitution will significantly affect protein function. Scores below 0.05 indicate a strong probability that protein function will be affected by the mutation [70] (Additional file 2).
Sequenom MassARRAY® assay
Total DNA was extracted from each genotype in the collection, from young leaves, by the method described in Doyle and Doyle [71] with minor modifications [4]. To improve the quality of the obtained DNA, 70% ethanol containing 15 mM ammonium acetate was used in the final wash, and the DNA was treated with RNAse A. DNA concentrations (in TE buffer) were measured on an ABI7900 (Applied Biosystems) using PicoGreen fluorescent dye and adjusted to 50 ng/μl. SNP genotyping was performed using an iPLEX® Gold MassARRAY® Sequenom system at the eEpigenetic and Genotyping unit of the University of Valencia (Unitat Central d´Investigació en Medicina (UCIM), University of Valencia, Valencia, Spain).
Melon fruit phenotyping
The whole collection was phenotyped in two locations. Three randomized plants per accession were grown in Valencia (Spain) (39°28′11″ N- 0°22′38″ W) at COMAV-UPV greenhouse facilities, and in VCO’s St Rémy station (France) (43°47′18″ N- 4°49′54″ E), during spring/summer 2008. The following fruit traits (one fruit per plant) were analyzed: (i) fruit weight; (ii) flesh color ( L*C*h*, lightness, chroma and hue angle were measured with a Minolta colorimeter, at COMAV facilities, while at VCO, color was scored visually on the following scale: 1 = green, 2 = green-white, 3 = green-yellow, 4 = white, 5 = cream, 6 = yellow, 7 = orange, 8 = orange-green, 9 = orange-white, 10 = other); (iii) total soluble solids, °Brix (measured from drops of juice using a Milwaukee MR32ATC refractometer); and (iv) flesh firmness (measured using a fruit pressure tester FT327 with a plunger diameter of 8 mm). Flesh color, firmness, and total soluble solids were measured at four points in the equatorial region of the mesocarp. Samples of flesh tissue from the same region were harvested for metabolite analysis from fruits grown at COMAV, immediately frozen in liquid nitrogen and stored at -80°C until analysis. Soluble sugars (glucose, fructose and sucrose) and malate were measured enzymatically in ethanolic extracts as described in Stitt et al. [72].
From the classical markers of climacteric ripening (peak in ethylene production, degreening of the rind, aroma production, formation of an abscission layer and fruit detachment), formation of an abscission layer and fruit detachment were selected as the most amenable for high-throughput phenotyping to classify the accessions as climacteric or non-climacteric. Formation of an abscission layer was scored visually in the assay performed at COMAV-UPV on a scale from zero (no abscission layer) to four (fully formed abscission layer). Fruit detachment was scored visually in the assay performed at VCO on the scale 1 = absent, 2 = no slip, 3 = half slip, 4 = full slip.
Genetic variability analysis
The genetic diversity of the germplasm collection as a whole was assessed by principal component analysis (PCA) using the ‘adegenet 1.4-0’ package in the R project for statistical computing (version R.14.1; www.r-project.org) [73,74]. The population genetic structure was also analyzed with STRUCTURE v.2.3.3 software [75]. Twenty independent runs for each K value ranging from 2 to 10 were performed with a burn-in length of 500,000 and 1 million iterations. The optimal subpopulation was calculated from the second order rate of change of likelihood (ΔK method) [48].
Basic genetic variability parameters, MAF, gene diversity (expected heterozygosity), observed heterozygosity (Ho) and Fisher’s fixation index (Fis) were calculated for each locus with PowerMarker 3.5 [76]. In order to investigate which SNPs could be responsible of the genetic structure, Fst and Analysis of Molecular Variance (AMOVA) were also studied for each SNP using the STRUCTURE defined groups with PowerMarker 3.5.
