

Abstract
Efficient clearance of apoptotic cells (efferocytosis) prevents inflammation and permits repair following tissue injury. Kidney injury molecule-1 (KIM-1) is a receptor for phosphatidylserine, an “eat-me” signal exposed on the surface of apoptotic cells that marks them for phagocytic clearance. KIM-1 is upregulated on proximal tubule epithelial cells (PTECs) during ischemic acute kidney injury (AKI), enabling efferocytosis by surviving PTECs. KIM-1 is spontaneously cleaved at its ectodomain region to generate a soluble fragment that serves a sensitive and specific biomarker for AKI, but the biological relevance of KIM-1 shedding is unknown. Here, we sought to determine how KIM-1 shedding might regulate efferocytosis. Using cells that endogenously and exogenously express KIM-1, we found that hydrogen peroxide-mediated oxidative injury or PMA treatment accelerated KIM-1 shedding in a dose-dependent manner. KIM-1 shedding was also accelerated when apoptotic cells were added. Accelerated shedding or the presence of excess soluble KIM-1 in the extracellular milieu significantly inhibited efferocytosis. We also identified that TNF-α-converting enzyme (TACE or ADAM17) mediates both the spontaneous and PMA-accelerated shedding of KIM-1. While accelerated shedding inhibited efferocytosis, we found that spontaneous KIM-1 cleavage does not affect the phagocytic efficiency of PTECs. Our results suggest that KIM-1 shedding is accelerated by worsening cellular injury, and excess soluble KIM-1 competitively inhibits efferocytosis. These findings may be important in AKI when there is severe cellular injury.
Keywords: kidney injury molecule-1 (KIM-1), efferocytosis, receptor, shedding, proximal tubule epithelial cells (PTECs)
acute kidney injury (AKI) occurs in up to 7% of hospitalized patients and is associated with high mortality and morbidity (13, 19). Ischemia-reperfusion injury is the most common cause of AKI, whereby tissue damage is caused by the generation of reactive oxygen species (ROS), as blood flow is reestablished after a period of ischemia (4, 71). Renal tubule epithelial cells represent the most abundant cell type in the kidney, with those in the proximal tubular region being particularly sensitive to oxygen deprivation and ROS-mediated injury (20). Proximal tubule epithelial cells (PTECs) die by one of two mechanisms: necrosis or apoptosis. Characteristics of necrosis are loss of cell membrane integrity, release of intracellular contents into the surrounding tissues, and activation of innate immune responses that can exacerbate tissue injury and limit tissue repair (18, 51, 62a, 70a, 73a). Conversely, apoptosis is a highly regulated form of cell death, leading to DNA fragmentation, cytoplasmic condensation, and production of apoptotic bodies that are efficiently cleared by phagocytes before cell membrane integrity is breached, thus avoiding significant inflammatory responses (41, 42). The phagocytic clearance of apoptotic cells, known as efferocytosis, is believed to be critical in regulating inflammation, a process likely of utmost importance after acute tissue injury (5, 16, 35, 40, 51). Furthermore, dying PTECs that are extruded from the basement membrane can obstruct the tubular lumen and contribute further to renal dysfunction during severe AKI (69).
Kidney injury molecule-1 (KIM-1) (36), also known as T cell immunoglobulin mucin domain-1 (TIM-1) (52), is a type 1 transmembrane glycoprotein that is undetectable in the healthy kidney, but whose expression is highly upregulated on the apical surface of PTECs during AKI (25, 32, 36). Recently, we (35) and others (44, 66) uncovered that KIM-1 is a receptor for phosphatidylserine (PtdSer). PtdSer is an “eat-me” signal displayed on the cell surface of apoptotic cells that marks them for removal by efferocytosis (24). Structurally, KIM-1 is made up of an immunoglobulin (Ig) domain that contains its metal-ion-dependent PtdSer binding site, a mucin domain, a transmembrane domain, and a cytosolic domain that contains a tyrosine kinase phosphorylation motif (45, 65). KIM-1 expression on the apical surface of PTECs enables them to recognize and engulf not only apoptotic but also necrotic material during AKI (35). In vivo, apoptotic cells and necrotic debris can be readily visualized during the process of engulfment within KIM-1-expressing PTECs in rodents subjected to renal ischemia-reperfusion injury (35). Although KIM-1-deficient mice have been generated and have no renal phenotype at baseline (76), their response to acute kidney injury has not been reported. Mice expressing a KIM-1 mucin domain deletion mutant, rendering it phagocytosis defective, are normal at baseline (79) but have been reported to exhibit severe inflammation and tissue injury after being subjected to renal ischemia-reperfusion injury (82a). It is believed that “nonprofessional” phagocytes such as PTECs may have important roles in removing apoptotic cells or cell fragments, in particular in tissues which macrophages cannot infiltrate (33), or under conditions such as AKI where existing professional phagocytes can be overwhelmed by a flood of dying cells (63). Thus the injury-specific upregulation of KIM-1 in the kidney likely serves to curtail inflammation during AKI.
