

Abstract
Background
Aneurysms affecting the aorta are a common condition associated with high mortality as a result of aortic dissection or rupture. Investigations of the pathogenic mechanisms involved in syndromic types of thoracic aortic aneurysms, such as Marfan and Loeys-Dietz syndromes, have revealed an important contribution of disturbed transforming growth factor (TGF)-β signaling.
Objectives
This study sought to discover a novel gene causing syndromic aortic aneurysms in order to unravel the underlying pathogenesis.
Methods
We combined genome-wide linkage analysis, exome sequencing, and candidate gene Sanger sequencing in a total of 470 index cases with thoracic aortic aneurysms. Extensive cardiological examination, including physical examination, electrocardiography, and transthoracic echocardiography was performed. In adults, imaging of the entire aorta using computed tomography or magnetic resonance imaging was done.
Results
Here, we report on 43 patients from 11 families with syndromic presentations of aortic aneurysms caused by TGFB3 mutations. We demonstrate that TGFB3 mutations are associated with significant cardiovascular involvement, including thoracic/abdominal aortic aneurysm and dissection, and mitral valve disease. Other systemic features overlap clinically with Loeys-Dietz, Shprintzen-Goldberg, and Marfan syndromes, including cleft palate, bifid uvula, skeletal overgrowth, cervical spine instability and clubfoot deformity. In line with previous observations in aortic wall tissues of patients with mutations in effectors of TGF-β signaling (TGFBR1/2, SMAD3, and TGFB2), we confirm a paradoxical up-regulation of both canonical and noncanonical TGF-β signaling in association with up-regulation of the expression of TGF-β ligands.
Conclusions
Our findings emphasize the broad clinical variability associated with TGFB3 mutations and highlight the importance of early recognition of the disease because of high cardiovascular risk.
Key Words: Loeys-Dietz syndrome, gene, TGF-β pathway, thoracic aortic aneurysm
Abbreviations and Acronyms: LAP, latency-associated peptide; LDS, Loeys-Dietz syndrome; LOF, loss of function; MFS, Marfan syndrome; MIM, Mendelian Inheritance in Man; SNP, single nucleotide polymorphism; TAAD, thoracic aortic aneurysms and dissections; TGF, transforming growth factor; TGFBR, transforming growth factor beta receptor
The transforming growth factor (TGF)-β pathway plays an important role in many medically relevant processes, including immunologic maturity, inflammation, cancer, and fibrosis, as well as skeletal, vascular, and hematopoietic homeostasis (1). With the discovery of dysregulated TGF-β signaling in Fbn1 knockout mice, the TGF-β pathway was revealed as a key player in the pathogenesis of thoracic aortic aneurysm development in Marfan syndrome (MFS; [Mendelian Inheritance in Man (MIM) 154700]) 2, 3. MFS is a multisystemic disease characterized by cardiovascular, ocular, and skeletal features caused by mutations in the FBN1 gene (4). More recently, we and others identified pathogenic mutations in the genes encoding the TGF-β receptor (TGFBR) subunits TGFBR1 and TGFBR2 5, 6, the signaling transducer SMAD3 (7), the ligand TGFB2 8, 9, and the inhibitor SKI (10), occurring predominantly in patients with syndromic presentations of thoracic aortic aneurysms and dissections (TAAD), designated Loeys-Dietz syndrome (LDS1 [MIM 609192] [11]; LDS2 [MIM 610168] [11]; LDS3 [MIM 613795] [also known as aneurysms-osteoarthritis syndrome] 7, 12, 13, LDS4 [MIM 614816] [8]), and Shprintzen-Goldberg syndrome (SGS [MIM 82212]) 13, 14. The finding of human mutations in a ligand, receptors, a signaling transducer, and an inhibitor of the TGF-β pathway confirms the essential role of TGF-β signaling in aortic aneurysm development.
Recently, de novo mutations in the gene encoding the TGFB3 ligand (TGFB3) were reported in 2 girls exhibiting a syndrome affecting body growth (either short or tall stature) accompanied by skeletal features reminiscent of MFS or LDS, but without significant vascular involvement 15, 16, 17. Here, we report that TGFB3 mutations cause a syndromic form of aortic aneurysms and dissections, characterized by cardiovascular, craniofacial, cutaneous, and skeletal anomalies that significantly overlap with other TGF-β vasculopathies, particularly those within the LDS clinical spectrum.
Methods
Patients
All patients or relatives provided written informed consent for participation in this study and, if applicable, publication of photographs. Family 1 was investigated by the department of Clinical Genetics (Erasmus University Medical Center, Rotterdam, the Netherlands) and Center for Medical Genetics (Antwerp University Hospital/University of Antwerp, Belgium) after previous surgical interventions. Clinical geneticists (M.W.W., B.L.L.) examined family members, with special attention to skeletal, joint, skin, and craniofacial features. Medical records from deceased patients were obtained for review. Extensive cardiological examination, including physical examination, electrocardiography, and transthoracic echocardiography, was performed. In adults, imaging of the entire aorta using computed tomography or magnetic resonance imaging was performed. Measurements of the aortic diameter were obtained at the level of the aortic annulus, sinuses of Valsalva, sinotubular junction, proximal ascending aorta, aortic arch, descending aorta, and suprarenal and infrarenal abdominal aorta. An aneurysm was defined as an arterial diameter >1.96 SDs above the predicted diameter 18, 19. Probands from families 2 through 8 and 9 through 11 were referred for molecular and/or clinical evaluation to Antwerp (Belgium) or Osaka (Japan), respectively.