LD estimation
TASSEL v 5.0 [77] (http://www.maizegenetics.net) was used to estimate the LD parameter r2 and the comparison-wise significance was computed from 1000 permutations. LD decay was drawn as a smooth line from r2 against distance in kb, fitting the data using a second-degree, locally-weighted scatterplot-smoothing LOESS [78], implemented in an Excel plug-in [79]. A critical value of r2 was derived from a distribution of the unlinked r2. The parametric 95th and 99th percentiles of the unlinked-r2 distribution were taken as a population specific critical value of r2.
Association analysis
Association analysis was investigated by general linear (GLM) and mixed linear models (MLM) implemented in TASSEL v. 5.0. In order to control genetic structure effects, a kinship matrix was calculated using reference SNPs Associations were considered statistically significant for an experimental-wise threshold p < 0.05 after adjusting by the Bonferroni’s correction based on the total number of tested markers (p < 0.000333 for individual test).
Acknowledgments
C.L. is recipient of a Marie Curie Career Integration Grant (CIG) and her visit to IBMCP-COMAV (Valencia) is supported by ESF Cost Action “FA1106 Quality Fruit” - Short-Term Scientific Mission (STSM). We thank Dr. Pau Javier-Montero (COMAV) and Dr. Javier Forment Millet (IBMCP) for their kind bioinformatics support, Nicole Krohn and Beatrice Encke (MPIMP) for their technical support in sugar and malic acid analyses, and Prof. Mark Stitt for helpful discussions. Genotyping was supported by SAFQIM project, AGL2012-40130-C02-02 from the Spanish Ministry of Economy and Competitivity (MINECO). Metabolite analysis at the MPIMP was supported by the German Federal Ministry of Education and Research (FP6 ERA-NET Plant Genomics project MELRIP; BMBF contract 0313987 to Mark Stitt).
Additional files
Melon germplasm analyzed in the current study. A given number, code, country of origin and local/commercial name is given for each accession. Accessions are classified according to the Cucumis melo subspecies (ssp. melo and ssp. agrestis) and cultivar group (cantalupensis, reticulatus, inodorus, ameri group (including ameri, adana and chandalack), flexuosus (including chate) and dudaim within ssp. melo and momordica, acidulus, conomon (including chinensis, conomon and makuwa), tibish, chito and agrestis, within ssp. agrestis), and others i.e., classification not clear.
List of genes considered in the association study. Their position in the genome and scaffold according to Garcia-Mas et al. [45], gene name and annotation according to Melogene (www.melogene.net) and Melonomics (www.melonomics.net), position in the genetic map according to Esteras et al. [4] are reported. AA represents the aminoacid change due to the SNP and the effect of the mutation screened by SIFT.
Estimated number of clusters obtained with STRUCTURE for K values from 1 to 10 using SNPs data for the all germplasm collection. a) Graphical representation of estimated mean L (k) and b) its derivative statistics ∆K. The graph below was obtained excluding k = 2 in order to evidence the subpopulations present in the germplasm. c) Table summarizing parameters of STRUCTURE simulations performed for each present K: mean likelihoods of models, their standard deviations, ∆K.
Genetic diversity parameters present in each group defined from STRUCTURE analysis. MAF indicates Major Alelle Frequency, gene diversity is the expected heterozygosity, Ho the observed heterozygosity, and Fis is Wright's fixation index.
Quantitative traits measured in the study for each accession (mean). The collection was phenotyped for Brix degree (measured from drops of juice using a refractometer) and for flesh firmness (measured using a fruit pressure tester with a plunger diameter of 8 mm) in two locations, in Valencia (Spain) at COMAV facilities, and in VCO’s St Rémy station (France). Additional fruit traits were analyzed at COMAV: fruit weight, flesh colour (measured with a Minolta colorimeter,L*C*h*, lightness, chroma and hue angle), and the formation of an abscission layer (scored visually on a scale from zero, no abscission layer, to four, fully formed abscission layer). Samples of flesh tissue were harvested for metabolite analysis (glucose, fructose and sucrose and malate) from fruits grown at COMAV. Additional fruit traits were analyzed at VCO: fruit detachment (scored visually on the scale 1 = absent, 2 = no slip, 3 = half slip, 4 = full slip) and flesh color (scored visually on the scale 1 = green, 2 = green-white, 3 = green-yellow, 4 = white, 5 = cream, 6 = yellow, 7 = orange, 8 = orange-green, 9 = orange-white, 10 = other). The subspecies, the cultivar group and the population or population mixture, according to STRUCTURE results in Figure 2, are indicated for each accession.
