

Abstract
Cell adhesion and spreading are regulated by complex interactions involving the cytoskeleton and extracellular matrix proteins. We examined the interaction of the intermediate filament protein vimentin with the actin cross-linking protein filamin A in regulation of spreading in HEK-293 and 3T3 cells. Filamin A and vimentin-expressing cells were well spread on collagen and exhibited numerous cell extensions enriched with filamin A and vimentin. By contrast, cells treated with small interfering RNA (siRNA) to knock down filamin A or vimentin were poorly spread; both of these cell populations exhibited >50% reductions of cell adhesion, cell surface β1 integrin expression, and β1 integrin activation. Knockdown of filamin A reduced vimentin phosphorylation and blocked recruitment of vimentin to cell extensions, whereas knockdown of filamin and/or vimentin inhibited the formation of cell extensions. Reduced vimentin phosphorylation, cell spreading, and β1 integrin surface expression, and activation were phenocopied in cells treated with the protein kinase C inhibitor bisindolylmaleimide; cell spreading was also reduced by siRNA knockdown of protein kinase C-ε. By immunoprecipitation of cell lysates and by pull-down assays using purified proteins, we found an association between filamin A and vimentin. Filamin A also associated with protein kinase C-ε, which was enriched in cell extensions. These data indicate that filamin A associates with vimentin and to protein kinase C-ε, thereby enabling vimentin phosphorylation, which is important for β1 integrin activation and cell spreading on collagen.
Keywords: actin, cell guidance, human embryonic kidney cells
the adhesion and spreading of fibroblasts are critical for development, tissue remodelling, and wound healing. Cell spreading is characterized by the formation of cell membrane protrusions, which include filopodia and lamellipodia (1, 29, 37, 44). The formation of protrusions in cells spreading on extracellular matrix proteins like collagen and fibronectin is governed by complex cell-matrix signaling pathways involving β1 integrins, small GTPases, and actin-binding proteins, which mediate remodelling of the actin cytoskeleton (47, 59, 79). Whereas the actin cytoskeleton is known to be crucially involved in cell adhesion and spreading (63, 72), the intermediate filament cytoskeleton, and in particular vimentin, may also play an important role in adhesion and spreading (20, 23, 30, 31, 70). From the colocalization of the actin and vimentin cytoskeletons at the leading edge of spreading cells (16), the two systems may be functionally coordinated during adhesion and spreading. However, the mechanics by which the two cytoskeletal systems may interact to regulate cell spreading are not defined.
Several proteins bind both actin filaments and intermediate filaments and may therefore functionally integrate the two cytoskeletal systems. Candidate proteins include plectin (62, 68), BPAG1 (76), fimbrin (16), and filamin A (8). Filamin A is an actin cross-linking protein that promotes gelation, membrane stability, adhesion, and motility (7, 22, 25, 58, 66). Filamin A is also well-positioned to regulate cell-matrix interactions because of its actin-binding and β1 integrin-regulating properties (48, 58, 66). Filamin A is enriched in focal adhesions (9, 17, 25) and impacts cell adhesion by controlling the cell surface expression (51) and activation (41) of β1 integrins.
Vimentin shares several functional features in common with filamin A, including regulation of cell adhesion and motility (20, 49, 53, 70, 74), enrichment in β1 integrin-containing focal adhesions (43), and mechanical stabilization of cells (20). Furthermore, the expression of filamin A and vimentin is increased simultaneously during shear stress in osteoblasts (34), suggesting that both of these proteins contribute to mechanoprotection. Like filamin A, several functions of vimentin are regulated by phosphorylation (15, 21, 33, 64). Specifically, the phosphorylation of vimentin by protein kinase C (PKC) is required for the recycling of β1 integrins to the plasma membrane and for cell migration (23, 31). Notably, filamin A binds PKC (22, 25, 69), which mediates vimentin phosphorylation (2, 33, 55, 77).
We demonstrate here that filamin A and vimentin coregulate the activation and cell surface expression of β1 integrin during cell spreading in fibroblasts. PKC-mediated phosphorylation of vimentin is involved in β1 integrin activation and cell spreading, and this phosphorylation is dependent on filamin A, which binds directly to vimentin.
MATERIALS AND METHODS
Reagents.
Antibodies against human filamin A (PM6/317), vimentin (3B4 and polyclonal), vinculin (FB11), cortactin (4F11), paxillin (349), PKC (M110), GAPDH (6C5), β1 integrin (12G10), Rac (polyclonal), and Cdc42 (polyclonal) were purchased from Chemicon (Temecula, CA). Antibodies against phospho-vimentin (serines-6, -33, and -50) were obtained from Stressgen (Victoria, BC, Canada). Antibodies against mouse filamin A (PM6/317), collagen (polyclonal), PKC-ε (polyclonal), phospho-PKC-ε (serine-729), and phospho-vimentin (serine 38) were purchased from Abcam (Cambridge, MA). Alexa 488-phalloidin was obtained from Molecular Probes (Eugene, OR). 4,6-diamidino-2-phenylindole (DAPI) was purchased from Roche (Mannheim, Germany). Bovine dermal collagen monomers (pepsin-digested, 95% type I collagen) were purchased from Inamed (Fremont, CA). Purified recombinant GST-tagged human vimentin protein (expressed in Eschericia coli cells) was obtained from Thermo Fisher Scientific (Fremont, CA). Purified glutathione-S transferase (GST) and FLAG proteins were purchased from Abcam (Cambridge, MA) and Sigma (Oakville, ON, Canada), respectively. Glutathione beads were obtained from GE Healthcare (Piscataway, NJ). Anti-FLAG-coated beads were purchased from Sigma (Oakville, ON, Canada). Magnetite beads were obtained from Polysciences (Warrington, PA). p21-activated kinase (PAK) binding domain (PAK-PBD) beads were obtained from Cytoskeleton (Denver, CO). Bisindolylmaleimide (BIM), calphostin C, and bryostatin were purchased from Calbiochem (San Diego, CA).
Cell culture.
Two different types of readily transfectable fibroblasts were studied to facilitate determination of the impact of cytoskeletal proteins on cell spreading. Human kidney (HEK-293) cells were cultured in DMEM (GIBCO) containing 10% fetal bovine serum and an antibiotic solution (0.17% wt/vol penicillin V, 0.01 μg/ml amphotericin B, and 0.1% gentamycin sulfate). Mouse 3T3 cells were cultured in DMEM containing 10% calf serum and antibiotics. Cell culture dishes were precoated with fibrillar bovine type I collagen.
Immunoprecipitation.
HEK cells were allowed to spread on smooth collagen-coated surfaces for 30 min and lysed with RIPA buffer. After being precleared with normal mouse serum, filamin A and associated proteins were immunoprecipitated using antibody to filamin A bound to agarose beads (ImmunoPure G, Pierce, Rockford, IL). All immunoprecipitation experiments employed controls using normal mouse serum. Immunoprecipitated proteins were separated by SDS-PAGE and immunoblotted with antibodies against vimentin, Rac, Cdc42, phospho-vimentin, PKC, or phospho-PKC-ε.
Isotope-coded affinity tag analysis.
Proteins associated with filamin A during cell spreading were identified with isotope-coded affinity tag (ICAT) analysis (Applied Biosystems; Foster City, CA). Filamin A immunoprecipitates (100 μg) were obtained from suspended and spreading human embryonic kidney (HEK) cells. The immunoprecipitates were denatured, reduced, labeled with either light or heavy (+9 Da) ICAT biotin-coupled reagents, combined, digested with trypsin, fractionated by cationic exchange, purified with avidin columns, cleaved, and analyzed by HPLC and tandem mass spectrometry. With the use of discriminant analysis (with positive protein identification set at >99%) and protein scores of 2.0 or greater, six different novel proteins from the two analyses were identified as being differentially expressed under the two experimentally different conditions.
Small interfering RNA knockdown.
HEK-293 cells were treated by RNA silencing of filamin A. Briefly, a filamin A-specific short-hairpin RNA (shRNA) was constructed from two inverted 21-base sequences (5′-GGGCTGACAACAGTGTGGTGC-3′) of the filamin-A cDNA and incorporated into a plasmid with the U6 promoter for shRNA expression and the pPUR vector for puromycin resistance (50). For filamin A-knockdown cells, 1 μg/ml of puromycin dihydrochloride (Sigma) was added to the culture medium. Mouse 3T3 cells stably transfected with a filamin A shRNA were from Dr. David Calderwood.
