

Abstract
We characterized the role of protein tyrosine phosphatase (PTP)-α in focal adhesion (FA) formation and remodeling using wild-type and PTPα-deficient (PTPα−/−) cells. Compared with wild-type cells, spreading PTPα−/− fibroblasts displayed fewer leading edges and formed elongated α-actinin-enriched FA at the cell periphery. These features suggest the presence of slowly remodeling cell adhesions and were phenocopied in human fibroblasts in which PTPα was knocked down using short interfering RNA (siRNA) or in NIH-3T3 fibroblasts expressing catalytically inactive (C433S/C723S) PTPα. Fluorescence recovery after photobleaching showed slower green fluorescence protein-α-actinin recovery in the FA of PTPα−/− than wild-type cells. These alterations correlated with reduced cell spreading, adhesion, and polarization and retarded contraction of extracellular matrices in PTPα−/− fibroblasts. Activation of Rac1 and its recruitment to FA during spreading were diminished in cells expressing C433S/C723S PTPα. Rac1−/− cells also displayed abnormally elongated and peripherally distributed FA that failed to remodel. Conversely, expression of constitutively active Rac1 restored normal FA remodeling in PTPα−/− cells. We conclude that PTPα is required for remodeling of FA during cell spreading via a pathway involving Rac1.
Keywords: cell spreading, integrins, extracellular matrix, actin cytoskeleton
cell adhesion and motility play pivotal roles in diverse processes, including embryonic development, tissue regeneration, wound healing, and neoplasia (41). As cells explore new regions of the substrate, they interact with extracellular matrix (ECM) molecules, in part through focal adhesions (FA). These integrin-based, multimolecular structures link the ECM to the actin cytoskeleton and comprise >50 structural and signaling molecules that regulate remodeling of FA, as well as FA-dependent signaling (4, 30). In the leading edges (lamellipodia) of motile cells, nascent FA, termed focal complexes, form and are then rapidly remodeled. Focal complexes display a punctuate appearance (32). As the rate of FA assembly and dissipation decreases near the edge of the lamellipodium, highly dynamic focal complexes mature into larger and more stable FA by the recruitment of additional proteins such as α-actinin (3, 19, 22). Mature FA facilitate the transmission of tensile forces to the substrate, and, during locomotion, these structures translocate toward the center of the cell, thus facilitating cell movement relative to the substrate (54). Our understanding of the mechanisms that regulate the assembly and disassembly of FA is incomplete.
Members of the Rho family of small GTPases, RhoA, Rac, and Cdc42, are key regulators of adhesion dynamics, in that they couple the formation and breakdown of FA to actin assembly (5). Activation of Cdc42 and Rac1 directs the formation of membrane protrusions, which are associated with increased FA turnover (31, 45). In contrast, active RhoA promotes the formation of actin stress fibers and more stable FA, which enhance cell attachment (20, 42). Despite recent progress, the molecular mechanisms that regulate activation of the Rho family of GTPases during cell spreading and migration remain incompletely understood. Notably, the protein tyrosine phosphatase (PTP)-α is enriched in FA (25) and is known to regulate cell motility (7, 21, 37). In the present study, we have considered that PTPα and Rho family GTPases act coordinately to regulate the formation and breakdown of FA.
PTPα is a transmembrane receptor-like PTP that is enriched in FA and has two cytoplasmic catalytic domains, D1 and D2. The D1 domain has significantly higher catalytic activity (56). During the early phases of cell spreading, interaction of PTPα with αv-integrins leads to downstream integrin-dependent signaling events involved in cell motility (52, 57). Among these events, PTPα promotes activation of Src family kinases via dephosphorylation of a COOH-terminal inhibitory tyrosine residue, which in turn leads to integrin-stimulated autophosphorylation and activation of FA kinase (7, 9). Fibroblasts lacking PTPα exhibit delayed spreading, which is associated with impaired formation of actin stress fibers and FA (46, 57). Thus PTPα appears to be pivotal in integrin-dependent signaling events that regulate FA formation.
The molecular mechanisms that link PTPα with FA formation and maturation are not well defined. In the present study, we have tested the hypothesis that PTPα promotes the turnover of FA required for remodeling at the leading edge of spreading cells. We demonstrate that, by enabling FA turnover, PTPα regulates the dynamics and distribution of FA and contractile stress fibers that are essential for cell adhesion, spreading, and transmission of tensile force to the substrate.
MATERIALS AND METHODS
Antibodies.
Mouse monoclonal antibodies to α-smooth muscle actin (α-SMA; clone 1A4), β-actin (clone AC-15), α-actinin (clone BM 75.2), and FITC-phalloidin were obtained from Sigma-Aldrich (St. Louis, MO); rabbit polyclonal anti-myc antibodies from Cell Signaling (Beverly, MA); rabbit anti-Rac1 antibody from BD Transduction Laboratories (San Jose, CA); rabbit anti-hemagglutinin (HA) and anti-paxillin antibodies from Santa Cruz Biotechnology (Santa Cruz, CA); and rabbit anti-PTPα polyclonal antibody from Upstate (Charlottesville, VA).
Cell culture.
Human gingival fibroblasts were grown in α-MEM as previously described (28). Wild-type (PTPαwt) and PTPα-null (PTPα−/−) murine embryonic fibroblasts were provided by Dr. Jan Sap (University of Copenhagen, Copenhagen, Denmark) (46). Genetically modified NIH-3T3 fibroblasts that express HA-tagged wild-type PTPα (PTPαwt ind) and PTPα C433S/C723S (PTPαCCSS ind) under control of a doxycycline-repressible promoter were generated as described elsewhere (58). The latter cells were grown in DMEM in the presence of 5 ng/ml doxycycline (Sigma). Doxycycline was removed 14–16 h before the experiments to allow expression of recombinant PTPα. Rac1 conditional null fibroblasts (Rac1C/C) were treated with HNTC (polyhistidine, HIV TAT fusion protein and nuclear localization sequences fused to the Cre cDNA) peptide as described elsewhere (35, 50) to generate Rac1-null cells or used as a control (without the peptide treatment). All media contained 10% fetal bovine serum and antibiotics.
Short interfering RNA.
Specific knockdown of PTPα expression was conducted using commercial short interfering RNA (siRNA; PTPRA, Qiagen). Human gingival fibroblasts were transfected with 50–100 nM PTPRA siRNA or control siRNA for 72 h using Lipofectamine according to the manufacturer's specifications.
Plasmid constructs and transient transfection.
HA-tagged PTPαwt (HA-PTPαwt), PTPα lacking the D2 domain (D1/ΔD2), and PTPα lacking the D1 and D2 domains (ΔD1/ΔD2) were kindly provided by Dr. J. den Hertog (Hubrecht Laboratory, Netherlands Institute for Developmental Biology, Utrecht, The Netherlands). Green fluorescent protein (GFP)-α-actinin was provided by Dr. Michael P. Sheetz (Department of Biological Sciences, Columbia University, New York, NY). GFP-Rac1, GFP- p21-activated kinase (PAK1)-binding domain (PBD), GFP-tagged dominant-negative (DN) mutant of Rac1 (N17 Rac1), and myc-tagged constitutively active (CA) mutant of Rac1 (L61) were a gift from Dr. Sergio Grinstein (Hospital for Sick Children, Toronto, ON, Canada). Transient transfection of fibroblasts was performed using 1:3 DNA-Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. At 24 h after transfection, Lipofectamine was removed and cells were subcultured onto eight-well chamber slides (Labtek) or 25-mm glass coverslips, both of which were coated with fibronectin (10 μg/ml in PBS), and allowed to spread for an additional 1, 3, 6, or 16 h. GFP-tagged proteins were analyzed by direct fluorescence microscopy after fixation with 4% paraformaldehyde. Additionally, cells transfected with myc- or HA-tagged constructs were also stained with antibodies directed against the epitope tag.
Immunofluorescence.