Genetic Diversity parameters for the candidate genes for ripening and sugar accumulation as well as for referencel markers. MAF indicates Major Alelle Frequency, gene diversity is the expected heterozygosity and Ho refers to observed heterozygosity.
Loading for PC1 and PC2 of candidate genes for ripening, including ethylene and cell wall related, (orange) and sugar accumulation (blue).
Contribution of the candidate genes (orange ripening, including ethylene and cell wall related, blue sugars) to the genetic differentiation among groups based on STRUCTURE analysis. The SNPs with higher contributions are listed on the first column followed by the category candidate gene. Fst and percentage of variance (R2) among groups explained for each locus after AMOVA are depicted in the last columns.
Genotyping results for the polymorphic SNPs in the germplasm collection. Colors indicate the 7 groups of STRUCTURE in Figure 2.
Significant markers associated to the trait of interest from GLM analysis by using TASSEL. The analyzed traits are described in Additional file 5.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BP, CE, JEL, RS and FdL phenotyped the collection in the different localities and performed the chemical analysis of sugars and organic acids. BP and CE genotyped the collection; CL and AM performed the data analysis and statistical computation; BP, CL and AM selected the SNPs, designed the experiment, and drafted the manuscript. CM, JEL, RS and FdL contributed to the critical review of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Carmen Leida, Email: carmen.leida@fmach.it.
Claudio Moser, Email: claudio.moser@fmach.it.
Cristina Esteras, Email: criesgo@upvnet.upv.es.
Ronan Sulpice, Email: ronan.sulpice@nuigalway.ie.
John E Lunn, Email: lunn@mpimp-golm.mpg.de.
Frank de Langen, Email: frank.delangen@hmclause.coml.
Antonio J Monforte, Email: amonforte@ibmcp.upv.es.
Belen Picó, Email: mpicosi@btc.upv.es.
References
- 1.Sebastian P, Schaefer H, Telford IRH, Renner SS. Cucumber (Cucumis sativus) and melon (C. melo) have numerous wild relatives in Asia and Australia, and the sister species of melon is from Australia. Proc Natl Acad Sci U S A. 2010;107:14269–73. doi: 10.1073/pnas.1005338107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jeffrey C. A review of the Cucurbitaceae. Bot J Linn Soc. 1980;81:233–247. doi: 10.1111/j.1095-8339.1980.tb01676.x. [DOI] [Google Scholar]
- 3.Pitrat M. Melon (Cucumis melo L.) In: Prohens J, Nuez F, editors. Handb Crop breeding, vol I Veg. New York, USA: Springer; 2008. pp. 283–315. [Google Scholar]
- 4.Esteras C, Formisano G, Roig C, Díaz A, Blanca J, Garcia-Mas J, et al. SNP genotyping in melons: genetic variation, population structure, and linkage disequilibrium. Theor Appl Genet. 2013;126:1285–303. doi: 10.1007/s00122-013-2053-5. [DOI] [PubMed] [Google Scholar]
- 5.Dhillon NPS, Ranjana R, Singh K, Eduardo I, Monforte AJ, Pitrat M, et al. Diversity among landraces of Indian snapmelon (Cucumis melo var. momordica) Genet Resour Crop Evol. 2006;54:1267–1283. doi: 10.1007/s10722-006-9108-2. [DOI] [Google Scholar]
- 6.Dhillon NPS, Singh J, Fergany M, Monforte AJ, Sureja AK. Phenotypic and molecular diversity among landraces of snapmelon (Cucumis melo var. momordica) adapted to the hot and humid tropics of eastern India. Plant Genet Resour. 2009;7:291–300. doi: 10.1017/S1479262109990050. [DOI] [Google Scholar]