For silencing of vimentin in HEK-293 cells, three different small interfering RNAs (siRNAs, Ambion, Austin, TX) were combined and cotransfected into HEK-293 cells. The three human vimentin siRNA sequences were as follows: (5′-GGAGAGCAGGAUUUCUCUGtt-3′), (5′-GGCGAGGAGAGCAGGAUUUtt-3′), and (5-GGGAA ACUAAUCUGGAUUCtt-3′). For silencing of vimentin in mouse 3T3 cells, the siRNA sequences were used: (5′-GAGUCAAACGAGUACCGGAtt-3′), (5′-GGUUGACACCCACUCAAAAtt-3′), and (5′-GCCGAGGAAUGGUACAAGUtt-3′). In some experiments, the vimentin siRNAs were introduced into HEK-293 and 3T3 cells stably transfected with the filamin shRNA to produce cells deficient in both filamin and vimentin. For silencing of PKC-ε, three siRNAs bearing the following sequences were cotransfected into HEK-293 cells: (5′-ACCACGCAUUAAAACCAAAtt-3′), (5′-GGAAAGCAGGGAUACCAGUtt3-′), and (5′-GAGUGUAUGUGAUCAUCGAtt-3′).
Preparation of purified FLAG-tagged filamin A, dot-blot, and pull-down assays.
FLAG-tagged filamin A was expressed using a baculovirus expression system (Invitrogen) in Sf9 insect cells and purified as previously described (52). To examine filamin-vimentin interactions, purified vimentin was spotted onto a nitrocellulose membrane, dried, blocked with 5% milk, and incubated for 2 h with a solution of purified FLAG-tagged filamin protein (0.05 mg/ml). As a control, purified fibronectin was also spotted onto the membrane. The membrane was washed four times and immunoblotted with antibodies against FLAG and filamin. For reciprocal dot-blots, purified filamin was spotted onto nitrocellulose membranes, which were subsequently blocked with milk and incubated with a solution of purified vimentin (0.1 mg/ml). After washing was completed, membranes were immunoblotted for vimentin.
In vitro pull-down assays were used to assess direct filamin-vimentin binding. Briefly, purified GST-tagged vimentin was incubated with glutathione beads (50% slurry) in the presence or absence of purified FLAG-tagged filamin A, in a binding/washing buffer (50 mM Tris·HCl, 150 mM NaCl, 0.1% Triton X-100, 0.1 mM β-mercaptoethanol, 0.1 mM EGTA, pH 7.4; 400 μl) containing 1% BSA for 1 h at 4°C. Glutathione beads incubated with purified GST protein served as a negative control. The beads were sedimented and washed four times in ice-cold binding/washing buffer (600 μl). Bead-associated proteins were solubilized in SDS sample buffer, separated by SDS-PAGE, and immunoblotted with antibodies against filamin and vimentin. The reciprocal procedure was accomplished by incubating purified FLAG-tagged filamin A with anti-FLAG M2 agarose (50% slurry) in the presence or absence of vimentin. Anti-FLAG-coated beads incubated with purified FLAG protein served as a negative control. Bead-associated proteins were eluted and immunoblotted as outlined above.
Immunoblotting of plasma membrane-associated proteins.
We studied the relative abundance of vinculin and paxillin at the free and leading edges of cells during normal spreading. Cells were incubated on collagen-coated substrates to allow attachment and initial spreading. Magnetite beads (modal diameter = 5 μm) were coated with fibrillar collagen (14, 26, 42, 45). BSA-coated beads were prepared by incubating beads with 1 ml of 3% (vol/vol) solution of BSA. Beads were added to cultures (6 beads/cell) to engage β1 integrins that were not collagen bound, including those β1 integrins involved in spreading. Cells were detached from the substrates by incubation with cytoskeletal stabilizing buffer (26), and the beads were isolated magnetically. Proteins from beads were eluted and analyzed by Western blotting (78) using antibodies to vinculin and paxillin.
Actin monomer incorporation.
The incorporation of actin monomers was assayed as previously described (11). In permeabilized cells incubated with rhodamine actin monomers (primarily β-actin), increases of rhodamine fluorescence due to incorporation into nascent actin filaments were measured. Cells were permeabilized with octyl glucoside. Freshly sedimented rhodamine actin monomer was added to the samples, followed by paraformaldehyde fixation. Rhodamine fluorescence in single cells was quantified by microscope fluorimetry.
Rac and Cdc42 activation assay.
Lysates of spreading control and vimentin-deficient HEK-293 cells were incubated with PAK-PBD beads to precipitate the active, GTP-bound form of Rac and Cdc42. Bead-associated proteins were subsequently eluted and immunoblotted for Rac. Western blots were then stripped and reprobed for Cdc42.
Immunohistochemistry.
HEK cells were allowed to spread on collagen for 15–30 min, washed with PBS, fixed for 30 min in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked for 1 h in 0.1% BSA. Cells were incubated with primary antibody and FITC-conjugated secondary antibody for 1 h each and counterstained with DAPI. Cell spreading was quantified on the basis of the number of cell extensions resembling filopodia and/or lamellipodia. A minimum of 50 cells were included in counts of cell extensions. Since vinculin, paxillin, and cortactin are found in filopodia and lamellipodia (54, 57), cells were also immunostained for vinculin, paxillin, and cortactin to validate the observed cell extensions as true filopodia/lamellipodia.
Flow cytometry.
To quantify β1 integrins present at the cell surface during the earliest phase of cell spreading, HEK cells were allowed to spread on collagen for 15 min before being harvested in ice-cold versene (GIBCO) and light fixation in ice-cold 1% paraformaldehyde. The suspended cells were then immunostained with rhodamine-conjuated anti-β1 integrin (4B4) antibody (Beckman-Coulter, Miami, FL) and analyzed by flow cytometry (Beckman-Coulter Altra). In some experiments, active, ligand-bound β1 integrin was quantified by immunostaining cells with 12G10 antibody before flow cytometric analysis.
Statistical analysis.
Analysis of variance and Bonferroni post-hoc multiple comparison tests were used to assess the effect of cell type/treatment on cell spreading. Data were obtained from at least three independent experiments. Statistical significance was set at P < 0.05.
RESULTS
Filamin A associates with vimentin during cell spreading.
Filamin A is an important determinant of early events in cell spreading (41). Isotope-coded affinity tag analysis was performed on filamin A immunoprecipitates to identify novel filamin A-binding proteins that may be involved in filamin A-dependent spreading processes. From the 21 peptides that were identified with >99% certainty, six different proteins were predicted to be differentially expressed in suspended (labeled with heavy ICAT reagent) and spreading cells (labeled with light ICAT reagent), including vimentin (Table 1). From the previously identified roles of filamin A and vimentin in cell adhesion and migration (6, 20, 53, 70, 71), we focused subsequent investigations on the roles of filamin A and vimentin in early phases of cell spreading.
Table 1.
Filamin A-associated proteins identified by ICAT
| Confidence | Protein | Heavy:Light Ratio |
|---|---|---|
| >99% | Chain L, monoclonal antibody | 1.160 |
| >99% | Anti-colorectal carcinoma heavy chain | n/d |
| >99% | Filamin-A | 0.5464 |
| >99% | 60S ribosomal protein L12 | 0.6216 |
| >99% | Unnamed protein | n/d |
| >99% | Immunoglobulin heavy chain | 1.5448 |
| >99% | Vimentin | 0.3905 |
| >99% | Ribosomal protein L10a | n/d |
| >99% | hCG1766793 | 1.4345 |
| >99% | Chromodomain helicase DNA binding protein 9 | n/d |
| >99% | Ketohexokinase variant a | n/d |
| 95%–99% | Telengiectasia mutated protein isoform 2 | n/d |
Differential expression of filamin A-associated proteins as identified by isotope-coded affinity tag (ICAT) peptide labeling. Six different proteins were predicted to be differentially expressed in suspended (peptides labeled with heavy ICAT reagent) and spreading cells (peptides labeled with light ICAT reagent), including vimentin (n/d: heavy:light ratio not determined).