Screening of the entire coding region of TGFB3 was performed in 470 additional probands (120 probands had whole-exome sequencing), presenting both with syndromic and nonsyndromic forms of TAAD. The majority of these patients had been screened previously for all known TAAD genes. Family members of mutation-positive patients were ascertained and submitted to clinical investigations.
Genotyping and linkage analysis
Genomic DNA was extracted from peripheral blood samples (Gentra Systems, Qiagen, Hilden, Germany). RNA from 2 patients (1-II:12 and III:11) (Figure 1) was extracted from peripheral blood (collected in PAXgene tubes, PreAnalyliX, Qiagen) according to the manufacturer’s protocol (PreAnalyliX, Qiagen).
Figure 1.

Overview of Families With TGFB3 Mutations
The causal TGFB3 mutation is shown for each family. Probands are indicated with an arrow. Circle: female; square: male; open symbol: unaffected; solid symbol: affected; diagonal line: deceased; brackets: adopted; question mark: clinical affection status unknown. Plus and minus signs indicate presence or absence of a TGFB3 mutation, respectively.
Genome-wide genotyping was conducted using DNA from 6 family members (Figure 1, family 1) with Illumina Human SNP-Cyto12 Arrays (Illumina, San Diego, California), containing >262,000 genomic markers, as recommended by the manufacturer. The statistical package, easyLINKAGE Plus v5.08 (20), Merlin v1.0.1 software (Abecasis Lab, University of Michigan), was used to perform single-point and multipoint parametric linkage analysis as previously described 21, 22. Logarithm of odds scores were obtained using a dominant model of inheritance, with 90% penetrance and disease allele frequency of 1:1,000. Allele frequencies of genotyped single nucleotide polymorphisms (SNPs) were set to codominant, and spacing of 0.25 Mb to 0.15 Mb between SNPs was used. Haplotype blocks containing 100 SNPs were constructed with Merlin (option BEST) and they were visualized using HaploPainter (v1.042, H. Thiele, University of Cologne, Germany).
Sequencing and mutation analysis
Exome sequencing was performed for 120 patients after TruSeq Exome enrichment on HiSeq (Illumina). In 350 other probands, bidirectional Sanger sequencing of exons and exon–intron boundaries was undertaken using polymerase chain reaction primers designed by Primer3 software (v. 4.0.0, S. Rozen, Howard Hughes Medical Institute and the National Institutes of Health, National Human Genome Research Institute) (Online Table 1). Polymerase chain reaction products were purified and sequenced using BigDye Terminator chemistry v3.1 on an ABI Prism3130xl (Applied Biosystems, Foster City, California). Sequences were aligned (SeqScape v2.5 software, Applied Biosystems) and compared with consensus sequences obtained from the human genome databases (Ensembl and NCBI). For annotation of DNA and protein changes, the Mutation Nomenclature guidelines from the Human Genome Variation Society were followed (23). To describe mutations at the cDNA level, the A from the ATG start codon of the reference sequences is numbered as 1 (mRNA NM_003239.2 and protein NP_003230.1).
In silico analysis of novel variants
The effects of the mutations on protein structure and function were predicted using SIFT BLink (v.5.2.2) and Mutation Taster2. Population frequencies in controls were obtained from dbSNP, Exome Variant Server (EVS) (24), 1000Genomes (25), and Genome of the Netherlands (26). To assess the putative effects on splicing, the Splice Site Prediction by Neural Network (27), the NetGene2 (28) and Alamut Software Suite were used. Protein IDs used for conservation were gi|148342461 (ABQ59024.1), gi|135685 (P17125.1), gi|18266825 (P16047.2), gi|135682 (P17247.1), gi|52138563 (NP_919367.2), gi|410898023 (XP_003962498.1), gi|351050916 (CCD74236.1), gi|17137520 (NP_477340.1), gi|135674 (P01137.2), and gi|48429157 (P61812.1).
Homology modeling
A homology model was built using the experimentally solved structure of TGFB1 (PDB file [29] 3rjr, 60% identity) as a template. The model was built using an automatic YASARA script (30) with standard parameters. The model contains a homodimer of residues 14 to 412.
Immunohistochemistry
The protocol for staining of formalin-fixed, paraffin-embedded sections was adapted from Baschong et al. (31) with modifications (Detailed Methods, Online Appendix). Slides were stained overnight at 4°C with anti-pSmad2 antibody (clone A5S, 1:100, Millipore, Billerica, Massachusetts) and anti-pERK1/2 (clone D13.14.4E, 1:100, Cell Signaling Technology, Danvers, Massachusetts) in 0.1% Triton/TBS buffer, washed 3 × 10 min in Perm/Staining buffer, and then stained with anti-rabbit Alexa594 (Molecular Probes, Life Technologies, Carlsbad, California) at 1:200 for 1 h at RT. Slides were then washed 3 × 10 min in Perm/Staining buffer and mounted with Hard Set VECTASHIELD Mounting Media (Vector Laboratories, Burlingame, California) with 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI). Images were acquired on a Zeiss AxioExaminer (Carl Zeiss, Oberkochen, Germany) with 710NLO-Meta multiphoton confocal microscope at 25× magnification.
In situ RNA with ACD RNAscope probes
The ACD RNAscope probe Hs-TGFB1 probe (Advanced Cell Diagnostics [ACD], Hayward, California) was used to detect human TGFB1 transcript in conjunction with the RNAscope 2.0 HD Reagent Kit (RED) from ACD (Detailed Methods, Online Appendix).