- 7.Mavlyanova R, Rustamov A, Khakimov R, Khakimov A, Turdieva M, Padulosi S. O'zbekiston Qovunlari [Melons of Uzbekistan] IPGRI: Rome; 2005. [Google Scholar]
- 8.Stepansky A, Kovalski I. Intraspecific classifcation of melons (Cucumis melo L .) in view of their phenotypic and molecular variation. Plant Syst Evol. 2000;217:313–332. doi: 10.1007/BF00984373. [DOI] [Google Scholar]
- 9.Burger Y, Sa’ar U, Paris HS, Lewinsohn E, Katzir N, Tadmor Y, et al. Genetic variability for valuable fruit quality traits in Cucumis melo. Isr J Plant Sci. 2006;54:233–242. doi: 10.1560/IJPS_54_3_233. [DOI] [Google Scholar]
- 10.Fernández-Trujillo JP, Picó B, Garcia-Mas J, Álvarez JM, Monforte AJ, Fernandez-Trujillo JP, Picó B, García-Mas J, Alvarez JM. Breeding for fruit quality in melon. In: Jenks M-A, Bebeli PJ, editors. Breed fruit Qual. Winley-Bla. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2011. pp. 261–278. [Google Scholar]
- 11.Diaz A, Fergany M, Formisano G, Ziarsolo P, Blanca J, Fei Z, et al. A consensus linkage map for molecular markers and Quantitative Trait Loci associated with economically important traits in melon (Cucumis melo L.) BMC Plant Biol. 2011;11:111–151. doi: 10.1186/1471-2229-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai N, Cohen S, Portnoy V, Tzuri G, Harel-Beja R, Pompan-Lotan M, et al. Metabolism of soluble sugars in developing melon fruit: a global transcriptional view of the metabolic transition to sucrose accumulation. Plant Mol Biol. 2011;76:1–18. doi: 10.1007/s11103-011-9757-1. [DOI] [PubMed] [Google Scholar]
- 13.Ariizumi T, Higuchi K, Arakaki S, Sano T, Asamizu E, Ezura H. Genetic suppression analysis in novel vacuolar processing enzymes reveals their roles in controlling sugar accumulation in tomato fruits. J Exp Bot. 2011;62(8):2773–2786. doi: 10.1093/jxb/erq451. [DOI] [PubMed] [Google Scholar]
- 14.Moreno E, Obando JM, Dos-Santos N, Fernandez-Trujillo JP, Monforte AJ, Garcia-Mas J, et al. Candidate genes and {QTLs} for fruit ripening and softening in melon. Theor Appl Genet. 2008;116:589–602. doi: 10.1007/s00122-007-0694-y. [DOI] [PubMed] [Google Scholar]
- 15.Deleu W, Esteras C, Roig C, González-To M, Fernández-Silva I, Gonzalez-Ibeas D, et al. A set of EST-SNPs for map saturation and cultivar identification in melon. BMC Plant Biol. 2009;9:90–99. doi: 10.1186/1471-2229-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harel-Beja R, Tzuri G, Portnoy V, Lotan-Pompan M, Lev S, Cohen S, et al. A genetic map of melon highly enriched with fruit quality QTLs and EST markers, including sugar and carotenoid metabolism genes. Theor Appl Genet. 2010;121:511–533. doi: 10.1007/s00122-010-1327-4. [DOI] [PubMed] [Google Scholar]
- 17.Perin C, Gomez-jimenez M, Hagen L, Dogimont C, Pech J, Pitrat M, et al. Molecular and Genetic Characterization of a Non- Climacteric Phenotype in Melon Reveals Two Loci Conferring Altered Ethylene Response in Fruit 1. Plant Physiol. 2002;129:300–309. doi: 10.1104/pp.010613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obando-Ulloa JM, Moreno E, García-Mas J, Nicolai B, Lammertyn J, Monforte AJ, et al. Climacteric or non-climacteric behavior in melon fruit: 1. Aroma volatiles. Postharvest Biol Technol. 2008;49:27–37. doi: 10.1016/j.postharvbio.2007.11.004. [DOI] [Google Scholar]