Filamin A and vimentin coregulate cell spreading.
The extension of filopodia and/or lamellipodia by cells is an important indicator of early cell spreading (47, 59, 79). We determined whether filamin A and/or vimentin were present in the earliest formed extensions of spreading cells. Immunofluorescence showed that in control HEK cells spreading on collagen, filamin A and vimentin colocalized at well-defined cell extensions (Fig. 1, A and B). Since genuine filopodial and lamellipodial extensions contain cortactin, vinculin, and paxillin in addition to actin filaments (5, 54, 57, 73), we confirmed the presence of these proteins in cell extensions by immunofluorescence (Fig. 1C). To assess the individual roles of filamin A and vimentin in cell spreading, we used RNA interference to reduce filamin A and vimentin expression in HEK cells. The efficacy of the filamin A and vimentin knockdown was assessed by immunoblotting (Fig. 1D). For evaluating the combined roles of filamin A and vimentin in cell spreading, we used siRNA for vimentin in cells stably transfected with a filamin A shRNA, thereby creating a double knockdown (Fig. 1D). Since the initial extension of filopodia and lamellipodia generally occurs within 30 min postplating (4, 18, 60), we chose time points between 15 and 120 min to assess differences in cell spreading between control and filamin/vimentin-deficient cells. Knockdown of either filamin A or vimentin expression resulted in a significant (P < 0.05) reduction in the number of cell extensions (Fig. 1, E and F). Concurrent knockdown of filamin A and vimentin did not result in further reductions in the number of cell extensions compared with individual knockdown of filamin A or vimentin (Fig. 1E), suggesting that the role of vimentin in regulating cell spreading is not additive to the role of filamin A. Similar reductions in cell extension numbers were observed upon silencing of filamin A and/or vimentin in mouse 3T3 fibroblasts (Fig. 1, G and H). Filamin A expression was successfully rescued by introducing a filamin cDNA into filamin-knockdown cells (Fig. 2A). Vimentin expression was naturally restored in vimentin-knockdown cells 4 days following transient siRNA transfection (Fig. 2B). Notably, the formation of cell extensions by filamin- and vimentin-deficient HEK-293 cells was effectively rescued following the restoration of filamin or vimentin expression (Fig. 2C).
Fig. 1.
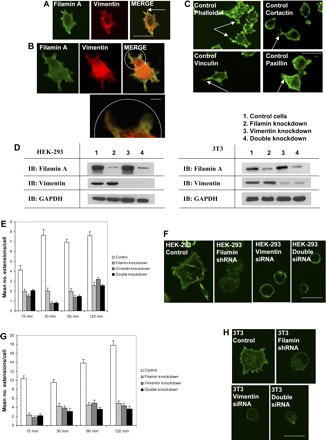
Filamin and vimentin coregulate cell spreading. A: confocal micrographs illustrate filamin A (left) and vimentin (center) recruited to the cell cortex during the earliest phases of cell spreading. Bar = 30 μm. B: high magnification confocal micrograph (bottom right, inset) illustrating the colocalization of filamin A and vimentin at the lamellipodium during cell spreading. Bar = 5 μm. C: confocal micrographs show presence of actin (top left), cortactin (top right), vinculin (bottom left), and paxillin (bottom right) in cell extensions. Cell extensions are evident in spreading cells (arrows). Bar = 30 μm. D: Western blots illustrating filamin A and/or vimentin knockdown by small interfering RNA (siRNA) in human embryonic kidney (HEK)-293 cells (left) and in mouse 3T3 cells (right). E: histogram showing differences in mean number of cell extensions ± SE among control, filamin-knockdown, vimentin-knockdown, and double-knockdown HEK-293 cells spreading on collagen. F: Alexa-488 phalloidin staining of control (far left), filamin-knockdown (left), vimentin-knockdown (right), and double-knockdown (far right) HEK-293 cells spreading on collagen. Bar = 30 μm. G: histogram showing differences in mean number of cell extensions ± SE between control, filamin-knockdown, vimentin-knockdown, and double-knockdown 3T3 cells spreading on collagen. H: Alexa-488 phalloidin staining of control (top left), filamin-knockdown (top right), vimentin-knockdown (bottom left), and double-knockdown (bottom right) 3T3 cells spreading on collagen. Bar = 30 μm.
Fig. 2.
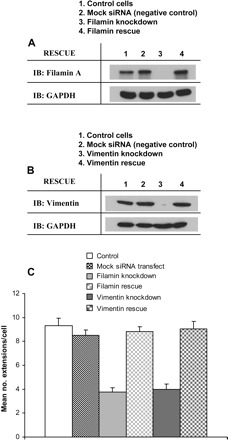
Restoration of filamin and vimentin expression rescues the formation of cell extensions. A: Western blots illustrate filamin A expression in control cells, mock siRNA-transfected cells, filamin knockdown cells, and filamin knockdown cells transfected with a filamin cDNA. B: Western blots illustrate vimentin expression in control cells, mock siRNA-transfected cells, vimentin knockdown cells, and vimentin knockdown cells following the transient siRNA period. C: histogram showing differences in mean number of cell extensions ± SE between control, mock-transfected, filamin-knockdown, filamin-rescue, vimentin-knockdown, and vimentin-rescue HEK-293 cells spreading on collagen.
Filamin A, but not vimentin, affects actin assembly in spreading cells.
We examined the individual and combined effects of filamin A and vimentin on the generation of actin filament free barbed ends in spreading cells. Filamin A regulates the GTPase activity of Rac and Cdc42 (6, 56, 71), which in turn control cell spreading, partly by determining the generation of free barbed ends required for actin filament assembly (67, 75). As expected, the incorporation of rhodamine-labeled actin monomers was significantly (P < 0.05) reduced in filamin A-deficient cells. However, in vimentin knockdown cells there was no effect on the number of free barbed ends (Fig. 3A). Interestingly, incorporation of actin monomers was nearly undetectable after concurrent knockdown of filamin A and vimentin (Fig. 3A). These data suggest that vimentin may contribute to the generation of free barbed ends in the absence of filamin A but that vimentin does not independently regulate actin assembly.
Fig. 3.
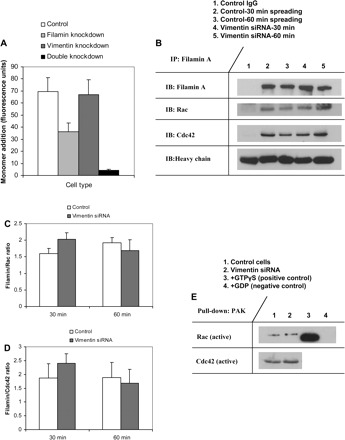
Vimentin does not affect filamin-mediated actin assembly. A: histogram illustrates differences in actin free barbed end formation in control, filamin-knockdown, vimentin-knockdown, and double-knockdown cells. B: coimmunopreciptation and Western blotting show filamin/Rac and filamin/Cdc42 association in spreading cells. Levels of filamin-associated Rac and Cdc42 are unaffected by vimentin knockdown. C: histogram compares ratios of filamin/Rac blot density ± SE between control and vimentin-deficient cells. D: histogram compares ratios of filamin/Cdc42 blot density ± SE between control and vimentin-deficient cells. E: Western blotting shows levels of active, GTP-bound Rac, and Cdc42 in control and vimentin-deficient cells. GTPγS was added to lysate as a positive control (lane 3); GDP was added to lysate as a negative control (lane 4).
Since filamin A has a documented role in binding and regulating the activity of Rac and Cdc42 (6, 56, 66, 71), we examined the possibility that vimentin mediates the association between filamin A and Rac and/or Cdc42. Filamin A was immunoprecipitated from spreading control and vimentin-deficient cells, and the immunoprecipitates were immunoblotted for Rac and Cdc42. The amount of filamin A-bound Rac and Cdc42 did not differ significantly between control and vimentin-deficient cells (Fig. 3, B–D). Moreover, levels of active, GTP-bound Rac, and Cdc42 were similar in control and vimentin-deficient cells (Fig. 3E). Taken together, the data indicate that vimentin regulates cell spreading independently of the filamin A-small GTPase-actin assembly pathway.
Filamin A and vimentin regulate β1 integrin expression and activation.