Anti-α-actinin (1:100 dilution), anti-paxillin (1:60 dilution), anti-myc (1:200 dilution), and anti-HA (1:200 dilution) antibodies were used to stain cells as described elsewhere (28). Fluorescence staining of actin filaments was performed with FITC-phalloidin (1 μM) for 40 min. Slides were viewed with a Zeiss LSM510 confocal microscope or a Leica DMIRB inverted fluorescence microscope equipped with a Q-Imaging camera. Digital images were processed with OpenLab software (Improvision). Cell area, cell length and width, and length of focal complexes/FA were estimated by measurements on paxillin- and α-actinin-stained cells using the same imaging software. Images of cells stained with anti-paxillin antibodies were magnified electronically, and the number of focal complexes/FA was counted visually in ≥100 cells. Inasmuch as FA in PTPαwt cells were very thin, quantitative analysis of their area was difficult because of difficulties in estimating their width. Therefore, we utilized length as the primary measurement of FA in these and PTPα−/− cells.
Isolation of focal complexes/FA and cellular insoluble fraction.
FA-like structures were isolated from fibroblasts using fibronectin-coated ferromagnetic beads as previously described (28). Briefly, cells were added to a poly-l-lysine-coated plate (500 μg/ml; Sigma), and 2-μm ferromagnetic beads (Sigma) coated with 0.02% fibronectin (Sigma) were then added to the dorsal surface of the cells and allowed to sediment by gravity (“parachuted”). Beads bound to the cells were counted electronically with a Coulter Counter to normalize the bead-to-cell ratio between different preparations. The cells were lysed, and the beads with bound FA-associated proteins were isolated using a magnetic separation stand (Promega). Finally, the proteins were eluted from the beads by boiling in Laemmli sample buffer, separated by SDS-PAGE, and analyzed by Western blot analysis. For isolation of the cytoskeletal fraction, cells were lysed in Triton X-100 buffer (see above) for 20 min on ice and sedimented. The supernatant was collected (soluble fraction), and the residual cellular components constituted the cytoskeletal fraction, which was resuspended in 1× SDS sample buffer before SDS-PAGE.
Cell adhesion and gel contraction assays.
To study adhesion, we incubated fibroblasts at 37°C with 2 μM calcein-AM (Calbiochem) on a rotator for 30 min. Cells (3.6 × 104/well) were seeded into 96-well plates precoated with fibronectin (10 μg/ml) and incubated for 1 h at 37°C. Plates were washed five times with PBS. Cells were fixed with paraformaldehyde (4% in PBS), and fluorescence was read on a multiplate reader (excitation at 494 nm and emission at 517 nm). For gel contraction assays (17), cells were maintained in 10% serum 16 h before they were seeded in a solution of type I collagen (1.8 mg collagen/ml, pH 7.4). For the floating model, mixtures of 2.29 × 104 cells/ml and collagen solution were divided into 200-μl aliquots and transferred to 35-mm non-tissue culture plastic plates. The gels were allowed to polymerize at 37°C in a 5% CO2 incubator for 1 h, and growth medium (2 ml of DMEM with 10% serum) was added. For the stressed model, 2 × 105 cells/ml in collagen solution were transferred to 24-well tissue culture plates. Cells were detached after 1 day. Initial baseline measurements and subsequent daily measurements were performed with a dissecting microscope fitted with an intraocular linear measurement eyepiece. Growth medium was changed every 2 days.
Rac activation assay.
Activated Rac1 was determined with a glutathione S-transferase-tagged fusion protein of the Rac1 binding domain of p21 kinase (provided by Dr. Sergio Grinstein) prebound to glutathione-Sepharose 4B beads. After the pull-down assay was performed, activated Rac1 was detected by immunoblot analysis. Total Rac1 was also assessed in whole cell lysates and served as loading control.
FA formation on fibronectin-coated beads.
Glass coverslips were coated with 500 μg/ml poly-l-lysine (Sigma) overnight at 4°C and then washed. The 2-μm latex beads (Sigma) were coated with 0.02% fibronectin (Sigma) solution overnight at 4°C and then washed. The beads were added to the polylysine-coated coverslips and allowed to adhere for 30 min at room temperature. The coverslips were gently washed once, and the transfected fibroblasts were added to the coverslips. Live images were acquired using an inverted Zeiss LSM 510 confocal microscope.
Fluorescence recovery after photobleaching.
Fluorescence recovery after photobleaching (FRAP) analysis was conducted essentially as described elsewhere (11). Briefly, cells transfected with GFP constructs of FA proteins were grown on fibronectin-coated coverslips and mounted for observation in a sealed chamber containing normal growth medium. To avoid possible artifacts of overexpression, only cells expressing low but detectable amounts of protein were chosen for further analysis. Selected FA were photobleached using the 488-nm laser line of the Zeiss LSM 510 confocal microscope at full power, resulting in ∼80% reduction in the fluorescence intensity. The bleached areas were ∼10–18 μm2. Fluorescence recovery was monitored at 12-s (see Fig. 2A) and 5-s (see Fig. 7I) intervals. For each time point, the intensity of the bleached area was normalized to that of a corresponding unbleached area. All FRAP measurements were performed at 37°C. All quantitative data for FRAP recovery kinetics represent averages ± SE from 9–10 cells imaged in three independent experiments. FRAP recovery curves were generated using StatView software. The paired Student's t-test was used to determine the statistical significance of these results. The values of intensity vs. time (min) were charted in a scatter plot. The half-recovery time (t1/2) was measured from the plots and represented as means ± SD from 9–10 cells imaged in three independent experiments.
Fig. 2.
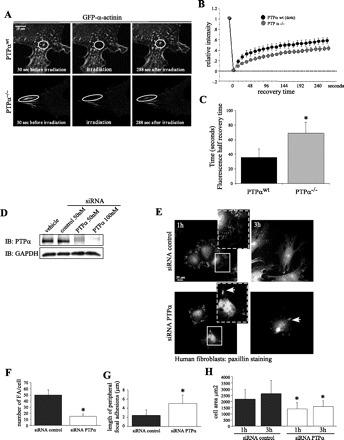
PTPα regulates α-actinin dynamics. GFP-α-actinin dynamics were analyzed by fluorescence recovery after photobleaching (FRAP) in transfected PTPαwt and PTPα−/− cells. A: time-lapse images of GFP-α-actinin at cell periphery before (30 s) and after (288 s) photobleaching. In PTPαwt cells, small “dots” and larger “mature” α-actinin were selected for photobleaching. PTPα−/− cells displayed mostly supermature FA (larger α-actinin aggregates), which were irradiated. B: FRAP of PTPαwt and PTPα−/− cells. Values are means ± SE. C: fluorescence half-recovery time. Values are means ± SD calculated from 9–10 curves for each category. *P < 0.01 vs. PTPαwt. D: Western immunoblot (IB) assessment of PTPα knockdown by 50–100 nM short interfering (siRNA). E: human gingival fibroblasts transfected with siRNA against PTPα or control siRNA and immunostained for paxillin. Cells were allowed to spread for 1 and 3 h. F and G: quantification of numbers and length of FA in fibroblasts treated with PTPα-siRNA and control siRNA. *P < 0.01 vs. siRNA control. H: cell area. Values are means ± SD of 40 independent measurements. *P < 0.05 vs. respective (1 and 3 h) siRNA control.
Fig. 7.