- 19.Zheng XY, Wolff DW. Ethylene production, shelf-life and evidence of RFLP polymorphisms linked to ethylene genes in melon (Cucumis melo L.) TAG Theor Appl Genet. 2000;101:613–624. doi: 10.1007/s001220051523. [DOI] [Google Scholar]
- 20.Eduardo I, Arus P, Monforte AJ, Obando J, Fernandez-Trujillo JP, Martinez JA, et al. Estimating the genetic architecture of fruit quality traits in melon using a genomic library of near isogenic lines. J Am Soc Hortic Sci. 2007;132:80–89. [Google Scholar]
- 21.Fita A, Picó B, Monforte AJ, Nuez F. Genetics of Root System Architecture Using Near-isogenic Lines of Melon. J Am Soc Hortic Sci. 2008;133:448–458. [Google Scholar]
- 22.Obando-Ulloa JM, Nicolai B, Lammertyn J, Bueso MC, Monforte AJ, Fernández-Trujillo JP. Aroma volatiles associated with the senescence of climacteric or non-climacteric melon fruit. Postharvest Biol Technol. 2009;52:146–155. doi: 10.1016/j.postharvbio.2008.11.007. [DOI] [Google Scholar]
- 23.Vegas J, Garcia-Mas J, Monforte AJ. Interaction between QTLs induces an advance in ethylene biosynthesis during melon fruit ripening. Theor Appl Genet. 2013;126:1531–1544. doi: 10.1007/s00122-013-2071-3. [DOI] [PubMed] [Google Scholar]
- 24.Pech JC, Bouzayen M, Latche A. Climacteric fruit ripening: Ethylene-dependent and independent regulation of ripening pathways in melon fruit. Plant Sci. 2008;175:114–120. doi: 10.1016/j.plantsci.2008.01.003. [DOI] [Google Scholar]
- 25.Chervin C, El-Kereamy A, Roustan J-P, Latché A, Lamon J, Bouzayen M. Ethylene seems required for the berry development and ripening in grape, a non-climacteric fruit. Plant Sci. 2004;167:1301–1305. doi: 10.1016/j.plantsci.2004.06.026. [DOI] [Google Scholar]
- 26.Trainotti L, Pavanello A, Casadoro G, Colombo VG, Padova I. Different ethylene receptors show an increased expression during the ripening of strawberries: does such an increment imply a role for ethylene in the ripening of these non-climacteric fruits? J Exp Bot. 2005;56:2037–46. doi: 10.1093/jxb/eri202. [DOI] [PubMed] [Google Scholar]
- 27.Gao F, Hao J, Yao Y, Wang X, Hasi A. Cloning and characterization of ethylene-insensitive 2 (EIN2) gene from Cucumis melo. Russian J Plant Physio. 2013;60:713–719. doi: 10.1134/S102144371305004X. [DOI] [Google Scholar]
- 28.Kendrick MD, Chang C. Ethylene signaling: new levels of complexity and regulation. Curr Opin Plant Biol. 2008;11:479–85. doi: 10.1016/j.pbi.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eriksson EM, Bovy A, Manning K, Harrison L, Andrews J, De Silva J, Tucker GA, Seymour GB. Effect of the Colorless non-ripening mutation on cell wall biochemistry and gene expression during tomato fruit development and ripening. Plant Physiol. 2004;136:4184–4197. doi: 10.1104/pp.104.045765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giovannoni JJ. Fruit ripening mutants yield insights into ripening control. Curr Opin Plant Biol. 2007;10:283–9. doi: 10.1016/j.pbi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Manning K, Tor M, Poole M, Hong Y, Thompson AJ, King GJ, Giovannoni JJ, Seymour GB. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet. 2006;38:948–952. doi: 10.1038/ng1841. [DOI] [PubMed] [Google Scholar]
