

Abstract
Chronic viral infections represent a major burden to human health, and modulation of the immune system is emerging as a novel approach to fighting such infections. Pellegrini et al. (2011) demonstrate that treatment with the cytokine IL-7 may reinvigorate the immune response to persistent infection by targeting immunosuppressive Socs3 proteins.
Persistent infection with viruses such as human immunodeficiency virus (HIV) and hepatitis C virus (HCV) causes debilitating illness associated with high rates of mortality and morbidity. While long-term viral persistence can often be attributed to viral evasion of the immune system, it is now evident that host-derived immunosuppressive processes also actively disrupt viral clearance. T cells represent a key effector arm of the immune system required for virus control. However, during certain chronic viral infections, some antiviral T cells fail to survive, leaving holes in the T cell repertoire, whereas others persist in a dysfunctional or “exhausted” state with impaired effector functions (Zajac et al., 1998). Strikingly, a host program of immunosuppression that involves immunological signaling molecules (cytokines) such as IL-10 and TGF-β, as well as inhibitory receptors like PD-1, directs such T cell dysfunction (Barber et al., 2006; Brooks et al., 2006; Ejrnaes et al., 2006; Tinoco et al., 2009).
Though it may seem counterintuitive to dampen immune responsiveness to an ongoing infection, this process likely evolved to limit the tissue destruction that would result from an unregulated immune response against a widely disseminated virus. Nevertheless, transient interference with these inhibitory pathways has clear therapeutic benefits, given that it improves T cell function and lowers viral titers in animal models (Barber et al., 2006; Brooks et al., 2006; Ejrnaes et al., 2006; Tinoco et al., 2009). However, lethal immunopathology can arise if the timing of treatment is wrong (Barber et al., 2006), demonstrating that boosting the immune response can come at a cost. Therefore, the ideal immunotherapy would act to boost the immune response while limiting any collateral damage to host tissues. In this issue, Pellegrini et al. (2011) demonstrate that administration of the cytokine IL-7 leads to viral control during chronic infection through its ability to simultaneously augment the T cell response and induce factors that limit tissue destruction. IL-7 treatment has the added benefit of boosting overall T cell numbers, a feature that could help to counter the low T cell numbers associated with HIV-induced acquired immunodeficiency syndrome (AIDS).
Though IL-7 is known primarily for its role in promoting survival and homeostasis of naive and memory T cells, past work by the authors demonstrated that IL-7 also boosts effector functions within T cells. The authors speculated that IL-7 administration during chronic viral infection might similarly improve antiviral T cell function and facilitate viral clearance. To test this hypothesis, they administer IL-7 to mice infected with lymphocytic choriomeningitis virus (LCMV) clone 13, a powerful animal model of chronic viral infection that recapitulates many aspects of persistent virus infection in humans. The authors observe a dramatic effect, with accelerated virus clearance due to a large boost in both the numbers and functionality of antiviral T cells. Surprisingly, the animals survive this immune onslaught without detectable organ damage, at least as assessed by examining liver damage. More impressively, the treatment is effective despite beginning at day 8 postinfection, when virus levels peak and antiviral T cell responses begin showing signs of exhaustion.
The profound impact of IL-7 treatment on the immune response is likely multifactorial. Given the known role of IL-7 in T cell survival, the authors first examine the effect of IL-7 on overall T cell numbers and find that IL-7 drives an expansion of the entire pool of T cells, in part due to an increase in T cell production by the thymus. However, their work suggests that the increase in thymic output of T cells probably does not contribute to the elevation in virus-specific T cell numbers, but rather, this stems from other effects. Nevertheless, the effect of IL-7 was dependent on T cells, given that depletion of T cells (but not B cells) ablates the response induced by IL-7.
The authors also examine changes in cytokine levels after IL-7 treatment and find a large shift in the cytokine profile, most notably an increase in the levels of IL-6 and IL-17, a decrease in immunosuppressive TGF-β, and an increase in the tissue-protective cytokine IL-22. IL-6 appears to be a key cytokine in this context, given that it is required for both the enhanced immune response and the cytoprotective effects of IL-7 treatment. Furthermore, the elevated IL-22 secretion is dependent on IL-6, and the authors subsequently find that IL-22 plays a key role in the prevention of liver destruction. These results thus explain how IL-6 prevents tissue destruction, but they don’t explain how IL-6 promotes the T cell response.
Next, the authors investigate potential mechanisms by which IL-6 may boost the immune response. They first show that the effects of IL-7 do not depend on regulatory T cells, a cell type known to suppress activated T cell function. Instead, they speculate that IL-7 may alter the responsiveness of T cells to IL-6. They test this idea by measuring the levels of suppressor of cytokine signaling 3 (Socs3), a protein known to modulate IL-6 responsiveness. Indeed, they find higher Socs3 levels in T cells derived from mice with chronic versus acute LCMV infection. Furthermore, IL-7 treatment lowers the amount of Socs3 within T cells, possibly via AKT and FOXO signaling. Perhaps most importantly, selective ablation of Socs3 in T cells causes early virus clearance and recapitulates many of the effects of IL-7 treatment. These data demonstrate a role for Socs3 in limiting T cell responsiveness during chronic viral infection and implicate IL-7 treatment as a potential therapeutic approach for interfering with this pathway (Figure 1).