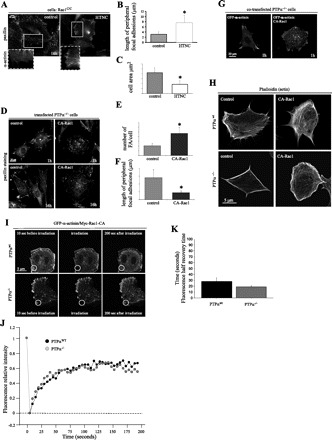
Rac1 rescues FA remodeling in PTPα−/− cells. A: fluorescent images of α-actinin and paxillin distribution in Rac1 conditional fibroblasts (Rac1C/C) treated with HNTC peptide (polyhistidine, HIV TAT fusion protein and nuclear localization sequences fused to the Cre cDNA) to generate Rac1-null cells or used as a control (same cells treated with vehicle alone, i.e., no HNTC peptide). Where indicated, cells were transfected with GFP-α-actinin and allowed to spread for 16 h and fixed with 4% paraformaldehyde. Cells were then immunostained using anti-paxillin antibodies. B and C: length of peripheral FA and cell area of control and HTNC-treated cells. *P < 0.05 vs. control. D: fluorescent images of PTPα−/− cells transfected with myc-tagged constitutively active (CA) L61 Rac1 (CA-Rac1) or vehicle control, allowed to spread for 1 and 16 h, and stained with anti-paxillin antibodies. E and F: number of FA per cell and length of peripheral FA in control and CA-Rac1 transfected cells. *P < 0.05 vs. control. G: PTPα−/− cells transfected with CA-Rac1 and FA and imaged by cotransfection with GFP-α-actinin. CA-Rac1 induces FA turnover and de novo FA formation. H: fluorescent images of PTPαwt and PTPα−/− cells transfected with CA-Rac1, stained with Alexa-phalloidin, and allowed to spread for 4 h. I: GFP-α-actinin dynamics were analyzed by fluorescence recovery after photobleaching in cotransfected (GFP-α-actinin and CA-Rac1) PTPαwt and PTPα−/− cells. Images represent time lapse of GFP-α-actinin at the cell periphery before (10 s) and after (0 and 200 s) photobleaching. Circles represent bleached area. J: rate of fluorescence recovery (measured every 5 s) in a representative experiment as analyzed by Zeiss LSM Image Examiner software. Intensity at each time point was normalized to a control area of equivalent size (unbleached area). K: half-recovery time of 3 curves for each group of cells (PTPαwt and PTPα−/−). Values are means ± SE of 3 experiments. Values are not statistically different (by Student's t-test).
Data analysis.
For continuous variables, means and standard deviation of means were computed. Data were analyzed by ANOVA with Dunnett's or Scheffé's correction for multiple comparisons or by paired or unpaired Student's t-test. Statistical significance was set at P < 0.05.
RESULTS
PTPα mediates FA remodeling.
Using paxillin and α-actinin immunofluorescence, we examined the subcellular distribution of FA in primary murine fibroblasts derived from PTPαwt and PTPα−/− mice (46) spreading on fibronectin. Paxillin, which is present in filopodia and initiates FA formation (34), was used as a marker to study the early stages of FA formation and maturation, and α-actinin was used as a marker to characterize the later stages of FA maturation (22, 51). Immunofluorescence staining of paxillin from PTPαwt and PTPα−/− fibroblasts revealed key differences in the structure and stability of peripheral FA (Fig. 1A). After 1 h of spreading, PTPαwt fibroblasts displayed predominantly peripheral filopodia with small, nascent FA (focal complexes) that matured over time. As cells spread (3–16 h), FA underwent remodeling that was associated with formation of membrane protrusions and cell polarization, consistent with the concept that FA formation during cell spreading is balanced by continuous disassembly at the leading edge (43). In contrast to wild-type cells, PTPα−/− fibroblasts displayed predominantly larger and unusually shaped FA that localized to the periphery of cells (Fig. 1, A and B). This morphology of FA has been termed “supermature,” denoting an area significantly larger than that of typical mature FA (10, 16, 22). Quantitative analysis confirmed that, even after 16 h of spreading, these supermature FA in PTPα−/− fibroblasts were significantly larger than those in PTPαwt cells (8.3 ± 3.4 vs. 3.0 ± 2.2 μm long; Fig. 1B). PTPα−/− cells were also significantly smaller (less spread) than their wild-type counterparts (Fig. 1C). In summary, supermature FA in PTPα−/− fibroblasts correlated with the absence of membrane protrusions, cell spreading, and formation of classical (smaller) FA at the cell border.
Fig. 1.
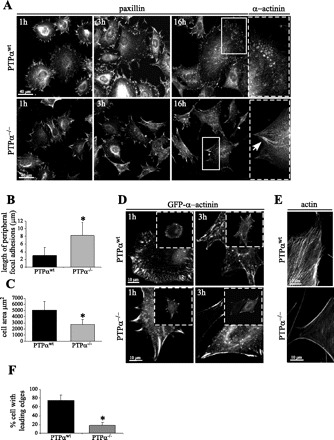
Supermature focal adhesions (FA) in protein tyrosine phosphatase-α (PTPα)-null (PTPα−/−) cells associated with lack of remodeling. Wild-type PTPα (PTPαwt) and PTPα−/− cells were plated on fibronectin (10 mg/ml) for 1, 3, and 16 h and then fixed with paraformaldehyde. A: FA visualized with anti-paxillin and anti-α-actinin antibodies. Arrow denotes enlarged and elongated (supermature) FA, characterized by inclusion of α-actinin. B and C: length of peripheral FA and cell area on paxillin-stained cells after 16 h of spreading. Values are means ± SD of 40–50 measurements in 3 independent experiments. *P < 0.01 vs. PTPαwt (by unpaired Student's t-test). D: α-actinin visualized in cells transfected with green fluorescent protein (GFP)-α-actinin. In PTPα−/− cells, supermature FA containing increased amounts of α-actinin failed to turn over. E: FITC-phalloidin staining showing immobile edges in PTPα−/− cells, characterized by long bundles of actin that originate from FA and delineate the cell periphery. F: quantification of leading edges in PTPαwt and PTPα−/− cells. Values are means ± SD of 40–50 measurements in 3 independent experiments. *P < 0.01 vs. PTPαwt (by unpaired Student's t-test).
Incorporation of α-actinin into FA occurs after paxillin at a stage of FA maturation when the adhesions are more stable (51). Immunofluorescence analysis of α-actinin distribution after 16 h revealed larger structures in PTPα−/− fibroblasts that colocalized with paxillin at the cell borders (Fig. 1A). These findings suggest that, in the absence of PTPα, additional FA proteins accumulate but cannot be redistributed from FA.
To obtain a more detailed kinetic analysis of α-actinin recruitment to FA, we transiently transfected PTPαwt and PTPα−/− fibroblasts with GFP-α-actinin and studied its subcellular distribution over 1–3 h of cell spreading (Fig. 1D). PTPαwt cells displayed relatively few (classical) mature FA but exhibited small aggregates of α-actinin that are typical of focal complexes in areas of active FA remodeling (44). By contrast, after 1 h of spreading, PTPα−/− cells displayed larger structures containing α-actinin that were restricted to the cell periphery (Fig. 1D). After 3 h of spreading, these α-actinin-containing structures did not disassemble but, rather, elongated further, delineating the border of the PTPα−/− cells. This phenotype is reminiscent of previously described nonmotile edges that are demarcated by concave bundles of actin filaments (44, 55).
To characterize the architecture of the actin cytoskeleton associated with these distinctive enlarged FA, we stained PTPαwt and PTPα−/− cells with FITC-phalloidin (Fig. 1E). Whereas PTPαwt cells displayed well-formed actin stress fibers throughout the cell, PTPα−/− cells exhibited the elongated bundles of F-actin typical of nonmotile edges (44). In accordance with the concept that FA form at the termini of stress fibers (44), the more generalized distribution of stress fibers in PTPαwt cells correlated with the presence of smaller and more dispersed FA. However, in PTPα−/− cells, the bundles of actin stress fibers were primarily restricted to the cell edges, mirroring the distribution of the supermature FA. Quantitative analysis revealed significant differences between the number of PTPαwt and PTPα−/− cells displaying leading edges (Fig. 1F). Thus, by regulating the dynamics and distribution of FA, PTPα also appears to affect the localization of stress fibers intimately associated with the FA (3).