- 32.Lin Z, Hong Y, Yin M, Li C, Zhang K, Grierson D. A tomato HD-Zip homeobox protein, LeHB-1, plays an important role in floral organogenesis and ripening. Plant J. 2008;55:301–10. doi: 10.1111/j.1365-313X.2008.03505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida H, Nagata M, Saito K, Wang KLC, Ecker JR. Arabidopsis ETO1 specifically interacts with and negatively regulates type 2 1-aminocyclopropane-1-carboxylate synthases. BMC Plant Biol. 2005;5:14. doi: 10.1186/1471-2229-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lincoln JE, Cordes S, Read E, Fischer RL. Regulation of gene expression by ethylene during Lycopersicon esculentum (tomato ) fruit development. Proc Natl Acad Sci. 1987;84:2793–2797. doi: 10.1073/pnas.84.9.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deikman J, Xu R, Kneissl ML, Ciardi JA, Kim KN, Pelah D. Separation of cis elements responsive to ethylene, fruit development, and ripening in the 5’-flanking region of the ripening-related E8 gene. Plant Mol Biol. 1998;37:1001–11. doi: 10.1023/A:1006091928367. [DOI] [PubMed] [Google Scholar]
- 36.Klee HJ, Giovannoni JJ. Genetics and control of tomato fruit ripening and quality attributes. Annu Rev Genet. 2011;45:41–59. doi: 10.1146/annurev-genet-110410-132507. [DOI] [PubMed] [Google Scholar]
- 37.Fujisawa M, Nakano T, Shima Y, Ito Y. A Large-Scale Identification of Direct Targets of the Tomato MADS Box Transcription Factor RIPENING INHIBITOR Reveals the Regulation of Fruit Ripening. Plant Cell. 2013;25:371–386. doi: 10.1105/tpc.112.108118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mascarell-Creus A, Cañizares J, Vilarrasa-Blasi J, Mora-García S, Blanca J, Gonzalez-Ibeas D, Saladié M, Roig C, Deleu W, Picó-Silvent B, López-Bigas N, Aranda MA, Garcia-Mas J, Nuez F, Puigdomènech P, Caño-Delgado AI. An oligo-based microarray offers novel transcriptomic approaches for the analysis of pathogen resistance and fruit quality traits in melon (Cucumis melo L.) BMC Genomics. 2009;10:467. doi: 10.1186/1471-2164-10-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esteras C, Lunn J, Sulpice R, Blanca J, Garcia-Mas J, Pitrat M et al. Phenotyping a highly diverse core melon collection to be screened using Ecotilling. Plant Genomics Eur Meet (Plant Gem), Lisbon (Portugal), 7–10 Oct 2009 2009:214.
- 40.González M, Xu M, Esteras C, Roig C, Monforte AJ, Troadec C, Pujol M, Nuez F, Bendahmane A, Garcia-Mas J, Picó B. Towards a TILLING platform for functional genomics in Piel de Sapo melons. BMC Res Notes. 2011;4:289. doi: 10.1186/1756-0500-4-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eduardo I, Arús P, Monforte AJ. Development of a genomic library of near isogenic lines (NILs) in melon (Cucumis melo L.) from the exotic accession PI161375. Theor Appl Genet. 2005;112:139–48. doi: 10.1007/s00122-005-0116-y. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalo MJ, Claveria E, Monforte AJ, Dolcet-sanjuan R. Parthenogenic Haploids in Melon : Generation and Molecular Characterization of a Doubled Haploid Line Population. J Amer Soc Hort Sci. 2011;136:145–154. [Google Scholar]
- 43.Blanca J, Cañizares J, Ziarsolo P, Esteras C, Mir G, Nuez F, García-Mas J, Picó B. Melon transcriptome characterization: Simple sequence repeats and Single nucleotide polymorphisms discovery for high throughput genotyping across the species. The Plant Genome. 2011;4(2):118–131. doi: 10.3835/plantgenome2011.01.0003. [DOI] [Google Scholar]