Since cell spreading is a β1 integrin-dependent process (24, 38, 59) and since filamin A-null cells reportedly express fewer cell surface β1 integrins (51), we examined the possibility that filamin A and vimentin may also coregulate cell surface β1 integrin expression. To assess the individual and collective effects of filamin A and vimentin knockdown on β1 integrin expression and activation, HEK cells were allowed to spread on collagen for 15 min to allow initial adhesion formation (79). Cells were subsequently suspended and incubated with the 4B4 antibody to label cell surface β1 integrins. Active, ligand-bound β1 integrins were labeled with the 12G10 antibody. Flow cytometric analysis showed that cell surface β1 integrin was reduced ∼50% following knockdown of filamin A and/or vimentin (Fig. 4, A and B). This defect was rescued following reexpression of filamin or vimentin, consistent with the cell spreading data (Fig. 4, A and B). Notably, 12G10 staining intensity was reduced fourfold following filamin A and/or vimentin knockdown (Fig. 4, C and D). These data indicate that filamin and vimentin are important determinants of β1 integrin expression but are especially important for regulating β1 integrin activation.
Fig. 4.
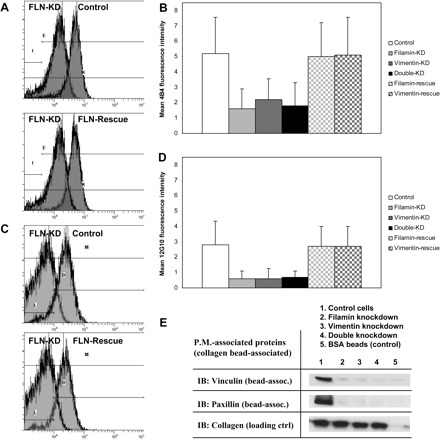
Filamin and vimentin regulate β1 integrin expression and activation. A: flow cytometry indicates decreased cell surface expression of β1 integrins following knockdown of filamin (FLN-KD, top). Cell surface levels of β1 integrin are restored by rescue of filamin expression (bottom). B: histogram shows mean 4B4 fluorescence intensity ± SD in control, filamin-KD, vimentin-KD, double-KD, filamin-rescue, and vimentin-rescue cells. C: flow cytometry indicates decreased β1 integrin activation following knockdown of filamin (top). Levels of β1 integrin activation are restored by rescue of filamin expression (bottom). D: histogram shows mean 12G10 fluorescence intensity ± SD in control, filamin-KD, vimentin-KD, double-KD, filamin-rescue, and vimentin-rescue cells. E: immunoblots (IB) of plasma-membrane (P.M.)-associated proteins show decreased recruitment of vinculin and paxillin to focal adhesions following knockdown of filamin and/or vimentin.
We examined proteins that were associated with nascent cell adhesions. Collagen-coated magnetite beads were incubated with cells, isolated and bead-associated proteins were immunoblotted (25, 26, 78). Bead counts and the abundance of collagen were used as loading controls to adjust for variations of the number of attached beads in different cell preparations. There was more bead-associated vinculin and paxillin in control cells than filamin A- or vimentin-deficient cells (Fig. 4E), indicating that filamin A and vimentin are both required to generate stable adhesions. Collectively, these data indicate that filamin A and vimentin regulate β1 integrin activation, cell adhesion, and early steps of cell spreading, possibly through a common mechanism.
Vimentin phosphorylation is required for β1 integrin expression, β1 integrin activity, and cell spreading.
The phosphorylation of vimentin mediated by PKC-ε is an important regulatory step for β1 integrin trafficking and cell motility (23, 31). We evaluated the role of PKC-mediated phosphorylation of vimentin in early cell spreading. Cells were treated for 90 min with BIM (1 μM), which inhibits PKC-ε (31, 32), and plated on collagen-coated substrates. To verify the specificity of BIM as a PKC inhibitor, experiments were also performed with cells pretreated with calphostin C (100 nM). In BIM- and calphostin C-treated cells undergoing early spreading, there was decreased phosphorylation of vimentin at serines-6, -38, and -50, which are known PKC phosphorylation sites (2, 33, 55, 77) (Fig. 8C). BIM-treated and calphostin C-treated cells also showed a significant (P < 0.05) reduction in the number of cell extensions (Fig. 5, A and C) compared with untreated controls. Notably, BIM- and calphostin C-induced effects on cell spreading were comparable in magnitude to those achieved by siRNA knockdown of vimentin (Fig. 5, A and C). To further validate the use of BIM as an inhibitor of PKC-ε, we compared the phenotype of BIM-treated and calphostin C-treated cells to that of PKC-ε-deficient cells generated by siRNA knockdown (Fig. 5B). PKC-ε-knockdown cells showed reduced numbers of cell extensions comparable to those observed in vimentin-knockdown, BIM-treated, and calphostin C-treated cells (Fig. 5, A and C). Although vimentin is not a direct substrate for PKC-ε (31), its phosphorylation is PKC-ε dependent. Therefore, these data indicate that the phosphorylation of vimentin mediated by PKC-ε may be one of the key regulatory steps involved in early cell adhesion and spreading.
Fig. 8.

Filamin associates with PKC to regulate vimentin phosphorylation. A: coimmunoprecipitation (IP) and Western blotting show close association, in spreading cells, between filamin A, PKC, phospho-PKC-ε, and phospho-vimentin. B, top: confocal micrographs showing colocalization of filamin A and PKC-ε in extensions of spreading cells. Bar = 40 μm. Bottom: confocal micrographs showing, at higher magnification, colocalization of filamin A and PKC-ε in extensions (arrow) of the indicated cell. Bar = 20 μm. C: Western blots of spreading cells illustrate levels of vimentin phosphorylated at serines-6, -38, and -50. Loss of filamin, treatment with BIM or calphostin C, or loss of PKC-ε results in reduced levels of vimentin phosphorylated at serines-6, -38, and -50.
Fig. 5.

Vimentin phosphorylation is required for β1 integrin expression, activation, and cell spreading. A: Alexa-488 phalloidin staining of control (far left), vimentin-knockdown (left), bisindolylmaleimide (BIM)-treated (center), calphostin C-treated (right), and PKC-ε-knockdown cells (far right) spreading on collagen. Bar = 30 μm. B: Western blot illustrates knockdown of PKC-ε by siRNA. C: histogram showing differences in mean number of cell extensions ± SE between control, vimentin-knockdown, BIM-treated, calphostin C-treated, and PKC-ε-knockdown cells spreading on collagen. D: flow cytometry indicates decreased cell surface expression of β1 integrins following BIM treatment (left) or knockdown of PKC-ε (right). E: histogram shows mean 4B4 fluorescence intensity ± SD in control, vimentin-knockdown, BIM-treated, calphostin C-treated, and PKC-ε-knockdown cells. F: flow cytometry indicates decreased β1 integrin activation following BIM treatment (left) or knockdown of PKC-ε (right). G: histogram shows mean % of 12G10 fluorescence intensity ± SD among control, vimentin-KD, BIM-treated, calphostin C-treated, and PKC-ε-KD cells.
PKC-ε-mediated vimentin phosphorylation is a critical determinant of cell surface β1 integrin expression (31). Based on flow cytometric analysis, nonpermeabilized cells undergoing initial spreading showed less active and total cell surface β1 integrin following knockdown of filamin A and/or vimentin, BIM treatment, calphostin C treatment, or PKC-ε knockdown (Fig. 5, D–G). These data suggest that the phosphorylation of vimentin attributable to PKC is an important determinant of cell surface β1 integrin expression and activation.
Filamin A binds vimentin directly in vitro and in spreading cells.
As filamin A can bind PKC (22, 25, 69), we considered that filamin A may mediate vimentin phosphorylation by interacting directly with vimentin. To investigate the nature of the filamin A-vimentin interaction, filamin A was immunoprecipitated from the lysates of spreading HEK-293 cells; the immunoprecipitates were subsequently blotted for vimentin (Fig. 6A). We circumvented interference from the mouse immunoglobulin heavy chain by using a chicken vimentin antibody for immunoblotting filamin A immunoprecipitates. Consistent with the ICAT data, which showed a small increase of vimentin-filamin A association in response to spreading (Table 1), we found a small increase of filamin A-associated vimentin in spreading versus suspended cells by immunoprecipitation. These data suggest that filamin A-vimentin interactions are largely constitutive and are only moderately dependent on cell adhesion.