PTPα is required for rapid remodeling of α-actinin in FA.
We studied the role of PTPα in the dynamics of FA remodeling in more detail by transiently transfecting PTPαwt and PTPα−/− fibroblasts with GFP-α-actinin. Using FRAP of GFP-α-actinin, we analyzed the stability of FA. An ∼10- to 18-μm2 area was photobleached, and fluorescence in this area was measured over the ensuing 290 s (Fig. 2A). Fluorescence recovery of GFP-α-actinin was substantially slower in PTPα−/− than in PTPαwt cells (t1/2 = 35.8 ± 12 vs. 68.8 ± 15 s; Fig. 2, B and C), indicating that the fraction of α-actinin associated with the enlarged FA is more stable in PTPα−/− than in PTPαwt cells. Thus small aggregates of α-actinin in wild-type cells undergo more rapid remodeling than α-actinin associated with the enlarged FA in PTPα−/− cells, suggesting that PTPα enables turnover of FA components, which favors their recycling and redistribution into small nascent focal complexes.
To validate the observations in PTPα−/− cells, we employed siRNA to knock down PTPα expression in human gingival fibroblasts and studied FA formation. The use of siRNA for PTPα resulted in substantial reduction of PTPα protein compared with cells treated with control siRNA (Fig. 2D) and led to the accumulation of paxillin in enlarged and elongated FA that localized at the periphery of spreading cells (Fig. 2E, arrows). Quantification revealed fewer FA in fibroblasts treated with siRNA against PTPα (Fig. 2F). These FA were also enlarged (Fig. 2G), indicating an inability of the cells to redistribute FA proteins into smaller focal complexes/FA. Furthermore, siRNA-treated cells were unable to spread and form membrane protrusions, in contrast to cells transfected with control siRNA (Fig. 2H). These results support the validity of our previous observations on the importance of PTPα in FA remodeling during cell spreading.
Membrane proximal (D1) catalytic domain of PTPα is required for turnover of FA.
As an independent approach to assess the involvement of PTPα in FA turnover and to evaluate the specific role of the D1 and D2 catalytic domains of PTPα in these processes, we transiently expressed the following fusion proteins in PTPα−/− fibroblasts: 1) wild-type PTPα (HA-PTPαwt), 2) PTPα lacking the D2 domain (D1/ΔD2), 3) PTPα lacking the D1 domain (ΔD1/D2), and 4) PTPα lacking D1 and D2 domains (ΔD1/ΔD2). Inasmuch as the constructs were tagged with HA, transfected cells were identified by HA immunostaining. To avoid possible artifacts of overexpression, only cells expressing low, but detectable, amounts of fusion protein were chosen for analysis. To study the effect of these PTPα mutants on FA formation and maturation, cells were transfected and stained with anti-paxillin antibodies (Fig. 3A). PTPα−/− cells transfected with GFP alone (not illustrated) or exposed to the vehicle control (Fig. 3A) without plasmid DNA displayed enlarged and arrowhead-shaped FA that did not remodel and were indistinguishable from the untreated PTPα−/− cells. By contrast, the expression of PTPα with a functional D1 catalytic domain (wild-type or the D1/ΔD2 mutant) in PTPα−/− cells was sufficient to restore turnover of FA proteins and prevent formation of supermature FA, as assessed by paxillin immunostaining (Fig. 3A). However, PTPα−/− cells in which mutant PTPα with deletion of the D1 domain (ΔD1/D2) or both catalytic domains (ΔD1/ΔD2) was expressed displayed enlarged FA with an arrowhead appearance that did not remodel, similar to the untransfected PTPα−/− cells (Fig. 3A).
Fig. 3.
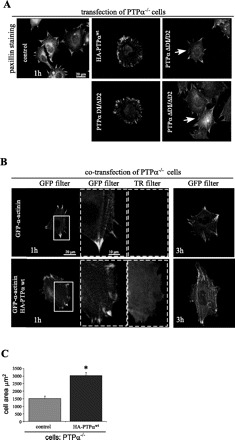
Transfection of PTPα rescues FA turnover in PTPα−/− cells. A: transient transfection of PTPα−/− cells with hemagglutinin (HA)-tagged PTPα constructs. Cells were plated on fibronectin for 1 h and then stained with anti-paxillin antibodies. Transfected cells were identified by staining with anti-HA antibodies using a specific Texas Red (TR) secondary antibody (not illustrated). Arrows indicate supermature FA. D1/ΔD2, PTPα lacking D2 domain; ΔD1/D2, PTPα lacking D1 domain; ΔD1/ΔD2, PTPα lacking D1 and D2 domains. B: transient transfection of PTPα−/− cells with HA-PTPαwt. FA were visualized by cotransfection with GFP-α-actinin. Fibroblasts were allowed to spread for 1 and 3 h. FA turnover in rescued PTPα−/− cells is reflected by formation of smaller and more widely distributed FA. C: cell area after 3 h of spreading. Values are means ± SD of 50 measurements in 3 independent experiments. *P < 0.01 vs. control (by unpaired Student's t-test).
Similarly, transfection of HA-PTPαwt into the PTPα−/− cells restored α-actinin incorporation into FA that were smaller than those in cells exposed to the vehicle control (Fig. 3B). Additionally, when the cells were allowed to spread for 3 h on fibronectin, expression of wild-type PTPα restored normal membrane protrusion and cell spreading (Fig. 3B). As anticipated, the mean cell area increased in the PTPα−/− cells transfected with HA-PTPαwt (Fig. 3C). We conclude that a functional (catalytically active) D1 domain of PTPα is required for FA remodeling and cell spreading.
PTPα phosphatase activity and FA remodeling, membrane protrusion, and cell polarization.
In addition to catalytic activity, the D1 domain of PTPα contains potential protein binding sites, raising the possibility that deletion of this domain might disrupt protein-protein interactions that are involved in FA dynamics. To examine more specifically the role of the catalytic (phosphatase) activity of PTPα in FA remodeling, we exploited previously characterized NIH-3T3 cells lines that inducibly express catalytically inactive PTPα with point mutations in the catalytic domains (C433S/C723S) (58). Wild-type PTPα (PTPαwt ind) was expressed as a control. When fully induced, the cells express up to 15 times the endogenous level of PTPα functioning as a DN (58). For the experiments described here, the PTPα transgene was induced ∼6–10 times the endogenous level as determined by Western blotting (not illustrated) to avoid artifacts from overexpression. We monitored alterations in cell morphology and area and FA length, number, and localization by paxillin immunostaining in cells spreading on fibronectin for 3 h (Fig. 4A). Cells overexpressing modest levels (6–10 times) of recombinant wild-type PTPα displayed classical FA assembly and dynamics that correlated with formation of membrane protrusions, cell spreading, and polarization comparable to untransfected wild-type cells (Fig. 4A). In contrast, the morphology of FA in wild-type cells expressing PTPαCCSS ind was reminiscent of the pattern in PTPα−/− cells, with predominantly large (supermature) and peripherally distributed FA that did not remodel (Fig. 4A). Measurement of the area of cells from images shown in Fig. 4A confirmed that fibroblasts expressing PTPαCCSS ind failed to spread (Fig. 4B). These cells also displayed enlarged peripherally distributed FA compared with cells expressing wild-type PTPα (Fig. 4, C and D). As an indicator of cell polarization, we measured cell length and width and calculated a shape factor (Fig. 4E) and found that cells expressing PTPαCCSS ind were rounder than cells expressing PTPαwt ind. Moreover, cells expressing PTPαCCSS ind and transfected with GFP-α-actinin (Fig. 4F) showed accumulation of α-actinin in enlarged and elongated FA, indicating slower remodeling of FA, consistent with the results obtained using paxillin immunostaining.
Fig. 4.