Histology
Slides were histologically examined after hematoxylin-eosin, Elastica van Gieson (elastin), Alcian blue (proteoglycans), or Masson’s trichrome (collagen) staining using standard techniques.
Results
We studied a large Dutch family (family 1) with clinical features overlapping with MFS and LDS consistent with an autosomal dominant inheritance pattern. Seven family members, between 40 and 68 years of age, presented with aneurysms and dissections, mainly involving the descending thoracic and abdominal aorta (Figure 1, Online Table 2). Three patients died from aortic dissection and rupture of the descending thoracic or abdominal aorta (1-II:4, II:5, and II:7) (Figure 1), confirmed by autopsy in 2 cases (1-II:4, age 57 years and II:5, 56 years). In addition, 4 members had mitral valve abnormalities, ranging from mild prolapse to severe regurgitation requiring surgical intervention. Craniofacial abnormalities were rather subtle, including a long face, high-arched palate, and retrognathia (Figure 2). Pectus deformity and scoliosis were frequently observed (Figure 2). Other recurrent findings included velvety skin, varices, and hiatal hernia. Several family members presented with autoimmune features including (HLA-B27 positive) spondyloarthritis, Graves' disease, and celiac disease.
Figure 2.

Phenotypic Characteristics of Patients With a TGFB3 Mutation
Observed clinical features include: long face (1-III:11, 5-II:1, 7-II:1, 8-II:1); pectus carinatum (1-IV:2); hypertelorism (2-III:7, 2-IV:2, 2-IV:3, 7-II:1, 8-II:1); bifid uvula (2-III:7, 2-IV:3, 7-II:1); joint hypermobility (2-IV:2); arachnodactyly (5-II:1); and metatarsus adductus (8-III:1). All affected individuals or parents gave permission to publish these photographs.
Sequencing of all known TAAD genes (ACTA2, COL3A1, EFEMP2, FBN1, FLNA, MYH11, MYLK, NOTCH1, PRKG1, SKI, SLC2A10, SMAD3, TGFB2, TGFBR1, TGFBR2) failed to identify a causal mutation. Linkage analysis using SNP genotypes from 6 patients of the family identified 2 large genomic regions on chromosomes 14 and 15 shared by all affected patients (Online Figure 1). Detailed inspection of the genes in the regions identified several candidates, most prominently the TGFB3 gene on 14q24. Subsequent Sanger sequencing of all 7 exons and intron boundaries identified a heterozygous intronic variant affecting the highly conserved (PhastCons: 1, PhyloP: 4.97) canonical donor splice site of exon 4 (c.754+2T>C), which is absent from variant databases (Variant Server, Genome of the Netherlands, 1000Genomes). Sequencing of the cDNA for 2 patients (1-II:12 and III:11) confirmed skipping of exon 4, leading to an in-frame deletion of 108 nucleotides (Online Figure 2). At the protein level, a deletion of 36 amino acids is expected (p.Glu216_Lys251del). This TGFB3 mutation (c.754+2T>C) segregated with the clinical phenotype and was also present in 1 young individual (1-IV:1, 17 years old) without documented cardiovascular features (Figure 1, Online Table 2), but with mild systemic manifestations including craniofacial features, easy bruising, and scoliosis.
To further investigate the role of TGFB3 in TAAD etiology, DNA samples from 350 syndromic and nonsyndromic TAAD probands were Sanger sequenced for mutations in all exon–intron boundaries and the coding region of TGFB3. Additionally, in 120 TAAD patients, a targeted analysis of TAAD candidate genes after whole-exome sequencing was performed. This revealed additional heterozygous TGFB3 mutations in 10 other probands (7 from Sanger sequencing and 3 from the exome sequencing cohort): 4 different missense mutations, p.Asp263His (family 3), p.Arg300Trp (families 2, 4, 5, 11), p.Ile322Thr (family 10), p.Leu401Pro (family 8); 1 nonsense mutation, p.Tyr365* (family 6); and 2 single-base deletions leading to a frameshift and premature stop codon, p.Leu386Argfs*21 (family 7) and p.Asn235Metfs*11 (family 9) (Figure 3). All missense mutations were predicted as deleterious by SIFT (32) and as disease causing by Mutation Taster (33). The 2 missense mutations in exon 5 (p.Asp263His and p.Arg300Trp) both affect highly conserved amino acids of the latency-associated peptide (LAP) domain, which are also conserved among the TGFB1, TGFB2, and TGFB3 proteins (Figure 3). The p.Asp263His alteration disrupts the Arg-Gly-Asp (RGD) motif, which is essential for binding to the αvß3, αvß6, αvß1, and αvß5 integrins 34, 35, 36. Mutations of the RGD motif in LAPβ3 were demonstrated to abolish binding to αvß3, αvß5, and αvß6 (36). The second missense mutation in exon 5, p.Arg300Trp, affects the last amino acid of the LAP domain, disrupting the last residue of the RKKR minimal recognition motif of the furin or related protease cleavage site (37). Mutations affecting similar amino acids in TGFB2 have been shown to be causal in syndromic forms of aortic aneurysms (8). The 2 other missense mutations, p.Ile322Thr and p.Leu401Pro, affect highly conserved amino acids located in the region of the active cytokine. The 3 other mutations create premature stop codons, either in the LAP domain or in the active TGFB3 (cytokine) domain, leading to nonsense-mediated decay or truncated proteins, which probably lose their cytokine activity.