Upon expression on PTECs, KIM-1 undergoes rapid and spontaneous membrane-proximal cleavage both in vivo (32, 72) and in vitro (7). Ectodomain proteolysis generates a 14-kDa membrane-anchored fragment (hereafter referred to as cleaved KIM-1) and a soluble 90-kDa extracellular fragment (hereafter referred to as soluble KIM-1) that is detectable by ELISA and Western blotting in the urine (in vivo) and in the conditioned medium, respectively (7, 84). KIM-1 is now recognized as a sensitive and specific biomarker of AKI in humans and animals (32, 47, 72). KIM-1 shedding has been well characterized in renal cell carcinoma (RCC) cells and LLC-PK1 porcine PTECs that express endogenous and recombinant KIM-1, respectively (7, 35, 84). Using these culture models, Zhang et al. (84) recently demonstrated that KIM-1 is shed constitutively into the conditioned media of cells and is accelerated by pervanadate (PV), a potent inhibitor of protein tyrosine phosphatases. Furthermore, they showed that the constitutive and PV-accelerate cleavage of KIM-1 are mediated by ERK and p38 MAP kinase respectively. Importantly, both the spontaneous and accelerated shedding of KIM-1 were inhibited when cells were pretreated with metalloprotease inhibitors such as GM6001 and BB94 (7, 84). These data suggest that shedding is likely to be a regulated process (84), but its relevance to KIM-1-mediated efferocytosis has not been addressed to date.
Shedding of cell-surface proteins is a well-recognized mechanism of regulating receptor function (9, 50). Soluble receptors, for instance, can exert biological effects by altering the interaction between ligand and membrane-bound receptors (21). Membrane-bound receptors and their soluble counterparts can act agonistically, as is the case with the TNF-α receptor-1 (75). The majority of soluble receptors, however, act antagonistically and function as decoys to sequester soluble ligands (62). Here, we tested whether exaggerated injury would accelerate KIM shedding and whether the resultant excess soluble KIM-1 would serve as an antagonist or agonist of KIM-1-dependent efferocytosis. Thus the primary aim of this study was to characterize and explore the functional significance of KIM-1 shedding with respect to its ability to transform PTECs into phagocytes for clearance of apoptotic cells (35, 37, 44).
MATERIALS AND METHODS
Reagents.
PMA (Sigma-Aldrich), GM6001 (Santa Cruz Biotechnology), and TAPI-0 (Enzo Life Sciences) were diluted in culture medium from a stock solution made in DMSO. PV was made fresh before each experiment by mixing sodium orthovanadate (Sigma-Aldrich) and hydrogen peroxide (H2O2) as previously described (84).
Animals.
Six- to eight-week-old male C57BL/6 mice were purchased from Charles River Laboratories and housed at the animal facility at Western University (London, ON). Experiments were conducted by following established guidelines for animal care approved by the University Council on Animal Care at Western University.
Cells and cell culture.
769-P (RCC), LLC-PK1 (porcine renal epithelial cell) cells were purchased from American Tissue Culture Collection (ATCC) and grown in DMEM supplemented with 10% FBS. HK-2 (human proximal tubule cell) cells were also purchased from ATCC and cultured in keratinocyte serum-free medium supplemented with bovine pituitary extract (0.05 mg/ml) and human recombinant epidermal growth factor (5 ng/ml, Life Technologies). 786-O cells (human RCC) stably expressing a short hairpin (sh) RNA to TNF-α-converting enzyme (TACE) was kindly provided to us by Dr. Stephen Lee (University of Ottawa, Ottawa, ON). The generation of the G418-resistant (Santa Cruz Biotechnology) stable cell lines transfected with pcDNA3 vector alone or encoding human KIM-1 is described elsewhere (7, 35, 36). The KIM-1-Tet-off MDCKII canine kidney epithelial cell line expressing KIM-1 was generated using a BD Tet-Off Gene Expression System kit (Clontech) as described elsewhere (35).
Western blotting.