Fig. 6.
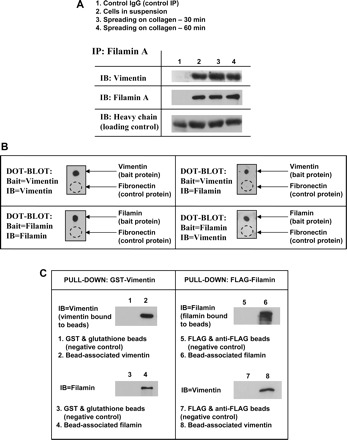
Filamin binds vimentin in spreading cells and in vitro. A: coimmunoprecipitation and Western blotting show close association, in spreading cells, between filamin A and vimentin. B: dot-blot assay illustrating binding between purified FLAG-tagged filamin and GST-tagged vimentin. Fibronectin is used as a negative control. Dashed circle represents location of fibronectin application. C: filamin-vimentin binding is illustrated with pull-down assays using GST-tagged vimentin bound to glutathione beads (left) and FLAG-tagged filamin bound to anti-FLAG-coated beads (right).
By dot-blot analysis we found that purified filamin A and vimentin may interact directly in vitro (Fig. 6B). We explored the possibility of a direct interaction further using purified GST-tagged vimentin bound to glutathione beads and incubated the beads with purified FLAG-tagged filamin A (Fig. 6C). A direct filamin A-vimentin interaction was confirmed by immunoblotting the bead-associated proteins for filamin A. Conversely, purified vimentin associated with purified FLAG-filamin A bound to beads as detected by immunoblotting for vimentin (Fig. 6C). Since the data showed that filamin A binds vimentin directly, we considered that filamin A may regulate the phosphorylation of vimentin by protein kinase C.
Filamin A is a determinant of vimentin phosphorylation and reorganization.
Phosphorylation by PKC is required for the assembly and disassembly of vimentin filaments and its distribution in the cytoplasm (21, 33, 64), processes that are known to affect cell adhesion and motility, possibly by regulating the recycling and trafficking of β1 integrins to the cell membrane (23, 31). By immunofluorescence we found spatial colocalization of vimentin and PKC-ε (Fig. 7A) and that vimentin and β1 integrins colocalize in spreading control cells (Fig. 7, B and C). This colocalization was disrupted after knockdown of filamin A or treatment with BIM to inhibit PKC (Fig. 7, D–F). Moreover, the cytoplasmic distribution of vimentin was markedly different among filamin A-expressing, filamin A-deficient, and BIM-treated cells. Control cells exhibited a well-defined vimentin filament network throughout the cytoplasm, in which vimentin was present in cell extensions (Fig. 1A and Fig. 7A). By contrast, in the cytoplasm of filamin A-deficient or BIM-treated cells, vimentin appeared as a single focal accumulation (Fig. 7, D and E). These data are consistent with the notion that filamin A expression and PKC activity are both required for vimentin reorganization and redistribution.
Fig. 7.
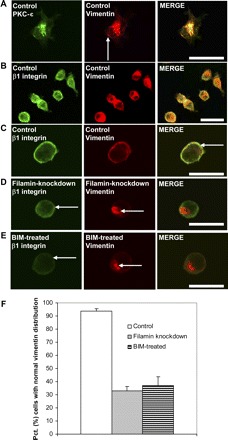
Filamin and PKC regulate vimentin distribution. A: confocal micrographs showing colocalization of PKC-ε and vimentin. Bar = 20 μm. B and C: confocal micrographs showing vimentin recruited to cell cortex during cell spreading, colocalized with β1 integrins (right, arrow). Bar = 40 μm (B), bar = 20 μm (C). D: confocal micrographs show decreased cell surface β1 integrin (left, arrow) and failure of vimentin translocation following knockdown of filamin (center, arrow). Bar = 20 μm. E: confocal micrographs showing decreased cell surface β1 integrin (left, arrow) and failure of vimentin translocation following inhibition of PKC-ε with BIM (center, arrow). Bar = 20 μm. F: histogram illustrates differences in mean percentage ± SE of cells with normal vimentin distribution (% cells with vimentin distributed throughout the cytosol) between control, filamin-knockdown, and BIM-treated cells.
We then directly evaluated a role for filamin A in PKC-mediated phosphorylation of vimentin by examining the association between PKC and filamin A by coimmunoprecipitation (Fig. 8A). Coimmunoprecipitation showed that filamin A associates with the phosphorylated form of vimentin as well as the phosphorylated form of PKC-ε (Fig. 8A), which is notable since the catalytic activity of PKC-ε is regulated by its phosphorylation at serine-729 (10). By coimmunofluorescence, we also found spatial colocalization of filamin A and PKC-ε in cells spreading for 30 min on collagen (Fig. 8B).
We also evaluated the effect of filamin A knockdown on PKC-mediated phosphorylation of vimentin using phospho-specific antibodies to serine-6, serine-33, serine-38, and serine-50, four vimentin residues known to be phosphorylated by PKC (2, 33, 55, 77). Knockdown of filamin A abrogated vimentin phosphorylation at serines-6, -38, and -50 in spreading cells (Fig. 8C), but there was no detectable change of phosphorylation at serine-33 (data not shown). Filamin A-deficient cells showed lower levels of the phosphorylated, catalytically active form of PKC-ε (data not shown). These findings are consistent with the notion that filamin A is pivotal for PKC-mediated phosphorylation of vimentin. Notably, the phosphorylation of vimentin at serines-6, -38, and -50 was restored following pretreatment of filamin-knockdown cells with the PKC activator bryostatin (Fig. 8C). Moreover, the impaired formation of cell extensions and reduced cell surface β1 integrin expression observed in filamin-deficient cells was reversed following bryostatin treatment (Fig. 9, A–F).
Fig. 9.

Filamin and PKC coregulate β1 integrin expression, activation, and cell spreading. A: Alexa-488-phallodin staining of control (left), filamin-knockdown (center), and filamin-knockdown cells treated with the PKC activator bryostatin (rightl). The formation of cell extensions is rescued by bryostatin treatment (arrow). Bar = 30 μm. B: histogram showing differences in mean number of cell extensions ± SE among control, filamin-knockdown, BIM-treated, filamin-rescue, and bryostatin-treated HEK-293 cells spreading on collagen. C: flow cytometry indicates restored levels of cell surface expression of β1 integrins in filamin-knockdown cells treated with the PKC activator bryostatin. D: histogram shows mean 4B4 fluorescence intensity ± SD in control, filamin-knockdown, BIM-treated, filamin-rescue, and filamin-knockdown cells treated with bryostatin. E: flow cytometry indicates restored levels of β1 integrin activation in filamin-knockdown cells treated with the PKC activator bryostatin. F: histogram shows mean 12G10 fluorescence intensity ± SD in control, filamin-knockdown, BIM-treated, filamin-rescue, and filamin-knockdown cells treated with bryostatin.
DISCUSSION
Our major finding is that in spreading fibroblasts, filamin A binds to vimentin and acts as a scaffold for PKC-mediated phosphorylation of vimentin. These functions serve to regulate the activation of β1 integrins and their trafficking to the cell surface. The direct interaction between filamin and vimentin provides a novel mechanism by which the actin and intermediate filament cytoskeletons coregulate cell adhesion and spreading (Fig. 10). Whereas the actin and intermediate filament cytoskeletons have distinct roles in regulating cell adhesion, spreading, and migration (30, 63, 72), the functions of these two cytoskeletal systems may be interdependent (3, 13, 28, 39). Proteins that bind both actin and intermediate filaments, such as plectin (62, 68), BPAG1 (76), or fimbrin (16), may integrate the functions of these cytoskeletal systems. Our data provide a new mechanism in which the actin binding protein filamin A acts as linker between vimentin and actin filaments.
Fig. 10.