Figure 3.

Mutation Overview of the TGFB3 Gene
Exons are represented by rectangles. Exon numbering is given, and different colors denote the different protein domains. Mutations found in this study are indicated below the gene in the respective domains. Evolutionary conservation in TGFB3 and its related proteins is given for the 4 missense mutations (p.Arg300Trp: Family 2, 4, 5 and 11; p.Asp263His: Family 3; p.Leu401Pro: Family 8; p.Ile322Thr: Family 10). Previously published mutations are shown above the gene, with a single degree symbol indicating mutation described in Matyas et al. (17) and double degree symbols indicating mutation from Rienhoff et al. (16).
The causal nature of the TGFB3 mutations was further supported by de novo occurrence (family 6) (Figure 1) and absence from controls (all mutations) in EVS, 1000Genomes, and the Genome of the Netherlands. Although p.Tyr365* in family 6 occurred de novo, we previously identified a SMAD3 variant (p.Ala250Thr) of unknown significance in the proband. This SMAD3 variant was also present in the proband’s mother, who presented with variable connective tissue findings and mild cardiovascular involvement, making its precise contribution to pathogenesis unclear.
We studied the molecular effects of these mutations in more detail using a homology model of the TGFB3 dimer. Asp263 is located in a surface loop where it is accessible for integrins (Figure 4). Mutation p.Glu216_Lys251del results in deletion of a central beta-strand and subsequent surface loop in the LAP domain (Figure 4). This will severely affect this domain’s conformation, including the position of the RGD motif, and thereby affect dimerization and inhibition of the TGFB3 domain.
Figure 4.

Overview of the TGFB3 Dimer Model and Mutations
One monomer is shown in grey, with the other monomer in cyan (LAP domain) and blue (TGFB3 domain). Residues deleted by the p.Glu216_Lys251del mutation are in purple. Residues affected by the p.Leu386Argfs21* mutation are in yellow (note that this mutation also adds 21 different residues that cannot be modeled). Residues deleted by the p.Tyr365* mutation are shown in orange and yellow (note that this mutation deletes all residues following Tyr365). Residues deleted by mutation p.Asn235Metfs*11 are shown in grey-olive in the second monomer (note that this mutation also adds 11 different new residues, which cannot be modeled). The point mutations p.Asp263His, p.Arg300Trp, p.Ile322Thr, and p.Leu401Pro are shown in red with their side chains visible as red spheres. A detailed close-up of these mutations is shown in 4 extra panels. In these panels, the wild-type residue side chain is in green, whereas the mutant side chain is in red. For the p.Arg300Trp mutation, side chains of nearby negative residues are also shown. For the p.Leu401Pro mutation, nearby hydrophobic residues are shown.
Missense mutations p.Arg300Trp, p.Ile322Thr, and p.Leu401Pro are also predicted to alter TGFB3 function. Besides participating in the cleavage site, Arg300 is involved in several ionic interactions that will be lost with the substitution to tryptophan, and this bulky residue will likely induce steric rearrangements. Ile322Thr is predicted to alter the positioning of Arg325, an important amino acid residue for binding with TGFBR2. In addition, hydrophobic contacts with the N-terminal helix in the LAP domain will be affected by the substitution with threonine (hydrophilic). Substitution from Leu401 to proline is predicted to change the hydrophobic interactions with residues of both the LAP and TGFB3 domain.
The clinical phenotypes in the 10 additional families demonstrate significant overlap with Loeys-Dietz syndrome (Online Tables 3 to 6). Vascular involvement ranges from no cardiovascular abnormalities at age 64 (3-II:2) to type A (median age of 51 years, range 40 to 80 years) or type B aortic dissection (median age of 44.5 years, range 30 to 57), abdominal aortic dissection and death as a result of cerebral aneurysm dissection at age 55 (2-II:1) (Table 1). So far, no examples of early arterial dissection or dissection at small aortic dimension were observed. Other cardiovascular features include mitral valve disease, ranging from mild insufficiency to chorda rupture necessitating surgery, and persistent foramen ovale and atrial or ventricular septal defects. Disease beyond the aorta, with iliac and subclavian artery aneurysms, was only identified in 2 patients (1-II:12 and 1-III:13). No striking aortic or arterial tortuosity was observed.
Table 1.
Patient Characteristics
| Affected Individuals (n = 43)∗ | |
|---|---|
| Sex, M/F | 23/20 |
| Age, yrs | 34 (3–74) |
| Age at death, yrs | 56 (40–80) |
| Age at dissection, yrs | 47.5 (30–80) |
| Cardiovascular findings | |
| Type A dissections, age, yrs | 4 (51; 40–80) |
| Type B dissections, age, yrs | 6 (44.5; 30–57) |
| Aortic aneurysm†, age, yrs | 6 (34; 3–68) |
| Abdominal aortic surgery‡ | 2 |
| Disease beyond aorta§ | 3 |
| Skeletal findings | |
| Tall stature | 12 |
| Arachnodactyly | 16 |
| Pectus deformity | 8 |
| Kyphoscoliosis | 11 |
| Joint hypermobility | 9 |
| Loeys-Dietz features | |
| Hypertelorism | 14 |
| Bifid uvula | 11 |
| Cleft palate | 5 |
Values are n, median (range), or n (median; range).
Not all patients were evaluated for all features.
Four aneurysms affected the sinuses of Valsalva, and 2 only affected the ascending aorta.