The conditioned media and total cell extracts were collected (in 4% SDS in PBS) from cell monolayers after appropriate treatments and separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes. Membranes (EMD Millipore) were blocked with 5% nonfat dry milk and incubated with primary antibodies against the human (monoclonal AKG) (7) or mouse (AF1817, R&D Systems) KIM-1 extracellular domain, respectively. The full-length and cleaved human KIM-1 fragments were detected using a polyclonal rabbit antiserum (1400) (7) raised against the cytosolic domain of KIM-1. The respective secondary anti-mouse or anti-rabbit antibodies were used as indicated. Reactive bands were observed by chemiluminescence (EMD Millipore). Where indicated, some Western blots were probed with an anti-TACE antibody (Santa Cruz Biotechnology) and anti-actin antibody (Santa Cruz Biotechnology). Actin was used as a loading control. Soluble mouse and human KIM-1 proteins migrated at approximate molecular weights of 60 and 90 kDa, respectively, reflecting differences in their protein sequence (36, 38).
Quantification of Western blots.
Densitometry (ImageJ 1.47 software) was used to quantify bands from the Western blot results where indicated. The relative increase in soluble or cleaved KIM-1 expression represented by the numbers listed under some of the Western blot image panels were obtained as follows. First, we calculated the ratio of the densitometric reading for soluble or cleaved KIM-1 to that for total KIM-1. Then, we divided each of the ratios by the ratio obtained for the control treatment to obtain a treatment value relative to the control value (i.e., relative to a control of 1).
Real-time quantitative RT-PCR.
For quantification of mRNA by RT-PCR, total RNA was isolated from cells using TRIzol Reagent (Life Technologies) according to the manufacturer's instructions. Five micrograms of total RNA was treated with DNAse I (Invitrogen) and reverse transcribed with the Maloney murine leukemia virus (M-MLV) reverse transcriptase kit and Oligo dT primers (New England Biolabs). Next, 100 ng of the cDNA stock was amplified using the Perfecta SYBR Green Fastmix with ROX (Quantas). Primers were purchased from Integrated DNA Technologies and used at 250 nM. The primer sequences were as follows (from 5′ to 3′): GAPDH forward: CTCTTCTGCTCC TCCTGTTCGAC; reverse: TGAGCAATGTGGCTCGGCT; and KIM-1 forward: GAAGTGGCTACTGGTTCATGG; reverse: ACGACTGTTCGAACGAGCAC. Amplification was done using the StepOne Plus thermocycler (Life Technologies) and was analyzed with the corresponding software for detection of mRNA expression. The cycling conditions were as follows: one cycle at 95°C for 30 s, followed by 40 cycles at a temperature between 0 and 5°C lower then the primer melting temperatures (Tm), and finally a melt curve dissociation step. The relative gene expression was calculated using the 2−ΔΔCT method, and GAPDH was used as the normalizing sample.
Apoptotic cells.
Primary thymocytes were isolated from thymi of 6- to 8 wk-old C57bL/6 mice by passing through a 40 μM cell strainer, and any red blood cells were lysed in ACK lysis buffer (Life Technologies). The single-cell suspension of thymocytes was labeled with 5-(and 6)-carboxyfluorescein di-acetate succinimidyl ester (CFSE) according to manufacturer's instructions (Life Technologies), exposed to UV radiation (254 nm) for 3 min, and then incubated overnight at 37°C and 5% CO2. Over 90% of cells were deemed to be early apoptotic cells based on positive staining with FITC-conjugated annexin V (Biolegend) and exclusion of propidium iodide as previously reported (35).
Phagocytosis assay and flow cytometry.
Cells were plated in 9.6-cm2 wells in six-well plates supplemented with the appropriate media until they reached 80–90% confluence. At the start of each assay, each well received fresh medium unless otherwise stated along with varying amounts of reagents and then left to incubate for various lengths of times at 37°C/5% CO2. Fluorescently labeled apoptotic cells were then added to the monolayer of cells for 30–90 min at 37°C/5% CO2. The conditioned medium was either collected from each well for Western blot analysis where indicated or discarded. The wells were then washed three times with ice-cold PBS to remove any unbound apoptotic cells. The adherent cells were then harvested with 300 μl of trypsin/EDTA (Life Technologies) solution and resuspended in FACS buffer. The cells were analyzed by flow cytometry as previously described (35). Briefly, the forward-scatter/side-scatter (FSC/SSC) ratio was used to exclude apoptotic cell debris, and uptake of labeled apoptotic cells was quantified by using the shift in fluorescence in the live cells. Samples were analyzed on a FACSCalibur cytometer (BD) equipped with 488- and 633-nm lasers, and emission filters for FITC, propidium iodide, Alexa Fluor 633, and allophycocyanin (APC). Data were analyzed with FlowJo Mac software (Tree Star). The percentage of maximum intensity was calculated by dividing the mean fluorescence intensity (MFI) of each sample by that of the maximally affected sample, (i.e., percent max = MFIsample/MFImax * 100).