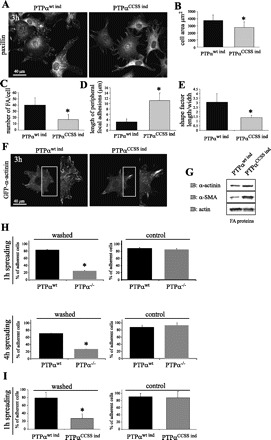
PTPα phosphatase activity is required for FA remodeling. A: genetically modified NIH-3T3 cells lines induced to express wild-type human PTPα (PTPαwt ind) and double-mutant Cys433 → Ser/Cys723 → Ser PTPα (PTPαCCSS ind). Cells were plated on fibronectin for 3 h. FA were observed by staining for paxillin (A) or by transfection with GFP-α-actinin (F). B–E: cell area, number of FA per cell, length of peripheral FA, and shape factor (length vs. width), which indicates degree of polarization, from images in A. Values are means ± SD of 30–40 measurements in 3 independent experiments. *P < 0.01 vs. PTPαwt ind (by unpaired Student's t-test). G: Western blot analysis of FA fraction (FA proteins). Note greater amounts of α-smooth muscle actin (α-SMA) and α-actinin in cells expressing PTPαCCSS ind. Levels of β-actin did not vary considerably and served as control. H and I: PTPα phosphatase activity is required for efficient adhesion to fibronectin. Cell adhesion was measured in primary PTPαwt and PTPα−/− murine fibroblasts (H) and genetically modified NIH-3T3 cells expressing PTPαwt ind and PTPαCCSS ind (I). Cells were stained with calcein-AM and plated on fibronectin for 1 or 4 h and then washed 5 times with PBS. Control cells were not washed. Calcein-AM fluorescence of the remaining cells was measured using a microplate fluorescence reader. Values of intensity were normalized to a percentage of control. Values are means ± SD of 16 tests per cell line in 3 independent experiments. *P < 0.05 vs. PTPαwt ind or PTPαwt.
To complement this morphological analysis, we utilized a biochemical approach. To isolate FA proteins, we used beads coated with fibronectin that were added to the dorsal surface of the cells (see materials and methods) to induce the formation of FA-like structures. Immunoblotting of bead-associated proteins revealed more α-actinin and α-SMA in cells expressing PTPαCCSS ind than in PTPαwt ind cells (Fig. 4G). These results illustrate the importance of the phosphatase activity of PTPα in the turnover of FA proteins, which facilitates their remodeling during cell spreading and polarization.
PTPα phosphatase activity is required for cell adhesion.
The abnormalities in FA structure and dynamic remodeling observed in cells expressing mutant PTPα prompted us to study the importance of the enzymatic (phosphatase) activity of PTPα in cell adhesion. We compared adhesion in PTPαwt and PTPα−/− cells (Fig. 4H) and in NIH-3T3 cells expressing recombinant wild-type PTPα (PTPαwt ind) or catalytically inactive PTPαCCSS ind (Fig. 4I). Fibroblasts were allowed to adhere to a fibronectin-coated surface for 1 h and washed, and the number of remaining cells was quantified (see materials and methods). Cells not subjected to washing were used as controls. After 1 h, 82 ± 1.87% of PTPαwt cells were adherent vs. 23.6 ± 0.82% of PTPα−/− cells. Similar results were obtained when the cells were allowed to adhere and spread for 4 h (76.8 ± 1.24% and 28.7 ± 0.57% of PTPαwt and PTPα−/− cells, respectively), indicating that the decreased adhesion was not simply due to a delay in the adhesive process. Similarly, cells expressing PTPαCCSS ind were less adherent than PTPαwt ind cells after 1 h of spreading (Fig. 4I). These observations indicate the importance of PTPα and, specifically, its catalytic activity in cell adhesion during spreading.
PTPα is required for contraction of ECM.
We considered that the alterations in the distribution of contractile bundles of actin stress fibers associated with the reduced turnover of FA in cells deficient in PTPα would be reflected in their capacity to contract ECM (3, 19). We initially used a “two-step stressed” model in which fibroblasts in collagen gels are first allowed to attach to plastic tissue culture wells while the cells spread and form FA. This is followed by release of the gels from the substrate. This model system allows cells to contract the gel when there is no longer a rigid substrate to maintain isometric tension via a mechanism involving stress fibers (17, 49). After release from the substrate, PTPαwt cells were able to contract the gels to a significantly greater extent than PTPα−/− cells (Fig. 5A), indicating that the aberrant formation of FA and stress fibers in PTPα−/− cells described above interfered with transmission of tensile forces to the ECM and, thus, with gel contraction. Alternatively, the magnitude of traction forces generated by cells can be assessed in “floating” collagen matrices (17). In this system, cells are initially rounded and contract the gels as they spread or migrate, thereby allowing assessment of the role of FA dynamics in ECM contraction. In these floating collagen gels, PTPαwt cells induced significantly more contraction than PTPα−/− cells (Fig. 5B). Together, these observations underscore the importance of PTPα in formation of contractile stress fibers and in the generation of traction forces that lead to ECM contraction.
Fig. 5.
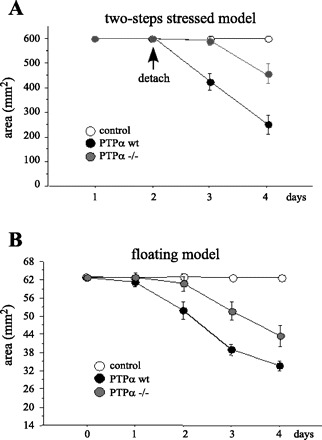
PTPα is required for collagen gel contraction. A: 2 × 105 PTPαwt and PTPα−/− cells were embedded in polymerized collagen gel, deposited as 1-ml aliquots over 24-well tissue culture dishes, and covered with normal medium. Gels were detached after 1 day. B: 2.29 × 104 PTPαwt and PTPα−/− cells were embedded in polymerized collagen gel, deposited as 200-μl aliquots over nontissue culture dishes, and covered with tissue culture medium. Diameter of each gel was measured on 4 consecutive days. Values are means ± SD of 3–5 collagen gels per cell line in 3 independent experiments.
PTPα regulates Rac1 activation and localization to FA during cell spreading.
The alterations of FA morphology and turnover observed in cells deficient in PTPα are similar to those described in cells deficient in the small GTPase Rac1, which is also known to regulate FA remodeling (18, 48) and to be a critical modulator of cell-matrix adhesion and cell migration (1, 32, 36). Given these similarities, we questioned whether the effects of PTPα on FA dynamics might be mediated via Rac1. To address this issue, we compared Rac1 activation in NIH-3T3 cells expressing recombinant wild-type (PTPαwt ind) or catalytically inactive PTPαCCSS ind that were allowed to spread for 1, 3, and 6 h. These studies revealed that Rac1 activation was markedly diminished in cells expressing PTPαCCSS ind (functioning in a DN fashion) compared with cells expressing wild-type PTPα (Fig. 6A). Interestingly, attachment and cell spreading induced a progressive translocation of Rac1 to the Triton X-100-insoluble (cytoskeletal) fraction in PTPαwt ind cells, but not in PTPαCCSS ind cells (Fig. 6A). Densitometry analysis in cells spreading for 6 h confirmed significant differences in Rac1 activation between PTPαwt ind and PTPαCCSS ind cells (Fig. 6B). The cytoskeletal fraction of cells that spread for 16 h (Fig. 6C) also contained higher amounts of Rac1 when the phosphatase activity of PTPα was intact (PTPαwt ind) than in cells expressing PTPαCCSS ind. Isolation of FA-associated proteins using fibronectin-coated beads (see materials and methods) confirmed that Rac1 was recruited to FA during cell spreading in PTPαwt ind cells, but not in cells expressing PTPαCCSS ind (Fig. 6c).
Fig. 6.