Figure 1. Effects of IL-7 during Chronic Viral Infection.
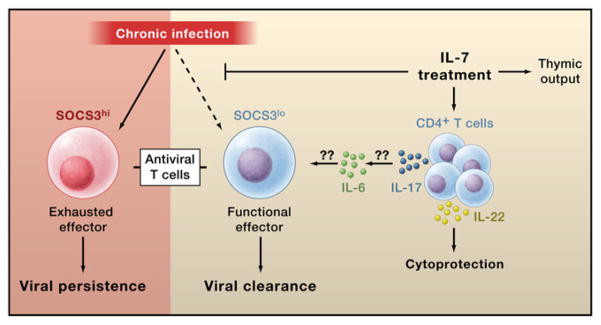
Pellegrini et al. (2011) demonstrate that chronic viral infection (left) promotes high expression of Socs3, a negative regulator of immune cytokine signaling, in antiviral T cells. Socs3 impairs T cell function and promotes T cell “exhaustion,” leading to viral persistence. Treatment with the cytokine IL-7 (right) blocks Socs3 induction in antiviral T cells, thereby promoting effector functions and viral clearance. IL-7 likely acts on CD4+ T cells to promote IL-17 secretion, which in turn induces IL-6 production. IL-6 promotes survival and function of antiviral T cell by an unknown mechanism. IL-7 also promotes IL-22 secretion, which protects against tissue destruction by the elevated immune response. IL-7 may also act directly on the antiviral CD4 and CD8 T cells (not shown). All of these factors lead to viral clearance without adverse immunopathology. In addition to its antiviral effect, IL-7 also boosts thymic production of naive T cells. The elevated thymic output could help to counter lower T cell levels during chronic HIV infection.
This study provides exciting clues as to how the immune response is regulated during chronic infection and how we may manipulate regulatory pathways therapeutically. A number of questions remain, however. First, the exact mechanism by which IL-7 treatment causes IL-6 upregulation remains unclear. The authors note an increase in both CD4+ T cells that produce IL-17 (Th17 cells) and serum IL-17 after IL-7 treatment. Given that Socs3 is an inhibitor of Th17 differentiation, they hypothesize that the reduction in Socs3 levels caused by IL-7 permits greater numbers of Th17 cells to develop. An increase in Th17 cells, in turn, likely induces IL-6 production, consistent with previous experiments (Ogura et al., 2008). Such a model predicts that IL-17 is required for the up-regulation of IL-6 upon IL-7 treatment, an idea that needs testing. Furthermore, it will be important to determine whether the Th17 cells are virus specific or derived from another source.
Second, it is unclear how IL-6 signaling and Socs3 deficiency conspire to elevate the antiviral T cell response. Socs3 loss could cause altered IL-6 signaling in antiviral T cells (Johnston and O’Shea, 2003), thereby boosting their survival and function. However, it is unlikely that IL-7 and IL-6 signaling is direct, given that IL-6 and IL-7 receptors are transcriptionally repressed in virus-specific T cells during clone 13 infection (Wherry et al., 2007). The cause of elevated Socs3 expression in untreated mice during chronic infection is also of interest. The inhibitory cytokine IL-10 is a likely candidate because IL-10 signaling induces Socs3 expression, and IL-10 deficiency has effects on chronic viral infection similar to those caused by IL-7 treatment (Brooks et al., 2006; Ejrnaes et al., 2006).
Finally, the study suggests that Th17 cells may protect against virus infection. Although Th17 cells have been previously associated with better control of influenza infection (McKinstry et al., 2009), Th17 cells are typically linked to antifungal and antibacterial immunity. Further work will be required to determine the exact role of these cells in antiviral immunity. Ultimately, this study suggests that IL-7 treatment holds great promise for controlling chronic viral infections, such as those caused by HIV and hepatitis B and C viruses, which together infect and afflict more than 10% of the world’s population.
References
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Nat Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, von Herrath MG. J Exp Med. 2006;203:2461–2472. doi: 10.1084/jem.20061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, O’Shea JJ. Nat Immunol. 2003;4:507–509. doi: 10.1038/ni0603-507. [DOI] [PubMed] [Google Scholar]
- McKinstry KK, Strutt TM, Buck A, Curtis JD, Dibble JP, Huston G, Tighe M, Hamada H, Sell S, Dutton RW, Swain SL. J Immunol. 2009;182:7353–7363. doi: 10.4049/jimmunol.0900657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, Iwakura Y, Hirano T. Immunity. 2008;29:628–636. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Calzascia T, Toe JG, Preston SP, Lin AE, Elford AR, Shahinian A, Lang PA, Lang KL, Morre M, et al. Cell. 2011;144:601–613. doi: 10.1016/j.cell.2011.01.011. this issue. [DOI] [PubMed] [Google Scholar]
- Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Immunity. 2009;31:145–157. doi: 10.1016/j.immuni.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]