Confocal microscopy.
Apoptotic thymocytes were labeled with pHrodo Red Succinmidyl Ester (Life Technologies) and incubated with confluent KIM-1 PK1 or Δ278–283-PK1 cells grown on glass coverslips on ice for 30 min. The plates were washed three times with ice-cold PBS and then fixed with 4% PFA for 15 min. After being washed twice more with PBS to remove any residual paraformaldehyde (PFA), the cells were then permeabilized by incubating the coverslips with 0.25% Triton X-100 in PBS for another 5 min. The coverslips were mounted on glass slides using Vectashield containing 4,6-diamidino-2-phenylindole (Vector Laboratories). The slides were viewed at 400× magnification using an Olympus FV1200 Biological Laser-Scanning Confocal Microscope.
Statistics and graphs.
All graphs were done in GraphPad Prism (GraphPad Software) and display relative densitometry values as determined through ImageJ software (Wayne Rasband, National Institute of Health). Student's t-test was used to determine significance between two unpaired groups. Error bars indicate SD. Unless stated otherwise, Western blot results shown are from a single experiment representative of at least three independent experiments. Significant difference (*P < 0.05) and no significant difference (NS) are shown accordingly.
RESULTS
KIM-1 shedding is accelerated by oxidative cellular injury.
Given that ROS are mediators of ischemia-reperfusion injury pathogenesis (17, 20, 46), we tested whether cellular injury mediated by ROS would augment KIM-1 shedding. We thus exposed primary mouse PTECs to increasing concentrations of H2O2 (0.0–10.0 mM) and examined the conditioned medium for appearance of (60 kDa) soluble mouse KIM-1 protein by Western blotting with an antibody raised against the extracellular domain of mouse KIM-1 (R&D Systems) (Fig. 1A). We observed a robust and dose-dependent increase in the 60-kDa band corresponding to soluble mouse KIM-1 in the conditioned media within 30 min of exposure to H2O2. As our primary goal was to study human KIM-1, we next transitioned to established cell models that have been used extensively to study both the spontaneous and accelerated shedding of human KIM-1. These include 769-P RCC cells, HK-2 (human kidney 2) proximal tubule cells that express KIM-1 endogenously (38, 73), and porcine (LLC-PK1) PTECs that have been stably transfected with human KIM-1 (7, 84). We exposed 769-P cells to H2O2 (1 mM) and examined the total cell lysate for cleaved (14 kDa) KIM-1 using Western blotting (Fig. 1B). There was a significant increase in the cleaved KIM-1 relative to the total cellular KIM-1. In parallel, we also treated 769-P cells to increasing concentrations of H2O2 (0.0–10 mM) and examined the conditioned media for appearance of (90 kDa) soluble KIM-1 protein using the AKG antibody(7, 36) raised against the extracellular mucin domain of human KIM-1 (Fig. 1C). Such a rapid increase in KIM-1 ectodomain shedding after H2O2 treatment suggested that this effect was not dependent on increased gene expression. To confirm this, we performed a similar experiment to that shown in Fig. 1B and determined the expression of KIM-1 mRNA relative to GAPDH mRNA using qRT-PCR. Data shown in Fig. 1D suggest that there was no significant change in KIM-1 mRNA before and after H2O2 treatment. These data suggest that H2O2-mediated cellular injury (34) can accelerate KIM-1 shedding.
Fig. 1.