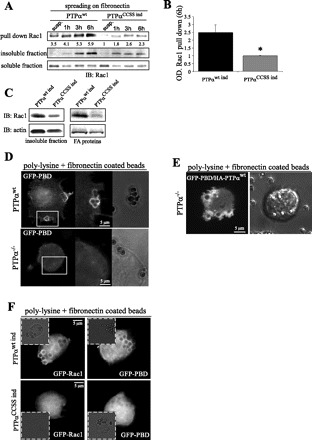
PTPα is required for Rac1 activation and localization at cell-matrix contacts. A: genetically modified NIH-3T3 cells expressing PTPαwt ind and PTPαCCSS ind were allowed to spread for 1, 3, and 6 h or kept in suspension (susp). Pull-down assay with glutathione S-transferase-tagged p21-activated kinase (PAK1)-binding domain (PBD) was used to purify active Rac1 from whole cell lysates, and Rac1 binding was analyzed by Western blotting. B: densitometry analysis of blot in A. OD, optical density. Values are means ± SD of 3 independent experiments. *P < 0.01 vs. PTPαwt ind (by unpaired Student's t-test). C: Western blot of Rac1 and actin in PTPαwt ind- and PTPαCCSS ind-expressing cells plated for 16 h. Triton X-100-insoluble fraction and FA-associated proteins are shown. D: images acquired from PTPαwt and PTPα−/− fibroblasts transiently transfected with GFP-tagged PBD and allowed to spread on fibronectin-coated beads for 30 min. E: images acquired from PTPα−/− cells cotransfected with GFP-PBD and HA-PTPαwt vectors and allowed to spread on fibronectin-coated beads for 30 min. F: images acquired from PTPαwt ind- and PTPαCCSS ind-expressing cells transfected with GFP-Rac1 or GFP-PBD and allowed to spread on fibronectin-coated beads for 30 min.
Activated Rac1 associates with the effector protein PAK1, inducing its activation and translocation to membrane ruffles and lamellipodia (33). We studied the localization of GFP-Rac1 and GFP-PBD (a marker for activated Rac1 and Cdc42) in PTPαwt and PTPα−/− cells and in PTPαwt ind and PTPαCCSS ind cells (Fig. 6, D and F). Fibroblasts were allowed to spread on a poly-l-lysine substrate [conditions that do not support FA formation (28)] containing fibronectin-coated beads. GFP-PBD localized to the sites of the cell-bead contact in PTPαwt, but not PTPα−/−, cells (Fig. 6D). The role of PTPα in this process was confirmed by reintroduction of HA-PTPαwt into PTPα−/− cells (Fig. 6E), which restored the wild-type phenotype (cf. Fig. 6, D and E). By contrast, GFP-Rac1 and GFP-PBD localization to the FA-bead sites was abolished in cells expressing PTPαCCSS ind compared with cells expressing PTPαwt ind (Fig. 6F) or PTPαwt cells (Fig. 6D). These data indicate that the phosphatase activity of PTPα is required for recruitment of Rac1 to FA and for its activation.
PTPα-induced FA remodeling requires Rac1.
If Rac1 induces FA remodeling, as previous studies have suggested, then cells deficient in Rac1 should also develop enlarged (supermature) FA that are suggestive of impaired FA turnover. We used conditional Rac1C/C cells that were treated with the peptide HNTC to generate Rac1-null fibroblasts or were exposed to the vehicle control (see materials and methods). After they were allowed to spread for 16 h on fibronectin, cells were immunostained with antibodies to paxillin and α-actinin as markers of FA distribution (Fig. 7A). Rac1C/C cells treated with the peptide HTNC (Rac1-null) developed elongated FA containing α-actinin (Fig. 7, A and B). Furthermore, these cells failed to spread, showing a phenotype analogous to that of cells deficient in PTPα (Fig. 7C).
To examine the involvement of Rac1 in the signaling pathway downstream of PTPα that regulates FA remodeling, we transfected PTPα−/− cells with constitutively active (CA) L61 Rac1 (Fig. 7D). The distribution of recombinant CA-Rac1 was diffusely cytosolic (not illustrated). Transfection of CA-Rac1 into PTPα−/− cells resulted in the redistribution of paxillin into smaller and more numerous focal complexes/FA (Fig. 7D). Quantification of FA (Fig. 7E) and the length of peripheral FA (Fig. 7F) confirmed the effect of CA-Rac1 in this response. Similarly, restoration of spreading-associated α-actinin (Fig. 7G) and actin (Fig. 7H) redistribution were observed in cells cotransfected with GFP-α-actinin and CA-Rac1 (Fig. 7G). This result indicates that active Rac1 can restore the normal formation and dynamic remodeling of FA and the actin cytoskeleton in PTPα−/− cells, which enables cells to spread and polarize.
To confirm the role of Rac1 in the turnover of FA, we repeated the FRAP analysis (similar to that in Fig. 2), but in this experiment the cells were transfected with CA-Rac1 (Fig. 7I). As predicted, t1/2 of the PTPα−/− cells was not different from that of PTPαwt cells under these conditions (cf. Fig. 7, J and K, and Fig. 2, B and C). These data provide additional evidence that Rac1 is downstream of PTPα in the signaling pathway, which modulates the turnover of FA proteins.
DISCUSSION
Cell spreading and motility require the dynamic remodeling of existing FA followed by their reassembly at a new location (42). Nascent FA (focal complexes) that form during initial attachment or after remodeling can undergo progressive maturation into larger and more stable (classical) FA. These adhesive structures form linkages with the ECM and facilitate the generation of traction forces necessary for locomotion (54). Cell motility requires de novo formation and maturation of FA, as well as remodeling of existing FA, which help direct the formation of membrane protrusions, establishment of cell polarity, adhesion to the substratum, and translocation of the cell body. The molecular mechanisms that regulate FA dynamics during these processes are incompletely understood. Here, we provide evidence that the enzymatic (phosphatase) activity of PTPα is requisite for dynamic remodeling of FA in the leading edge of motile cells. Our data indicate that, in the absence of functional PTPα, FA accumulate proteins such as α-actinin and α-SMA, likely because of slower turnover, and form enlarged adhesive structures, thereby impairing cell spreading and motility. Finally, we demonstrate that the mechanism by which PTPα modulates FA turnover involves the small GTPase Rac 1.
PTPα phosphatase activity is required for FA turnover.
Previous studies have documented that cells deficient in PTPα exhibit impaired formation of nascent FA (focal complexes) during the initial phases of attachment, as well as a reduced ability to spread and migrate (52, 57). Our data extend these observations and explain in part how PTPα deficiency interferes with the formation of classical FA. We provide evidence that the phosphatase activity of PTPα modulates FA primarily by regulating their dynamic remodeling. We monitored the early (0–60 min) phase of FA formation during cell spreading, as well as the later stages (up to 16 h), during which FA mature and reorganize. Although PTPα−/− cells, human gingival fibroblasts treated with PTPα (PTPRA) siRNA, and fibroblasts expressing phosphatase-deficient PTPα C433S/C723S were able to form FA, these multimolecular structures failed to turn over and continued to enlarge and accumulate additional FA proteins, including paxillin, α-actinin, and α-SMA. Additionally, these enlarged FA assumed an elongated morphology and became localized to the periphery of the cells. FA proteins in these enlarged FA failed to redistribute, even after prolonged times (up to 16 h), revealing a high degree of stability, as assessed by FRAP analysis of GFP-α-actinin. We interpret these data to mean that the absence of PTPα results in stabilization of actin-containing stress fibers and enlargement of FA, possibly by retarding their turnover (13, 38, 53, 54). Furthermore, we were able to rescue FA turnover by introduction of recombinant wild-type PTPα or recombinant PTPα with a functional D1 catalytic domain (D1/ΔD2) into PTPα−/− cells (Fig. 3). Collectively, these observations indicate that the phosphatase activity of the D1 domain of PTPα is required for the formation of motile leading edges and establishment of cell polarization, events that are dependent on FA remodeling (Figs. 1 and 4).
Dynamic FA remodeling enabled by PTPα is required for adhesion and generation of traction forces.