Surgery was performed on 1 patient at age 43 years and 1 at 50 years.
Cerebral, iliac, or subclavian arteries (n = 1 for each location).
Typical LDS findings such as hypertelorism, bifid uvula and cleft palate, cervical spine instability, and club foot deformity are commonly observed (Figure 2, Table 1, Table 2, and Online Tables 3 to 6). Other recurrent features include dolichocephaly, high-arched palate, retrognathia (with surgery in case 3-III:1), tall stature, joint hypermobility, arachnodactyly, pectus deformity, and inguinal hernia (Table 1, Figure 2). No evidence for ectopia lentis was found in the medical records. Early-onset osteoarthritis was only reported in 2 individuals (10-II:1 and 11-II:1). The clinical features from 43 identified patients belonging to 11 families are summarized in Table 1. We observed a striking intrafamilial and interfamilial clinical variability with typical LDS features in some, but complete absence in others.
Table 2.
Comparison of Phenotypical Characteristics of Patients With TGFBR1/2, SMAD3, TGFB2, and TGFB3 Mutations
| Phenotype |
TGFBR1 TGFBR2 |
SMAD3 | TGFB2 | TGFB3 |
|---|---|---|---|---|
| Hypertelorism | ✓ | ✓ | ✓ | ✓ |
| Bifid uvula/cleft palate | ✓ | ✓ | ✓ | ✓ |
| Exotropia | ✓ | ✓ | ✓ | ✓ |
| Craniosynostosis | ✓ | ✓ | × | × |
| Cervical spine instability | ✓ | ✓ | × | ✓ |
| Retrognathia surgery | ✓ | ✓ | ✓ | ✓ |
| Scoliosis/spondylolisthesis | ✓ | ✓ | ✓ | ✓ |
| Clubfoot | ✓ | ✓ | ✓ | ✓ |
| Osteoarthritis | ✓ | ✓ | × | ✓ |
| Dural ectasia | ✓ | ✓ | ✓ | ? |
| Pneumothorax | ✓ | ✓ | ✓ | × |
| Hernia | ✓ | ✓ | ✓ | ✓ |
| Dissection at young age | ✓ | ✓ | ✓ | ? |
| Disease beyond root | ✓ | ✓ | ✓ | ✓ |
| Cerebral hemorrhage | ✓ | ✓ | ✓ | ✓ |
| Arterial tortuosity | ✓ | ✓ | ✓ | × |
| Autoimmune findings | ✓ | ✓ | ✓ | ✓ |
A check mark indicates presence of the clinical feature, an X indicates absence of the clinical feature, and a question mark indicates presence of a clinical feature is unknown.
We subsequently investigated the effect of the p.Asp263His mutation on aortic wall architecture and TGF-β signaling. Microscopic examination of the dissected aortic wall, obtained at the time of surgery (3-III:1), showed elastic fiber fragmentation with higher collagen and proteoglycan deposition (Figures 5A to 5C). These histopathological findings are highly reminiscent of both MFS and LDS (8). Retrieved pathology reports from 2 patients (1-II:4 and II:5) carrying the p.Glu216_Lys251del mutation (family 1) also described extensive elastic fiber fragmentation with “pseudo cyst formation” in the medial layer of the dissected aorta and “aortic medial degeneration.” In families 9 and 10, only mild elastic fiber fragmentation was observed.
Figure 5.

Cardiovascular Pathology and Immunohistochemical Analysis of TGFB Family Proteins in a Human Subject With TGFB3 Mutation (3-III:1; p.Asp263His)
(A) Masson trichrome staining shows increased deposition of collagen (dark blue) and loss of smooth muscle fibers (red) in the media. (B) Elastin stain (Elastica van Gieson) shows loss of elastin fibers (black). (C) Hematoxylin-eosin staining shows deposition of proteoglycan (light blue) in the media. (A–C) Scale bar indicates 2 mm. (D–F) Cross sections of the media of the aortic wall of patient 3-III:1 and a matched control. Red staining corresponds to pSmad2 (D); pERK (E); and TGFB1 (F). Scale bars indicate 50 μm (D–E), 20 μm (F). Blue staining shows cell nuclei (DAPI), colocalization is purple. Red staining not colocalized with DAPI is nonspecific.
To investigate TGF-β signaling in the aortic wall of a patient carrying a TGFB3 mutation (p.Asp263His), we performed immunohistochemical analysis of aortic tissue (Figures 5D to 5F). Very similar to what has been detected in TGFB2-deficient aortic walls of humans and mice (8), we observed evidence of paradoxically enhanced TGF-β signaling in the aortic wall of a TGFB3 mutant patient, as shown by increased pSMAD2 (canonical TGF-β signaling), pERK (noncanonical TGF-β signaling), and elevated TGFB1 messenger RNA (Figures 5D to 5F).
Discussion
During mouse embryonic development, Tgfb3 is expressed in several tissues, including cardiovascular, pulmonary, skin, and craniofacial structures. Although Tgfb3 is expressed in overlapping fashion with Tgfb2 in the cardiovascular system, most attention has been paid to its role in palatogenesis, as Tgfb3 knockout mice die at birth because of cleft palate 38, 39. No major cardiac developmental defects have been reported in Tgfb3-deficient mice 38, 40, 41. Although minor abnormalities at the aortic arch level, as well as in position and curvature of the aortic arches and myocardial architecture, were described in the Tgfb3 knockout mice, no data are available on the aortic sizes of conditional knockout or haploinsufficient animals 39, 40. Of interest, the presence of aortic aneurysms and ruptures recapitulating the human phenotype were previously overlooked upon the initial phenotypic description of Tgfb2 haploinsufficient and Smad3 knockout mouse models 8, 30.