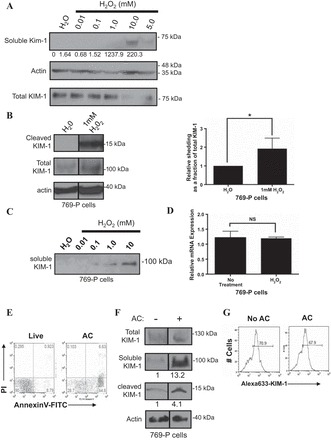
H2O2 and excess apoptotic cells accelerate Kidney injury molecule-1 (KIM-1) shedding. A: primary mouse proximal tubule epithelial cells (PTECs) were treated with increasing concentrations of H2O2, and both conditioned media and total cell lysates were collected. Soluble mouse KIM-1 was detected by SDS-PAGE and Western blotting. Soluble KIM-1 relative to total KIM-1 was quantified by densitometry. B: 769-P cells were exposed to H2O (1 μl) or H2O2 (1 mM) for 30 min in serum-free DMEM and the total cell lysates were collected and analyzed by Western blotting for cleaved and total KIM-1. The graph summarizes cleaved KIM-1 relative to total KIM-1 as determined by densitometry from 3 independent experiments (right). C: confluent monolayers of 769-P cells plated in 6-well tissue culture plates were exposed to H2O (1 μl) or various concentrations of H2O2 (0.01, 0.1, 1.0, and 10 mM) for 30 min in serum-free DMEM before analysis of the conditioned medium for soluble KIM-1. D: H2O2-induced KIM-1 shedding occurs independently of changes in KIM-1 mRNA expression. 769-P cells were exposed to H2O (1 μl) or H2O2 (1 mM) for 30 min in serum-free DMEM. Total cellular RNA was harvested, and relative KIM-1 mRNA expression was quantified using quantitative RT-PCR. E: UV-induced apoptotic(AC) or live mouse thymocytes that were stained with annexin V-FITC and propidium iodide (PI) and analyzed by flow cytometry. F: confluent monolayers of 769-P cells were not exposed or exposed to 107 apoptotic cells for 1 h. Relative shedding as a fraction of total KIM-1 was quantified by densitometry. A–E: soluble KIM-1 was detected from media samples with AKG antibody. Cleaved and total KIM-1 were detected in the total cellular lysates with 195 antibody. Actin was used as a loading control. The Western blot result is representative of 3 independent experiments. G: after being not fed or fed 5-(and 6)-carboxyfluorescein di-acetate succinimidyl ester (CFSE)-labeled apoptotic cells (AC) as in D, single-cell suspensions of the 769-P cells were analyzed for change in surface KIM-1 expression (Alexa 633-KIM-1) after staining with anti-KIM-1 primary antibody (AKG) and Alexa 633-conjugated secondary antibody. NS, not significant. *P < 0.05.
The eat-me signal PtdSer is a natural ligand for KIM-1 and specifically binds to its IgV-like ectodomain (35, 44, 65). During AKI, PTECs that succumb to irreversible injury can undergo apoptosis and are extruded into the tubular lumen can cause tubular obstruction (58). It has been proposed that KIM-1 exposed on the apical surface of surviving PTECs mediate efferocytosis to prevent tubular obstruction by apoptotic cells (35). Therefore, we tested whether apoptotic cells (displaying PtdSer) regulate KIM-1 shedding. Mouse thymocytes exposed to UV light (254 nm) predictably display PtdSer on their outer plasma membrane within 8 h of UV exposure and are classified as early apoptotic cells based on positive staining with FITC-conjugated annexin V and negative staining for propidium iodide as shown in Fig. 1E. On the other hand, live cells did not display PtdSer on their surface or take up propidium iodide. We then exposed confluent monolayers of 769-P cells to ∼107 early apoptotic cells and examined the conditioned media and total cell lysates for KIM-1 cleavage products (Fig. 1F). Relative to total KIM-1 expression, there was increased KIM-1 shedding after addition of apoptotic cells to 769-P cells. Next, we determined whether addition of apoptotic cells to 769-P cells was associated with changes in surface KIM-1 expression. Cell-surface KIM-1 was detected by flow cytometry using an AKG primary antibody (7, 36) together with an Alexa 633-conjugated secondary antibody. We did not observe a major change in the surface expression of KIM-1 (Fig. 1G). These data suggested that pathophysiological cues observed during AKI (11, 35, 48, 86) that are mimicked by H2O2 (34) or excess apoptotic cells can accelerate KIM-1 shedding.
KIM-1 shedding is accelerated by PMA.