Our experiments reveal an important role for PTPα and, specifically, its phosphatase activity in cell adhesion and transmission of traction forces to the substratum leading to ECM contraction. Previous studies have suggested that PTPα regulates the stretching of integrin-cytoskeleton bonds and the transduction of mechanical forces through the activation of the Src kinase family member Fyn (52). However, this must be reconciled with the evidence that Src, which is also activated by PTPα, inhibits reinforcement of FA and promotes their turnover (12, 13, 38). Our observations provide a potential explanation for these apparently contradictory results: we propose that generation of traction forces requires continued cycles of FA assembly and maturation that are coupled with their turnover. These cycles are enabled by PTPα. Dynamic maturation of FA is required to attain a critical level of tension that facilitates incorporation of α-SMA into FA, leading to further maturation and contraction of ECM (16). We observed that although cells deficient in PTPα were able to form large FA that contained α-SMA and α-actinin along the cell borders, their inability to turn over correlated with decreased ECM contraction, indicating that dynamic remodeling of FA is necessary for these processes. Deficiency of PTPα prevented the contraction of collagen gels either related to an inability to form and turn over classical FA and stress fibers during spreading (in the 2-step stressed model) or to the generation of traction forces associated with cell movement [in the floating gel model (Fig. 5)] (17). In addition, our experiments reveal that, in the presence of functional PTPα, FA are more diffusely distributed on the ventral cell surface in association with contractile stress fibers; this distribution appears to be fundamental to the ability of cells to exert tractional forces on the ECM, resulting in gel contraction (23). In the absence of functional PTPα, enlarged FA and stress fibers are restricted to the cell periphery. In support of our observations, Gupton and Waterman-Storer (19) recently proposed that the effects of tensile force (stretch) on FA and the distribution of FA combined with local myosin II activity in the contractile module direct the specific spatiotemporal organization states of F-actin.
PTPα regulation of FA involves Rac1.
The assembly and disassembly of the actin-based structures lamellipodia and filopodia at the leading edge of the cells are under the control of Rac1 and Cdc42 (32, 40). Rac1 regulates FA turnover or remodeling (6, 14, 42) and may act upstream of RhoA in the formation of actin stress fibers and stabilization of FA (18, 40). Here we demonstrate that PTPα regulates Rac1 activation during cell spreading, which, in turn, is linked to FA remodeling. Consequently, transfection of constitutively active Rac1 into PTPα−/− cells restored the remodeling and turnover of the enlarged (supermature) FA present in these cells and the reconstitution of classical FA formation and dynamics. Despite the wide cellular distribution of Rac1 effectors such as PAK1 and of active Cdc42/Rac1, the physical association of Cdc42 and Rac1 occurs only at specific sites in the cell periphery (8). In our experiments, PTPα was required for the translocation of Rac1 to the cytoskeleton and to FA. It has been shown that, during the early phases of cell spreading, interaction of PTPα with αv-integrins triggers downstream signaling events that may lead to Rac1 activation (52, 57). Our studies indicate that the catalytic activity of PTPα during cell spreading may mediate the recruitment of Rac1 to FA, releasing Rac1 from its association with Rho-GDP dissociation inhibitor (15, 33) and inducing its subsequent binding to PAK1. We observed that GFP-Rac1 and GFP-PAK1 binding domain colocalized with fibronectin-coated beads only in cells expressing functional PTPα.
Although the mechanism by which PTPα induces Rac1 activation remains to be clarified, it is likely that it occurs through the upstream activation of Src family kinases and FAK. The role of PTPα in the activation of Src family kinases, FAK and p130Cas, is well known (7, 21, 24, 39, 46, 58). Interaction of p130CAS with Crk can lead to Rac1 activation via translocation of the Rac1 activator DOCK180 to FA (27). Alternatively, phosphorylation of paxillin induced by Src leads to Crk binding, which enhances association of paxillin with the Rac1 exchange factor β-PIX (26, 47). Recently, it has been shown that FAK phosphorylates β-PIX on tyrosine and, thereby, increases its binding to Rac1 (6). Moreover, the paxillin-G protein-coupled receptor kinase-interacting protein type 2-β-PIX complex also binds PAK1, which in turn is recruited to the FA, where it can promote FA disassembly (2, 29). Further studies are required to determine whether these signaling pathways link PTPα with Rac1.
In summary, we provide evidence that the enzymatic activity of PTPα is required for the dynamic remodeling of FA, which in turn directs cell polarization and spreading. Furthermore, we propose that the ability of PTPα to direct the localization of Rac1 to FA is required for the remodeling of static FA sites into smaller more dynamic focal complexes. Thus, via activation of Rac1, PTPα promotes the dynamic remodeling of FA and their widespread distribution on the ventral surface, which are essential for cell spreading, motility, and transmission of force to the ECM.
GRANTS
This study was supported by an operating grant from the Canadian Institutes of Health Research to G. P. Downey and C. A. McCulloch.
Acknowledgments
We thank Drs. Joeren den Hertog and Jan Sap for providing critical reagents and for helpful discussions.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Allen WE, Jones GE, Pollard JW, Ridley AJ. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J Cell Sci 110: 707–720, 1997. [DOI] [PubMed] [Google Scholar]
- 2.Bagrodia S, Taylor SJ, Jordon KA, Van Aelst L, Cerione RA. A novel regulator of p21-activated kinases. J Biol Chem 273: 23633–23636, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Bershadsky AD, Ballestrem C, Carramusa L, Zilberman Y, Gilquin B, Khochbin S, Alexandrova AY, Verkhovsky AB, Shemesh T, Kozlov MM. Assembly and mechanosensory function of focal adhesions: experiments and models. Eur J Cell Biol 85: 165–173, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol 12: 463–518, 1996. [DOI] [PubMed] [Google Scholar]
- 5.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell 116: 167–179, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Chang F, Lemmon CA, Park D, Romer LH. FAK potentiates Rac1 activation and localization to matrix adhesion sites: a role for βPIX. Mol Biol Cell 18: 253–264, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen M, Chen SC, Pallen CJ. Integrin-induced tyrosine phosphorylation of protein-tyrosine phosphatase-α is required for cytoskeletal reorganization and cell migration. J Biol Chem 281: 11972–11980, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Del Pozo MA, Kiosses WB, Alderson NB, Meller N, Hahn KM, Schwartz MA. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol 4: 232–239, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Den Hertog J, Pals CE, Peppelenbosch MP, Tertoolen LG, de Laat SW, Kruijer W. Receptor protein tyrosine phosphatase-α activates pp60c-src and is involved in neuronal differentiation. EMBO J 12: 3789–3798, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dugina V, Fontao L, Chaponnier C, Vasiliev J, Gabbiani G. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J Cell Sci 114: 3285–3296, 2001. [DOI] [PubMed] [Google Scholar]
- 11.El Sayegh TY, Arora PD, Fan L, Laschinger CA, Greer PA, McCulloch CA, Kapus A. Phosphorylation of N-cadherin-associated cortactin by Fer kinase regulates N-cadherin mobility and intercellular adhesion strength. Mol Biol Cell 16: 5514–5527, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felsenfeld DP, Schwartzberg PL, Venegas A, Tse R, Sheetz MP. Selective regulation of integrin-cytoskeleton interactions by the tyrosine kinase Src. Nat Cell Biol 1: 200–206, 1999. [DOI] [PubMed] [Google Scholar]