Because we observed 3 truncating mutations and an in-frame splice site mutation, we hypothesize that the TGFB3 mutations lead to loss of function (LOF) of TGFB3. In addition, 2 missense mutations, located in the LAP domain, alter critical residues that are relevant for TGFB3 activation by integrins and TGFB3 processing 35, 42. Mice carrying a missense mutation affecting the RGD integrin-binding motif of Tgfb1 recapitulate the phenotype of Tgfb1 knockout mice (43), suggesting that the TGFB3 mutation disrupting the RGD (p.Arg263His) might also lead to LOF. Similarly, molecular analyses and predictions based on the TGFB3 dimer model confirm that most TGFB3 mutations reported here cause LOF. Although it was previously hypothesized that patients with LOF mutations in TGFB3 lack cardiovascular phenotypes (16), we clearly demonstrate that TGFB3 LOF mutations associate with aortic and other arterial aneurysms/dissections and mitral valve disease, and recognize an extremely variable cardiovascular phenotype in the TGFB3 cohort described here. The relatively young age of previously reported patients with TGFB3 mutations (8 [16] and 10.5 years of age [17]) might explain the lack of obvious cardiovascular disease. On the basis of expression studies of the mutant TGFB3 protein in a Xenopus model, Rienhoff et al. (16) hypothesized that the mutated, inactivated allele (p.Cys409Tyr) leads to a nonfunctional protein, decreasing both canonical and noncanonical TGF-β signaling. By contrast, our experiments on human aortic tissue reveal a signature of increased TGF-β signaling. These findings confirm our prior experience that mutational hits in the TGFBR1/2 receptors, the SMAD3 signal transducer, or the TGFB2 ligand lead to a paradoxical increase in TGF-β signaling, as evidenced here by increased immunohistochemical signals for pSMAD2, pERK, and TGFB1 5, 7, 8. Shifts in balances between canonical (pSMAD2) and noncanonical (pERK) cascades, classic, and alternative (BMP-driven) TGF-β superfamily cascades, as well as shifts in ligand expression (TGFB1 vs. TGFB2 or TGFB3) seem likely to be important contributing factors 44, 45.
TGFB3 mutations also appear to have opposing effects on height, as 1 patient in this study (3-III:1, p.Asp263His) has short stature and received growth hormone therapy during puberty, and the patient reported by Rienhoff et al. (16) (p.Cys409Tyr) presented with short stature (5th percentile), whereas others (several patients in this study and the patient reported by Matyas et al. [17]) presented with tall stature. TGFB3 mutations affecting residue Arg300 are associated with cleft palate and/or bifid uvula in our patients (Online Tables 3, 4, and 6) and in the patient reported by Matyas et al. (17). Our study confirms the association of TGFB3 mutation with overt cleft palate in humans and endorses its important role in palatogenesis.
Although our experience is limited to 43 patients in 11 families, our findings warrant comprehensive cardiovascular imaging of the patients. Thus far, no strong evidence has emerged for early aortic dissection in TGFB3 mutant patients, but as the phenotypical spectrum associated with TGFBR1/2, SMAD3, and TGFB2 has now been demonstrated to be extremely wide, we cannot rule out the occurrence of early catastrophic events. We recommend yearly echocardiographic evaluation of the aortic root in all mutation carriers, complemented with at least 1 baseline imaging of the complete aorta and side branches. Frequency of follow-up should be guided by initial findings, family history, and experience still to be gained. Depending on family history and future knowledge, additional imaging of the brain vessels might be indicated. Furthermore, the true incidence and full spectrum of autoimmune manifestations in TGFB3 mutation carriers should be determined in follow-up studies.
Study limitations
Not all clinical features are acquired in all patients. Further studies are needed to fully characterize the phenotypical spectrum we identified here. The predicted effects of the mutations in the homology model of TGFB3 are theoretical and should be complemented with additional protein studies, and the immunohistochemistry studies are hampered by limited availability of patients' aortic wall tissues.
Conclusions
We demonstrate that mutations in the TGFB3 ligand are responsible for a syndromic form of aortic aneurysmal disease. Consistent with our previous findings in TGFBR1/2, SMAD3, and TGFB2 mutation carriers, our study also provides evidence for a paradoxical increase in TGF-β signaling in the aorta. The clinical histories of the patients in our cohort warrant lifelong and widespread cardiovascular surveillance in patients with TGFB3 mutations (Central Illustration). Further research explaining the wide clinical variability is strongly indicated.
Central Illustration.