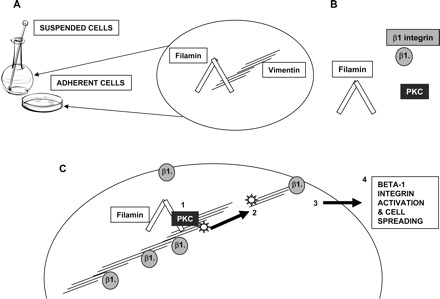
Filamin A regulates β1 integrin expression and activation via its association with vimentin and PKC. A: schematic diagram illustrating association between filamin A and vimentin. Filamin-vimentin binding is constitutive and only moderately adhesion dependent. B: filamin is a potential scaffold for signaling and adhesion molecules such as PKC and β1 integrin. C: schematic diagram of proposed model for filamin-vimentin-PKC interactions. 1) Filamin binds PKC and vimentin, leading to vimentin phosphorylation at serines-6, -38, and -50. 2) Phosphorylation and subsequent reorganization of vimentin allows recycling of β1 integrins to the cell surface. 3) Cell surface β1 integrin activation is upstream of actin polymerization, the formation of cell extensions and cell spreading (step 4).
Filamin A colocalizes with vimentin (8), and our data demonstrate that filamin A and vimentin bind directly in vitro. Furthermore, we found that the two proteins colocalize at the extensions of spreading cells, consistent with colocalization of filamin A and vimentin in tumor cell pseudopodia (35). Because of the direct interaction between these proteins and their recognized roles in regulating cell motility (20, 36, 49, 66), we investigated the possibility that filamin A and vimentin coregulate early events in cell spreading.
Filamin A and vimentin coregulate cell spreading.
Cell spreading and migration require the formation of cell extensions such as filopodia and lamellipodia (29, 37, 44). We quantified the formation of cell extensions and validated them as authentic filopodia/lamellipodia by immunostaining for vinculin, paxillin, and cortactin. Formation of these actin-rich cell extensions is mediated by β1 integrins, small GTPases, and actin-binding proteins (47, 79) such as filamin A, which binds actin filaments and regulates β1 integrin activity and small GTPases (6, 66, 71). We found that knockdown of filamin A and/or vimentin blocks the formation of cell extensions, consistent with reports of impaired migration in vimentin-deficient cells (20, 49). Notably, knockdown of filamin A and vimentin inhibited cell spreading equivalent to cells in which either protein alone was knocked down, indicating that filamin A and vimentin may coregulate early processes in cell spreading.
We found that actin free barbed ends were reduced following knockdown of filamin A but not knockdown of vimentin. Unexpectedly, simultaneous knockdown of filamin and vimentin reduced free barbed end formation to a greater extent than knockdown of filamin alone, indicating that vimentin may contribute to actin assembly in the absence of filamin A. Vimentin associates with PAK, a protein that regulates actin free barbed end formation through the LIM kinase-cofilin pathway (12, 27). As mutation of vimentin's PAK-binding site inhibits PAK activation (46), vimentin may play a role in PAK activation and actin free barbed end formation. However, our data show that vimentin does not affect free barbed end formation independent of filamin A, and that knockdown of vimentin does not affect the binding between filamin A and Rac or Cdc42, the small GTPases responsible for filopodia and lamellipodia formation (54). Collectively, these data suggest that the filamin A-vimentin interactions do not affect actin assembly directly but instead may regulate β1 integrin function at an upstream point in the cell spreading process.
Filamin A and vimentin coregulate β1 integrin expression and activation.
At early stages of cell spreading and migration, cell surface β1 integrins are endocytosed and are ultimately trafficked back to the plasma membrane (19, 38, 65). As the abundance and activation of cell surface integrins are important factors in cell adhesion and spreading (29), we considered that filamin A and vimentin may regulate β1 integrin activation in spreading. β1 Integrin activation assays indicated that initial β1 integrin-ligand binding was dependent on the presence of both filamin A and vimentin. Consistent with the cell spreading data, ligand-bound β1 integrins were reduced equivalently after knockdown of filamin A, vimentin, or both proteins. Thus filamin A and vimentin may coregulate β1 integrin activation. Immunoblots of focal adhesion proteins showed that filamin A and vimentin are both required for the recruitment of vinculin and paxillin to focal adhesions. Whereas other studies have shown the importance of vimentin for cell adhesion (20, 53, 70), we have identified filamin A as a novel regulator of this process. Whereas previous reports suggest that cell surface expression of β1 integrins is influenced by filamin A (51) or vimentin (53, 61), our data indicate that filamin A and vimentin coregulate β1 integrin trafficking, β1 integrin activation, cell adhesion, and cell spreading.
Vimentin phosphorylation by PKC is required for β1 integrin activity and cell spreading.
PKC-ε-mediated vimentin phosphorylation, β1 integrin recycling, and directed cell migration may be linked processes (31), but the role of vimentin phosphorylation in regulating early cell spreading is not defined. We found that vimentin knockdown and inhibition of PKC prevented the formation of cell extensions and reduced β1 integrin-ligand binding and cell surface β1 integrins. Furthermore, cell extensions in early stages of spreading were enriched with filamin A, vimentin, and PKC-ε. Thus filamin A, vimentin and phospho-vimentin may share a common pathway in regulating cell spreading. Since β1 integrins associate with filamin A (40, 48) and with vimentin (31, 43) while filamin A associates with PKC (25, 69), we considered that filamin A controls the activity of β1 integrins and cell spreading by regulating the phosphorylation of vimentin, which is mediated by PKC.
Filamin A is required for PKC-mediated vimentin phosphorylation.
We found a close spatial relationship between filamin A and PKC, consistent with the notion that filamin A is a substrate for PKC (25, 69). We showed that the catalytically active, phosphorylated form of PKC-ε coimmunoprecipitated with filamin A, which is important since the PKC-ε isoform is essential for integrin recycling (31). Furthermore, we found that knockdown of filamin A inhibits vimentin phosphorylation at serines-6, -38, and -50, which are known PKC targets (2, 33, 55). Whereas these data do not show a direct interaction between filamin A and PKC-ε, they strongly point to a role for filamin A in mediating vimentin phosphorylation by PKC. Moreover, knockdown of filamin A prevented translocation of vimentin to the cell periphery; the same result was observed following inhibition of PKC by BIM. These data are consistent with a model in which phosphorylation by PKC-ε is required for vimentin reorganization and for the trafficking of β1 integrins to the cell membrane (31). Our study extends their findings by demonstrating that filamin A regulates this pivotal phosphorylation step. We conclude that filamin A, vimentin, and PKC regulate β1 integrin activity, cell adhesion, and the formation of early cell extensions as a result of the binding of filamin A to vimentin.
GRANTS
This study was supported by Canadian Institutes of Health Research operating (MGP-37783), Group and Research Resource grants to C. A. McCulloch. H. Kim acknowledges fellowship support from the Heart and Stroke Foundation of Canada, the Canadian Arthritis Network, the University of Toronto Harron Fund, and the Canadian Institutes of Health Research Cell Signals Program (STP-53877).
DISCLOSURES
No conflicts of interest are declared by the author(s).
ACKNOWLEDGMENTS
The authors gratefully acknowledge Dr. Boris Hinz for critical reading of the manuscript, Dr. David Calderwood for providing filamin-deficient 3T3 fibroblasts, and Claire Hong for technical assistance.