- 44.Blanca J, Esteras C, Ziarsolo P, Pérez D, Fernandez-Pedrosa V, Collado C, Rodriguez de Pablos R, Ballester A, Roig C, Cañizares J, Picó B. Transcriptome sequencing for SNP discovery across Cucumis melo. BMC Genomics. 2012;13:280. doi: 10.1186/1471-2164-13-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Mas J, Benjak A, Sanseverino W, Bourgeois M, Mir G, González VM, Hénaff E, Câmara F, Cozzuto L, Lowy E, Alioto T, Capella-Gutiérrez S, Blanca J, Cañizares J, Ziarsolo P, Gonzalez-Ibeas D, Rodríguez-Moreno L, Droege M, Du L, Alvarez-Tejado M, Lorente-Galdos B, Melé M, Yang L, Weng Y, Navarro A, Marques-Bonet T. The genome of melon (Cucumis melo L.) Proc Natl Acad Sci. 2012;109:11872–7. doi: 10.1073/pnas.1205415109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esteras C, Nuez F, Picó B. Genetic diversity studies in Cucurbits using molecular tools. In: Behera T, Wang Y, Kole C, editors. Genet Genomics Breed Cucurbits. New Hampshire: CRC Press; 2012. pp. 140–198. [Google Scholar]
- 47.Monforte AJ, Oliver M, Gonzalo MJ, Alvarez JM, Dolcet-Sanjuan R, Arús P. Identification of quantitative trait loci involved in fruit quality traits in melon (Cucumis melo L.) Theor Appl Genet. 2004;108:750–758. doi: 10.1007/s00122-003-1483-x. [DOI] [PubMed] [Google Scholar]
- 48.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 49.Provvidenti R. A Source of a High Level of Tolerance to Squash Mosaic Virus in a Melon from China. Cucurbit Genet Coop Rep. 1998;21:29–30. [Google Scholar]
- 50.Yu X, Wang X, Zhang W, Qian T, Tang G, Guo Y, Zheng C. Antisense suppression of an acid invertase gene (MAI1) in muskmelon alters plant growth and fruit development. J Exp Bot. 2008;59:2969–77. doi: 10.1093/jxb/ern158. [DOI] [PubMed] [Google Scholar]
- 51.Yoo S-D, Cho Y, Sheen J. Emerging connections in the ethylene signaling network. Trends Plant Sci. 2009;14:270–9. doi: 10.1016/j.tplants.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cara B, Giovannoni JJ. Molecular biology of ethylene during tomato fruit development and maturation. Plant Sci. 2008;175:106–113. doi: 10.1016/j.plantsci.2008.03.021. [DOI] [Google Scholar]
- 53.Devoto A, Hartmann H, Piffanelli P, Elliott C, Simmons C, Taramino G, Goh G, Cohen F, Emerson B, Schulze-Lefert P, Panstruga R. Molecular phylogeny and evolution of the plant-specific seven-transmembrane MLO family. J Mol Evol. 2003;56:77–88. doi: 10.1007/s00239-002-2382-5. [DOI] [PubMed] [Google Scholar]
- 54.Clayberg CD. Interaction and linkage tests of flesh color genes in Cucumis melo L. Cucurbit Genet Coop Rep. 1992;15:53. [Google Scholar]
- 55.Dhillon N, Monforte A, Pitrat M, Pandey S, Singht P, Reitsma KR, Garcia-Mas J, Sharma A, McCreight JD. Melon landraces of India: contributions and importance. Plant Breed Rev. 2012;35:85–150. [Google Scholar]
- 56.Tomason Y, Nimmakayala P, Levi A, Reddy UK. Map-based molecular diversity, linkage disequilibrium and association mapping of fruit traits in melon. Mol Breed. 2013;31:829–841. doi: 10.1007/s11032-013-9837-9. [DOI] [Google Scholar]
- 57.Esteras C, Pascual L, Saladie M, Dogimont C, Garcia-Mas J, Nuez F et al. Use of Ecotilling to identify natural allelic variants of melon candidate genes involved in fruit ripening. In Plant Genomics Eur Meet (Plant Gem), Lisboa (Portugal), PLANT GEM 7-10 Oct 2009; 2009:213.