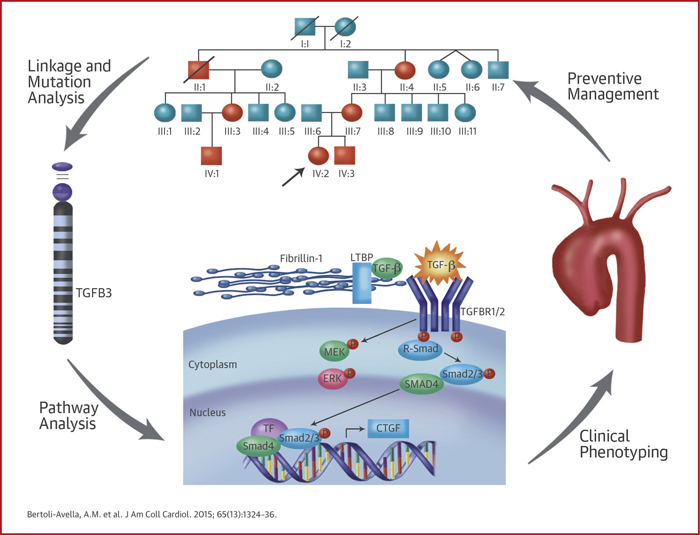
The Pathway From Patient to Gene and Back
The figure summarizes how initial identification of patients and families, followed by linkage and mutation analysis led to the discovery of TGFB3 mutations. Further exploration of the TGF-β pathway allowed a better phenotypical delineation and characterization that will have implications in personalized clinical management.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Mutations in genes encoding components of the TGF-β signaling pathway can cause aortic and arterial aneurysm and dissection.
COMPETENCY IN PATIENT CARE 1: Screening for TGFB3 mutations should be added to the expanding array of genetic testing for patients with unexplained aortic aneurysmal disease or arterial dissection.
COMPETENCY IN PATIENT CARE 2: Vascular imaging should be extended beyond the aortic root in patients with genetic mutations affecting TGF-β signaling because aneurysmal disease may involve more distal portions of the aorta and its arterial branches.
TRANSLATIONAL OUTLOOK: Further studies are needed to assess the safety and efficacy of such treatments as angiotensin receptor blocking drugs, which inhibit TGF-β activity, for prevention of aortic and arterial aneurysm expansion and dissection in patients with mutations involving TGF-β signaling pathways.
Acknowledgments
The authors are grateful to the families and patients that participated in this study. The authors acknowledge Christoph Hermans (Department of Pathology, Antwerp University Hospital) for technical support, Josephina Meester (Center for Medical Genetics, University of Antwerp) and Tom de Vries Lentsch (Erasmus University Medical Center) for helping with the artwork, Akiko Yoshida and Razia Sultana (Departments of Bioscience and Genetics, National Cerebral and Cardiovascular Center, Suita, Osaka, Japan) for genetic analysis, and Tatsuya Oda, Hiroshi Tanaka and Hiroaki Sasaki (Department of Cardiovascular Surgery, National Cerebral and Cardiovascular Center, Suita, Osaka, Japan) for patient management.
Footnotes
This research was supported by funding from the University of Antwerp (Lanceringsproject), the Fund for Scientific Research, Flanders (FWO, Belgium) [G.0221.12], The Dutch Heart Foundation, the Fondation Leducq, the Howard Hughes Medical Institute, the William S. Smilow Center for Marfan Syndrome Research, the Marfan Foundation and the National Institutes of Health (R01-AR41135), the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the Ministry of Health, Labour and Welfare of Japan. Dr. Loeys is senior clinical investigator of the Fund for Scientific Research, Flanders (FWO, Belgium); and holds a starting grant from the European Research Council (ERC). Ms. Gillis holds a grant from the Special Research Funding of the University of Antwerp (BOF-UA). Dr. Lammens has collaborated in studies funded by AstraZeneca, BioMérieux-Novartis, ArgenX, and Labcorps; and has received educational grants from Biocartis and Pfizer. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. Dr. Bertoli-Avella, Ms. Gillis, and Dr. H. Morisaki are joint first authors. Drs. T. Morisaki, Wessels, and Loeys are joint senior authors.
Appendix
For supplemental tables and figures, and an expanded methods section, please see the online version of this article.
Contributor Information
Aida M. Bertoli-Avella, Email: a.bertoliavella@erasmusmc.nl.
Bart L. Loeys, Email: Bart.Loeys@uantwerp.be.
Appendix
References
- 1.Massague J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neptune E.R., Frischmeyer P.A., Arking D.E., et al. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 3.Habashi J.P., Judge D.P., Holm T.M., et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dietz H.C., Cutting G.R., Pyeritz R.E., et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 5.Loeys B.L., Chen J., Neptune E.R., et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 6.Mizuguchi T., Collod-Beroud G., Akiyama T., et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van de Laar I.M., Oldenburg R.A., Pals G., et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 8.Lindsay M.E., Schepers D., Bolar N.A., et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boileau C., Guo D.C., Hanna N., et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doyle A.J., Doyle J.J., Bessling S.L., et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. 2012;44:1249–1254. doi: 10.1038/ng.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loeys B.L., Schwarze U., Holm T., et al. Aneurysm syndromes caused by mutations in the TGF-β receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 12.van de Laar I.M., van der Linde D., Oei E.H., et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet. 2012;49:47–57. doi: 10.1136/jmedgenet-2011-100382. [DOI] [PubMed] [Google Scholar]
- 13.van der Linde D., van de Laar I.M., Bertoli-Avella A.M., et al. Aggressive cardiovascular phenotype of aneurysms-osteoarthritis syndrome caused by pathogenic SMAD3 variants. J Am Coll Cardiol. 2012;60:397–403. doi: 10.1016/j.jacc.2011.12.052. [DOI] [PubMed] [Google Scholar]
- 14.Shprintzen R.J., Goldberg R.B. A recurrent pattern syndrome of craniosynostosis associated with arachnodactyly and abdominal hernias. J Craniofac Genet Dev Biol. 1982;2:65–74. [PubMed] [Google Scholar]
- 15.Rienhoff H.Y., Jr. Response to “De novo mutation of the TGFB3 latency-associated peptide domain in a patient with overgrowth and Loeys-Dietz syndrome features”. Am J Med Genet A. 2014;164A:2144–2145. doi: 10.1002/ajmg.a.36603. [DOI] [PubMed] [Google Scholar]