- 13.Fincham VJ, Frame MC. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J 17: 81–92, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank SR, Adelstein MR, Hansen SH. GIT2 represses Crk- and Rac1-regulated cell spreading and Cdc42-mediated focal adhesion turnover. EMBO J 25: 1848–1859, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gandhi PN, Gibson RM, Tong X, Miyoshi J, Takai Y, Konieczkowski M, Sedor JR, Wilson-Delfosse AL. An activating mutant of Rac1 that fails to interact with Rho GDP-dissociation inhibitor stimulates membrane ruffling in mammalian cells. Biochem J 378: 409–419, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, Hinz B. Focal adhesion size controls tension-dependent recruitment of α-smooth muscle actin to stress fibers. J Cell Biol 172: 259–268, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grinnell F. Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol 13: 264–269, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Guo F, Debidda M, Yang L, Williams DA, Zheng Y. Genetic deletion of Rac1 GTPase reveals its critical role in actin stress fiber formation and focal adhesion complex assembly. J Biol Chem 281: 18652–18659, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell 125: 1361–1374, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Hall A. Rho GTPases and the actin cytoskeleton. Science 279: 509–514, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Harder KW, Moller NP, Peacock JW, Jirik FR. Protein-tyrosine phosphatase-α regulates Src family kinases and alters cell-substratum adhesion. J Biol Chem 273: 31890–31900, 1998. [DOI] [PubMed] [Google Scholar]
- 22.Herrera Abreu MT, Wang Q, Vachon E, Suzuki T, Chow CW, Wang Y, Hong O, Villar J, McCulloch CA, Downey GP. Tyrosine phosphatase SHP-2 regulates IL-1 signaling in fibroblasts through focal adhesions. J Cell Physiol 207: 132–143, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Hu K, Ji L, Applegate KT, Danuser G, Waterman-Storer CM. Differential transmission of actin motion within focal adhesions. Science 315: 111–115, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Kostic A, Sheetz MP. Fibronectin rigidity response through Fyn and p130Cas recruitment to the leading edge. Mol Biol Cell 17: 2684–2695, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lammers R, Lerch MM, Ullrich A. The carboxyl-terminal tyrosine residue of protein-tyrosine phosphatase-α mediates association with focal adhesion plaques. J Biol Chem 275: 3391–3396, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Lamorte L, Rodrigues S, Sangwan V, Turner CE, Park M. Crk associates with a multimolecular paxillin/GIT2/β-PIX complex and promotes Rac-dependent relocalization of paxillin to focal contacts. Mol Biol Cell 14: 2818–2831, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Guris DL, Okura M, Imamoto A. Translocation of CrkL to focal adhesions mediates integrin-induced migration downstream of Src family kinases. Mol Cell Biol 23: 2883–2892, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacGillivray M, Herrera-Abreu MT, Chow CW, Shek C, Wang Q, Vachon E, Feng GS, Siminovitch KA, McCulloch CA, Downey GP. The protein tyrosine phosphatase SHP-2 regulates interleukin-1-induced ERK activation in fibroblasts. J Biol Chem 278: 27190–27198, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, Tan I, Leung T, Lim L. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol Cell 1: 183–192, 1998. [DOI] [PubMed] [Google Scholar]
- 30.McCulloch CA, Downey GP, El-Gabalawy H. Signalling platforms that modulate the inflammatory response: new targets for drug development. Nat Rev Drug Discov 5: 864–876, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol 144: 1235–1244, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81: 53–62, 1995. [DOI] [PubMed] [Google Scholar]
- 33.Parrini MC, Matsuda M, de Gunzburg J. Spatiotemporal regulation of the Pak1 kinase. Biochem Soc Trans 33: 646–648, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Partridge MA, Marcantonio EE. Initiation of attachment and generation of mature focal adhesions by integrin-containing filopodia in cell spreading. Mol Biol Cell 17: 4237–4248, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peitz M, Pfannkuche K, Rajewsky K, Edenhofer F. Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: a tool for efficient genetic engineering of mammalian genomes. Proc Natl Acad Sci USA 99: 4489–4494, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pestonjamasp KN, Forster C, Sun C, Gardiner EM, Bohl B, Weiner O, Bokoch GM, Glogauer M. Rac1 links leading edge and uropod events through Rho and myosin activation during chemotaxis. Blood 108: 2814–2820, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrone A, Battaglia F, Wang C, Dusa A, Su J, Zagzag D, Bianchi R, Casaccia-Bonnefil P, Arancio O, Sap J. Receptor protein tyrosine phosphatase-α is essential for hippocampal neuronal migration and long-term potentiation. EMBO J 22: 4121–4131, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene 23: 7928–7946, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Ponniah S, Wang DZ, Lim KL, Pallen CJ. Targeted disruption of the tyrosine phosphatase PTPα leads to constitutive downregulation of the kinases Src and Fyn. Curr Biol 9: 535–538, 1999. [DOI] [PubMed] [Google Scholar]
- 40.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70: 401–410, 1992. [DOI] [PubMed] [Google Scholar]
- 41.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science 302: 1704–1709, 2003. [DOI] [PubMed] [Google Scholar]
- 42.Rottner K, Hall A, Small JV. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr Biol 9: 640–648, 1999. [DOI] [PubMed] [Google Scholar]
- 43.Small JV, Resch GP. The comings and goings of actin: coupling protrusion and retraction in cell motility. Curr Opin Cell Biol 17: 517–523, 2005. [DOI] [PubMed] [Google Scholar]
- 44.Small JV, Rottner K, Kaverina I, Anderson KI. Assembling an actin cytoskeleton for cell attachment and movement. Biochim Biophys Acta 1404: 271–281, 1998. [DOI] [PubMed] [Google Scholar]
- 45.Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol 12: 112–120, 2002. [DOI] [PubMed] [Google Scholar]
- 46.Su J, Muranjan M, Sap J. Receptor protein tyrosine phosphatase-α activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr Biol 9: 505–511, 1999. [DOI] [PubMed] [Google Scholar]
- 47.Ten Klooster JP, Jaffer ZM, Chernoff J, Hordijk PL. Targeting and activation of Rac1 are mediated by the exchange factor β-Pix. J Cell Biol 172: 759–769, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Timpson P, Jones GE, Frame MC, Brunton VG. Coordination of cell polarization and migration by the Rho family GTPases requires Src tyrosine kinase activity. Curr Biol 11: 1836–1846, 2001. [DOI] [PubMed] [Google Scholar]
- 49.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349–363, 2002. [DOI] [PubMed] [Google Scholar]
- 50.Vidali L, Chen F, Cicchetti G, Ohta Y, Kwiatkowski DJ. Rac1-null mouse embryonic fibroblasts are motile and respond to platelet-derived growth factor. Mol Biol Cell 17: 2377–2390, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Von Wichert G, Haimovich B, Feng GS, Sheetz MP. Force-dependent integrin-cytoskeleton linkage formation requires downregulation of focal complex dynamics by Shp2. EMBO J 22: 5023–5035, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Von Wichert G, Jiang G, Kostic A, De Vos K, Sap J, Sheetz MP. RPTP-α acts as a transducer of mechanical force on αv/β3-integrin-cytoskeleton linkages. J Cell Biol 161: 143–153, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol 6: 154–161, 2004. [DOI] [PubMed] [Google Scholar]
- 54.Webb DJ, Parsons JT, Horwitz AF. Adhesion assembly, disassembly and turnover in migrating cells—over and over and over again. Nat Cell Biol 4: E97–E100, 2002. [DOI] [PubMed] [Google Scholar]
- 55.Wehrle-Haller B, Imhof B. The inner lives of focal adhesions. Trends Cell Biol 12: 382–389, 2002. [DOI] [PubMed] [Google Scholar]
- 56.Wu L, Buist A, den Hertog J, Zhang ZY. Comparative kinetic analysis and substrate specificity of the tandem catalytic domains of the receptor-like protein-tyrosine phosphatase-α. J Biol Chem 272: 6994–7002, 1997. [DOI] [PubMed] [Google Scholar]
- 57.Zeng L, Si X, Yu WP, Le HT, Ng KP, Teng RM, Ryan K, Wang DZ, Ponniah S, Pallen CJ. PTPα regulates integrin-stimulated FAK autophosphorylation and cytoskeletal rearrangement in cell spreading and migration. J Cell Biol 160: 137–146, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng XM, Resnick RJ, Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPα. EMBO J 19: 964–978, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]