REFERENCES
- 1. Albrecht-Buehler G, Lancaster RM. A quantitative description of the extension and retraction of surface protrusions in spreading 3T3 mouse fibroblasts. J Cell Biol 71: 370–382, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ando S, Tanabe K, Gonda Y, Sato C, Inagaki M. Domain- and sequence-specific phosphorylation of vimentin induces disassembly of the filament structure. Biochemistry 28: 2974–2979, 1989. [DOI] [PubMed] [Google Scholar]
- 3. Arocena M. Effect of acrylamide on the cytoskeleton and apoptosis of bovine lens epithelial cells. Cell Biol Int 30: 1007–1012, 2006. [DOI] [PubMed] [Google Scholar]
- 4. Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol Biol Cell 12: 2711–2720, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res 66: 3034–3043, 2006. [DOI] [PubMed] [Google Scholar]
- 6. Bellanger JM, Astier C, Sardet C, Ohta Y, Stossel TP, Debant A. The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat Cell Biol 2: 888–892, 2000. [DOI] [PubMed] [Google Scholar]
- 7. Brotschi EA, Hartwig JH, Stossel TP. The gelation of actin by actin-binding protein. J Biol Chem 253: 8988–8993, 1978. [PubMed] [Google Scholar]
- 8. Brown KD, Binder LI. Identification of the intermediate filament-associated protein gyronemin as filamin. Implications for a novel mechanism of cytoskeletal interaction. J Cell Sci 102: 19–30, 1992. [DOI] [PubMed] [Google Scholar]
- 9. Campbell ID. Studies of focal adhesion assembly. Biochem Soc Trans 36: 263–266, 2008. [DOI] [PubMed] [Google Scholar]
- 10. Cenni V, Doppler H, Sonnenburg ED, Maraldi N, Newton AC, Toker A. Regulation of novel protein kinase C epsilon by phosphorylation. Biochem J 363: 537–545, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan MW, Arora PD, McCulloch CA. Cyclosporin inhibition of collagen remodeling is mediated by gelsolin. Am J Physiol Cell Physiol 293: C1049–C1058, 2007. [DOI] [PubMed] [Google Scholar]
- 12. Chan W, Kozma R, Yasui Y, Inagaki M, Leung T, Manser E, Lim L. Vimentin intermediate filament reorganization by Cdc42: involvement of PAK and p70 S6 kinase. Eur J Cell Biol 81: 692–701, 2002. [DOI] [PubMed] [Google Scholar]
- 13. Chang L, Goldman RD. Intermediate filaments mediate cytoskeletal crosstalk. Nat Rev Mol Cell Biol 5: 601–613, 2004. [DOI] [PubMed] [Google Scholar]
- 14. Chong SA, Lee W, Arora PD, Laschinger C, Young EW, Simmons CA, Manolson M, Sodek J, McCulloch CA. Methylglyoxal inhibits the binding step of collagen phagocytosis. J Biol Chem 282: 8510–8520, 2007. [DOI] [PubMed] [Google Scholar]
- 15. Chou YH, Flitney FW, Chang L, Mendez M, Grin B, Goldman RD. The motility and dynamic properties of intermediate filaments and their constituent proteins. Exp Cell Res 313: 2236–2243, 2007. [DOI] [PubMed] [Google Scholar]
- 16. Correia I, Chu D, Chou YH, Goldman RD, Matsudaira P. Integrating the actin and vimentin cytoskeletons. Adhesion-dependent formation of fimbrin-vimentin complexes in macrophages. J Cell Biol 146: 831–842, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Critchley DR. Focal adhesions–the cytoskeletal connection. Curr Opin Cell Biol 12: 133–139, 2000. [DOI] [PubMed] [Google Scholar]
- 18. Defilippi P, Olivo C, Venturino M, Dolce L, Silengo L, Tarone G. Actin cytoskeleton organization in response to integrin-mediated adhesion. Micro Res Tech 47: 67–78, 1999. [DOI] [PubMed] [Google Scholar]
- 19. Dunphy JL, Moravec R, Ly K, Lasell TK, Melancon P, Casanova JE. The Arf6 GEF GEP100/BRAG2 regulates cell adhesion by controlling endocytosis of beta1 integrins. Curr Biol 16: 315–320, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eckes B, Dogic D, Colucci-Guyon E, Wang N, Maniotis A, Ingber D, Merckling A, Langa F, Aumailley M, Delouvee A, Koteliansky V, Babinet C, Krieg T. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J Cell Sci 111: 1897–1907, 1998. [DOI] [PubMed] [Google Scholar]
- 21. Eriksson JE, He T, Trejo-Skalli AV, Harmala-Brasken AS, Hellman J, Chou YH, Goldman RD. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J Cell Sci 117: 919–932, 2004. [DOI] [PubMed] [Google Scholar]
- 22. Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol 6: 1034–1038, 2004. [DOI] [PubMed] [Google Scholar]
- 23. Fortin S, Le Mercier M, Camby I, Spiegl-Kreinecker S, Berger W, Lefranc F, Kiss R. Galectin-1 is implicated in the protein kinase C epsilon/vimentin-controlled trafficking of integrin-beta1 in glioblastoma cells. Brain Pathol In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frame M, Norman J. A tal(in) of cell spreading. Nat Cell Biol 10: 1017–1019, 2008. [DOI] [PubMed] [Google Scholar]
- 25. Glogauer M, Arora P, Chou D, Janmey PA, Downey GP, McCulloch CA. The role of actin-binding protein 280 in integrin-dependent mechanoprotection. J Biol Chem 273: 1689–1698, 1998. [DOI] [PubMed] [Google Scholar]
- 26. Glogauer M, Arora P, Yao G, Sokholov I, Ferrier J, McCulloch CA. Calcium ions and tyrosine phosphorylation interact coordinately with actin to regulate cytoprotective responses to stretching. J Cell Sci 110: 11–21, 1997. [DOI] [PubMed] [Google Scholar]
- 27. Goto H, Tanabe K, Manser E, Lim L, Yasui Y, Inagaki M. Phosphorylation and reorganization of vimentin by p21-activated kinase (PAK). Genes Cells 7: 91–97, 2002. [DOI] [PubMed] [Google Scholar]
- 28. Green KJ, Geiger B, Jones JC, Talian JC, Goldman RD. The relationship between intermediate filaments and microfilaments before and during the formation of desmosomes and adherens-type junctions in mouse epidermal keratinocytes. J Cell Biol 104: 1389–1402, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guillou H, Depraz-Depland A, Planus E, Vianay B, Chaussy J, Grichine A, Albiges-Rizo C, Block MR. Lamellipodia nucleation by filopodia depends on integrin occupancy and downstream Rac1 signaling. Exp Cell Res 314: 478–488, 2008. [DOI] [PubMed] [Google Scholar]
- 30. Ivaska J, Pallari HM, Nevo J, Eriksson JE. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp Cell Res 313: 2050–2062, 2007. [DOI] [PubMed] [Google Scholar]
- 31. Ivaska J, Vuoriluoto K, Huovinen T, Izawa I, Inagaki M, Parker PJ. PKCepsilon-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J 24: 3834–3845, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ivaska J, Whelan RD, Watson R, Parker PJ. PKC epsilon controls the traffic of beta1 integrins in motile cells. EMBO J 21: 3608–3619, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Izawa I, Inagaki M. Regulatory mechanisms and functions of intermediate filaments: a study using site- and phosphorylation state-specific antibodies. Cancer Sci 97: 167–174, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jackson WM, Jaasma MJ, Tang RY, Keaveny TM. Mechanical loading by fluid shear is sufficient to alter the cytoskeletal composition of osteoblastic cells. Am J Physiol Cell Physiol 295: C1007–C1015, 2008. [DOI] [PubMed] [Google Scholar]
- 35. Jia Z, Barbier L, Stuart H, Amraei M, Pelech S, Dennis JW, Metalnikov P, O'Donnell P, Nabi IR. Tumor cell pseudopodial protrusions. Localized signaling domains coordinating cytoskeleton remodeling, cell adhesion, glycolysis, RNA translocation, and protein translation. J Biol Chem 280: 30564–30573, 2005. [DOI] [PubMed] [Google Scholar]
- 36. Johansen LD, Naumanen T, Knudsen A, Westerlund N, Gromova I, Junttila M, Nielsen C, Bottzauw T, Tolkovsky A, Westermarck J, Coffey ET, Jaattela M, Kallunki T. IKAP localizes to membrane ruffles with filamin A and regulates actin cytoskeleton organization and cell migration. J Cell Sci 121: 854–864, 2008. [DOI] [PubMed] [Google Scholar]
- 37. Johnston SA, Bramble JP, Yeung CL, Mendes PM, Machesky LM. Arp2/3 complex activity in filopodia of spreading cells. BMC Cell Biol 9: 65, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jovic M, Naslavsky N, Rapaport D, Horowitz M, Caplan S. EHD1 regulates beta1 integrin endosomal transport: effects on focal adhesions, cell spreading and migration. J Cell Sci 120: 802–814, 2007. [DOI] [PubMed] [Google Scholar]