- 58.Lasserre E, Bouquin T, Hernandez JA, Bull J, Pech JC, Balagué C. Structure and expression of three genes encoding ACC oxidase homologs from melon (Cucumis melo L.) Mol Gen Genet. 1996;251:81–90. doi: 10.1007/BF02174348. [DOI] [PubMed] [Google Scholar]
- 59.Chervin C, Deluc L. Ethylene signalling receptors and transcription factors over the grape berry development: gene expression profiling. Vitis. 2010;49:129–136. [Google Scholar]
- 60.Kieber JJ, Rothenberg M, Roman G, Feldmann KA, Ecker JR. CTR1, a negative regulator of the ethylene response pathway in Arabidopsis, encodes a member of the raf family of protein kinases. Cell. 1993;72:427–41. doi: 10.1016/0092-8674(93)90119-B. [DOI] [PubMed] [Google Scholar]
- 61.Wang A, Tan D, Takahashi A, Li TZ, Harada T. MdERFs, two ethylene-response factors involved in apple fruit ripening. J Exp Bot. 2007;58:3743–3748. doi: 10.1093/jxb/erm224. [DOI] [PubMed] [Google Scholar]
- 62.Bassett C, Artlip T, Nelson M. Characterization of an ETR homologue from peach (Prunus persica). Plant Biol 1999.
- 63.Barry CS, Mcquinn RP, Thompson AJ, Seymour GB, Grierson D, Giovannoni JJ, et al. Ethylene Insensitivity Conferred by the Green-ripe and Never-ripe 2 Ripening Mutants of Tomato 1. Plant Physiol. 2005;138:267–275. doi: 10.1104/pp.104.057745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giovannoni JJ. Genetic Regulation of Fruit Development and Ripening. Plant Cell. 2004;16:170–181. doi: 10.1105/tpc.019158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lanahan MB. The Never Ripe Mutation Blocks Ethylene Perception in Tomato. Plant Cell. 1994;6:521–530. doi: 10.1105/tpc.6.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilkinson J, Lanahan M, Yen H, Giovannoni J, Klee H. An ethylene-inducible component of signal transduction encoded by NEVER-RIPE. Science. 1995;270:1807–1809. doi: 10.1126/science.270.5243.1807. [DOI] [PubMed] [Google Scholar]
- 67.Yang YW, Wu Y, Pirrello J, Regad F, Bouzayen M, Deng W, Li ZG. Silencing Sl-EBF1 and Sl EBF2 expression causes constitutive ethylene response phenotype, accelerate plant senescence and fruit ripening in tomato. J Exp Bot. 2010;61:697–708. doi: 10.1093/jxb/erp332. [DOI] [PubMed] [Google Scholar]
- 68.Vrebalov J, Ruezinsky D, Padmanabhan V, White R, Medrano D, Drake R, Schuch W, Giovannoni J. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (Rin) locus. Science. 2002;296:343–346. doi: 10.1126/science.1068181. [DOI] [PubMed] [Google Scholar]
- 69.Saladié M, Rose JKC, Cosgrove DJ, Catalá C. Characterization of a new xyloglucan endotransglucosylase/hydrolase (XTH) from ripening tomato fruit and implications for the diverse modes of enzymic action. Plant J. 2006;47:282–95. doi: 10.1111/j.1365-313X.2006.02784.x. [DOI] [PubMed] [Google Scholar]
- 70.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Doyle JJDJ. Isolation of plant DNA from fresh tissue. Focus. 1990;12:13–15.74. [Google Scholar]
- 72.Stitt M, Lilley R, Gerhardt R, Heldt H. Determination of metabolite levels in specific cells and subcellular compartments of plant leave. Methods Enzym. 1989;174:518–522. doi: 10.1016/0076-6879(89)74035-0. [DOI] [Google Scholar]
- 73.Jombart T, Ahmed I. adegenet 1.3–1: new tools for the analysis of genome-wide SNP data. Bioinformatics. 2011;27:3070–1. doi: 10.1093/bioinformatics/btr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–5. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 75.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21:2128–2129. doi: 10.1093/bioinformatics/bti282. [DOI] [PubMed] [Google Scholar]
- 77.Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 78.Breseghello F, Sorrells ME. Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics. 2006;172:1165–77. doi: 10.1534/genetics.105.044586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peltier J: LOESS Smoothing in Excel. 2009.