- 16.Rienhoff H.Y., Jr., Yeo C.Y., Morissette R., et al. A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with Marfan and Loeys-Dietz syndrome. Am J Med Genet A. 2013;161A:2040–2046. doi: 10.1002/ajmg.a.36056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matyas G., Naef P., Tollens M., et al. De novo mutation of the latency-associated peptide domain of TGFB3 in a patient with overgrowth and Loeys-Dietz syndrome features. Am J Med Genet A. 2014;164A:2141–2143. doi: 10.1002/ajmg.a.36593. [DOI] [PubMed] [Google Scholar]
- 18.Campens L., Demulier L., De Groote K., et al. Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories. Am J Cardiol. 2014;114:914–920. doi: 10.1016/j.amjcard.2014.06.024. [DOI] [PubMed] [Google Scholar]
- 19.Rogers I.S., Massaro J.M., Truong Q.A., et al. Distribution, determinants, and normal reference values of thoracic and abdominal aortic diameters by computed tomography (from the Framingham Heart Study) Am J Cardiol. 2013;111:1510–1516. doi: 10.1016/j.amjcard.2013.01.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffmann K., Lindner T.H. easyLINKAGE-Plus—automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 21.van de Laar I., Wessels M., Frohn-Mulder I., et al. First locus for primary pulmonary vein stenosis maps to chromosome 2q. Eur Heart J. 2009;30:2485–2492. doi: 10.1093/eurheartj/ehp271. [DOI] [PubMed] [Google Scholar]
- 22.Thiele H., Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 23.den Dunnen J.T., Antonarakis S.T. Mutation nomenclature extensions and suggestions to describe complex mutations. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 24.Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, Washington. Available at: http://evs.gs.washington.edu/EVS/. Accessed January 27, 2015.
- 25.The 1000 Genomes Project Consortium. Abecasis G.R., Auton A., Brooks L.D., et al. An integrated map of human genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swertz M.A., Dijkstra M., Adamusiak T., et al. The MOLGENIS toolkit: rapid prototyping of biosoftware at the touch of a button. BMC Bioinformatics. 2010;11(Suppl 12):S12. doi: 10.1186/1471-2105-11-S12-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reese M.G., Eeckman F.H., Kulp D., et al. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 28.Brunak S., Engelbrecht J., Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- 29.Shi M., Zhu J., Wang R., et al. Latent TGF-β structure and activation. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krieger E., Vriend G. YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics. 2014;30:2981–2982. doi: 10.1093/bioinformatics/btu426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baschong W., Suetterlin R., Laeng R.H. Control of autofluorescence of archival formaldehyde-fixed, paraffin-embedded tissue in confocal laser scanning microscopy (CLSM) J Histochem Cytochem. 2001;49:1565–1572. doi: 10.1177/002215540104901210. [DOI] [PubMed] [Google Scholar]
- 32.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwarz J.M., Cooper D.N., Schuelke M., et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 34.Munger J.S., Harpel J.G., Giancotti F.G., et al. Interactions between growth factors and integrins: latent forms of transforming growth factor-β are ligands for the integrin αvβ1. Mol Biol Cell. 1998;9:2627–2638. doi: 10.1091/mbc.9.9.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Annes J.P., Rifkin D.B., Munger J.S. The integrin alphaVbeta6 binds and activates latent TGFbeta3. FEBS Lett. 2002;511:65–68. doi: 10.1016/s0014-5793(01)03280-x. [DOI] [PubMed] [Google Scholar]
- 36.Ludbrook S.B., Barry S.T., Delves C.J., et al. The integrin αvβ3 is a receptor for the latency-associated peptides of transforming growth factors β1 and β3. Biochem J. 2003;369:311–318. doi: 10.1042/BJ20020809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Constam D.B. Regulation of TGFβ and related signals by precursor processing. Semin Cell Dev Biol. 2014;32C:85–97. doi: 10.1016/j.semcdb.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 38.Kaartinen V., Voncken J.W., Shuler C., et al. Abnormal lung development and cleft palate in mice lacking TGF-β3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 39.Azhar M., Schultz Jel J., Grupp I., et al. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391–407. doi: 10.1016/s1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doetschman T., Georgieva T., Li H., et al. Generation of mice with a conditional allele for the transforming growth factor beta3 gene. Genesis. 2012;50:59–66. doi: 10.1002/dvg.20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azhar M., Runyan R.B., Gard C., et al. Ligand-specific function of transforming growth factor beta in epithelial-mesenchymal transition in heart development. Dev Dyn. 2009;238:431–442. doi: 10.1002/dvdy.21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Worthington J.J., Klementowicz J.E., Travis M.A. TGFβ: a sleeping giant awoken by integrins. Trends Biochem Sci. 2011;36:47–54. doi: 10.1016/j.tibs.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 43.Yang Z., Mu Z., Dabovic B., et al. Absence of integrin-mediated TGFβ1 activation in vivo recapitulates the phenotype of TGFβ1-null mice. J Cell Biol. 2007;176:787–793. doi: 10.1083/jcb.200611044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lindsay M.E., Dietz H.C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308–316. doi: 10.1038/nature10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallo E.M., Loch D.C., Habashi J.P., et al. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest. 2014;124:448–460. doi: 10.1172/JCI69666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.