- 39. Kasas S, Wang X, Hirling H, Marsault R, Huni B, Yersin A, Regazzi R, Grenningloh G, Riederer B, Forro L, Dietler G, Catsicas S. Superficial and deep changes of cellular mechanical properties following cytoskeleton disassembly. Cell Motil Cytoskel 62: 124–132, 2005. [DOI] [PubMed] [Google Scholar]
- 40. Kiema T, Lad Y, Jiang P, Oxley CL, Baldassarre M, Wegener KL, Campbell ID, Ylanne J, Calderwood DA. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell 21: 337–347, 2006. [DOI] [PubMed] [Google Scholar]
- 41. Kim H, Sengupta A, Glogauer M, McCulloch CA. Filamin A regulates cell spreading and survival via beta1 integrins. Exp Cell Res 314: 834–846, 2008. [DOI] [PubMed] [Google Scholar]
- 42. Knowles GC, McKeown M, Sodek J, McCulloch CA. Mechanism of collagen phagocytosis by human gingival fibroblasts: importance of collagen structure in cell recognition and internalization. J Cell Sci 98: 551–558, 1991. [DOI] [PubMed] [Google Scholar]
- 43. Kreis S, Schonfeld HJ, Melchior C, Steiner B, Kieffer N. The intermediate filament protein vimentin binds specifically to a recombinant integrin alpha2/beta1 cytoplasmic tail complex and co-localizes with native alpha2/beta1 in endothelial cell focal adhesions. Exp Cell Res 305: 110–121, 2005. [DOI] [PubMed] [Google Scholar]
- 44. Ladwein M, Rottner K. On the Rho'd: the regulation of membrane protrusions by Rho-GTPases. FEBS Lett 582: 2066–2074, 2008. [DOI] [PubMed] [Google Scholar]
- 45. Lee W, Sodek J, McCulloch CA. Role of integrins in regulation of collagen phagocytosis by human fibroblasts. J Cell Physiol 168: 695–704, 1996. [DOI] [PubMed] [Google Scholar]
- 46. Li QF, Spinelli AM, Wang R, Anfinogenova Y, Singer HA, Tang DD. Critical role of vimentin phosphorylation at Ser-56 by p21-activated kinase in vimentin cytoskeleton signaling. J Biol Chem 281: 34716–34724, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li S, Guan JL, Chien S. Biochemistry and biomechanics of cell motility. Annu Rev Biomed Eng 7: 105–150, 2005. [DOI] [PubMed] [Google Scholar]
- 48. Loo DT, Kanner SB, Aruffo A. Filamin binds to the cytoplasmic domain of the beta1-integrin. Identification of amino acids responsible for this interaction. J Biol Chem 273: 23304–23312, 1998. [DOI] [PubMed] [Google Scholar]
- 49. McInroy L, Maatta A. Down-regulation of vimentin expression inhibits carcinoma cell migration and adhesion. Biochem Biophys Res Commun 360: 109–114, 2007. [DOI] [PubMed] [Google Scholar]
- 50. Meng X, Yuan Y, Maestas A, Shen Z. Recovery from DNA damage-induced G2 arrest requires actin-binding protein filamin-A/actin-binding protein 280. J Biol Chem 279: 6098–6105, 2004. [DOI] [PubMed] [Google Scholar]
- 51. Meyer SC, Sanan DA, Fox JE. Role of actin-binding protein in insertion of adhesion receptors into the membrane. J Biol Chem 273: 3013–3020, 1998. [DOI] [PubMed] [Google Scholar]
- 52. Nakamura F, Osborn TM, Hartemink CA, Hartwig JH, Stossel TP. Structural basis of filamin A functions. J Cell Biol 179: 1011–1025, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nieminen M, Henttinen T, Merinen M, Marttila-Ichihara F, Eriksson JE, Jalkanen S. Vimentin function in lymphocyte adhesion and transcellular migration. Nat Cell Biol 8: 156–162, 2006. [DOI] [PubMed] [Google Scholar]
- 54. Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81: 53–62, 1995. [DOI] [PubMed] [Google Scholar]
- 55. Ogawara M, Inagaki N, Tsujimura K, Takai Y, Sekimata M, Ha MH, Imajoh-Ohmi S, Hirai S, Ohno S, Sugiura H, et al. Differential targeting of protein kinase C and CaM kinase II signalings to vimentin. J Cell Biol 131: 1055–1066, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ohta Y, Suzuki N, Nakamura S, Hartwig JH, Stossel TP. The small GTPase RalA targets filamin to induce filopodia. Proc Natl Acad Sci USA 96: 2122–2128, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Perrin BJ, Amann KJ, Huttenlocher A. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol Biol Cell 17: 239–250, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Popowicz GM, Schleicher M, Noegel AA, Holak TA. Filamins: promiscuous organizers of the cytoskeleton. Trends Biochem Sci 31: 411–419, 2006. [DOI] [PubMed] [Google Scholar]
- 59. Price LS, Leng J, Schwartz MA, Bokoch GM. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell 9: 1863–1871, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J 18: 578–585, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rizki A, Mott JD, Bissell MJ. Polo-like kinase 1 is involved in invasion through extracellular matrix. Cancer Res 67: 11106–11110, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Seifert GJ, Lawson D, Wiche G. Immunolocalization of the intermediate filament-associated protein plectin at focal contacts and actin stress fibers. Eur J Cell Biol 59: 138–147, 1992. [PubMed] [Google Scholar]
- 63. Serrels B, Serrels A, Brunton VG, Holt M, McLean GW, Gray CH, Jones GE, Frame MC. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat Cell Biol 9: 1046–1056, 2007. [DOI] [PubMed] [Google Scholar]
- 64. Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC. Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res 313: 2098–2109, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Skalski M, Coppolino MG. SNARE-mediated trafficking of alpha5beta1 integrin is required for spreading in CHO cells. Biochem Biophys Res Commun 335: 1199–1210, 2005. [DOI] [PubMed] [Google Scholar]
- 66. Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol 2: 138–145, 2001. [DOI] [PubMed] [Google Scholar]
- 67. Sun CX, Magalhaes MA, Glogauer M. Rac1 and Rac2 differentially regulate actin free barbed end formation downstream of the fMLP receptor. J Cell Biol 179: 239–245, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol 135: 991–1007, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tigges U, Koch B, Wissing J, Jockusch BM, Ziegler WH. The F-actin cross-linking and focal adhesion protein filamin A is a ligand and in vivo substrate for protein kinase C alpha. J Biol Chem 278: 23561–23569, 2003. [DOI] [PubMed] [Google Scholar]
- 70. Tsuruta D, Jones JC. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J Cell Sci 116: 4977–4984, 2003. [DOI] [PubMed] [Google Scholar]
- 71. Vadlamudi RK, Li F, Adam L, Nguyen D, Ohta Y, Stossel TP, Kumar R. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol 4: 681–690, 2002. [DOI] [PubMed] [Google Scholar]
- 72. Vicente-Manzanares M, Choi CK, Horwitz AR. Integrins in cell migration–the actin connection. J Cell Sci 122: 199–206, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Webb BA, Jia L, Eves R, Mak AS. Dissecting the functional domain requirements of cortactin in invadopodia formation. Eur J Cell Biol 86: 189–206, 2007. [DOI] [PubMed] [Google Scholar]
- 74. Whipple RA, Balzer EM, Cho EH, Matrone MA, Yoon JR, Martin SS. Vimentin filaments support extension of tubulin-based microtentacles in detached breast tumor cells. Cancer Res 68: 5678–5688, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Winokur R, Hartwig JH. Mechanism of shape change in chilled human platelets. Blood 85: 1796–1804, 1995. [PubMed] [Google Scholar]
- 76. Yang Y, Dowling J, Yu QC, Kouklis P, Cleveland DW, Fuchs E. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell 86: 655–665, 1996. [DOI] [PubMed] [Google Scholar]
- 77. Yasui Y, Goto H, Matsui S, Manser E, Lim L, Nagata K, Inagaki M. Protein kinases required for segregation of vimentin filaments in mitotic process. Oncogene 20: 2868–2876, 2001. [DOI] [PubMed] [Google Scholar]
- 78. Zhao XH, Laschinger C, Arora P, Szaszi K, Kapus A, McCulloch CA. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci 120: 1801–1809, 2007. [DOI] [PubMed] [Google Scholar]
- 79. Zimerman B, Volberg T, Geiger B. Early molecular events in the assembly of the focal adhesion-stress fiber complex during fibroblast spreading. Cell Motil Cytoskel 58: 143–159, 2004. [DOI] [PubMed] [Google Scholar]