We previously demonstrated that KIM-1 shedding is accelerated in vitro by treating the cells with PV, a process driven by activation of the p38 MAPK pathway (84). Both constitutive and PV-accelerated shedding were found to be efficiently blocked by pretreating cells with hydroxamate-based metalloprotease inhibitors such as GM6001 and BB-94 (7, 84). PMA is a PKC activator that is customarily used to enhance ectodomain shedding of surface proteins, particularly those mediated by TACE (2, 8, 59, 80). PMA treatment of 769-P cells resulted in the rapid appearance of a large amount of soluble KIM-1 in the culture medium (Fig. 2A). Relative to the total KIM-1, there was also a significant increase in the corresponding cell-retained (cleaved) fragment of KIM-1, in the cell lysate upon exposure of to PMA (Fig. 2A, right). The effect of PMA treatment on KIM-1 shedding was more pronounced when higher doses of PMA and with longer exposure times (Fig. 2B). Notably, there were no significant differences in KIM-1 mRNA expression with either PMA or PV treatments, respectively (Fig. 2C). Similarly, there were minimal changes to the surface expression of KIM-1 on 769-P cells after PMA and PV treatment, respectively (Fig. 2D). In keeping with this, we observed that pretreatment of 769-P cells with cycloheximide (CHX), a protein synthesis inhibitor, had no effect on PMA-induced KIM-1 shedding (Fig. 2E). Together, these data suggest that PMA and PV accelerate KIM-1 shedding independently of total KIM-1 expression.
Fig. 2.
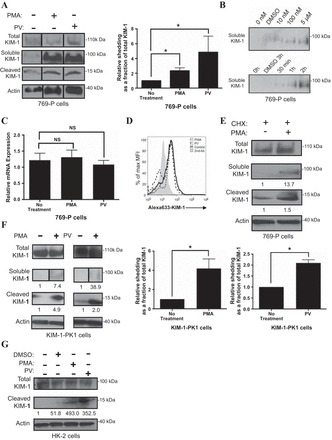
PMA and pervanadate (PV) accelerate KIM-1 shedding. A: confluent monolayers of 769-P cells were incubated in serum-free DMEM with DMSO (control) or PMA, (1 μM) for 1 h or PV (50 μM) for 30 min. A graph summarizing soluble KIM-1 relative to total KIM-1 in 769-P cells as determined by densitometry from 3 independent experiments is shown. B: 769-P cells were pretreated with PMA at various concentrations (0, 10, and 100 nM, and 5 μM) for 30 min (left) and for various durations of time (30 min, 1 h, or 2 h) at a concentration of 1 μM (right). C: PMA-induced KIM-1 shedding occurs independently of mRNA synthesis. 769-P cells were exposed to DMSO (control) or PMA for 1 h in serum-free DMEM. Total cellular RNA was harvested, and relative KIM-1 mRNA expression was analyzed using quantitative RT-PCR. D: 769-P cells were incubated for 30 min with or without PMA (1 μM) or PV (50 μM) before detection of surface KIM-1 expression by flow cytometry using AKG antibody and Alexa Fluoro 633-conjugated as secondary antibodies. KIM-1 expression is displayed in the form of a single-parameter (Alex 633-KIM-1) histogram against the % maximum mean fluorescent intensity. E: PMA-induced KIM-1 shedding occurs independently of protein synthesis. 769-P cells were preincubated for 30 min with cycloheximide (CHX; 25 μg/ml) in DMEM medium and then, with DMSO (control) or PMA (1 μM) for an additional 1 h before detection of soluble, cleaved, and total KIM-1 by Western blotting. F: KIM-1-PK1 cells were incubated in DMEM with DMSO (control) or PMA (1 μM) for 1 h, or with or without PV (50 μM) for 30 min. Graph summarizing cleaved KIM-1 relative total KIM-1 in KIM-1-PK-1 cells as determined by densitometry from 3 independent experiments is shown. G: HK-2 cells were incubated in DMEM with DMSO (control) or PMA (1 μM) for 1 h, or PV (50 μM) for 30 min. A–G: soluble KIM-1 was detected from media samples with AKG antibody. Cleaved and total KIM-1 were detected in the total cellular lysates with 195 antibody. Actin was used as a loading control. Cleaved KIM-1 relative to total KIM-1 was quantified by densitometry. Western blot results described above are representative of results obtained from 3 independent experiments. *P < 0.05.
Next, we extended our findings from 769-P cells to LLC-PK1 cells stably overexpressing human KIM-1 (KIM-1-PK1) (7, 84). Incubation of KIM-1-PK1 cells with PMA resulted in KIM-1 cleavage at the same concentrations of PMA used for 769-P cells (Fig. 2F). In keeping with the findings of Zhang et al. (84), we too observed robust shedding in response to PV (Fig. 2, A and F). Moreover, PMA- and PV-mediated acceleration of KIM-1 shedding occurred independently of any changes in the expression of total KIM-1 (Fig. 2F, right). PMA and PV also triggered accelerated KIM-1 cleavage in human kidney tubular cells (HK-2 cells) that express endogenous KIM-1 (Fig. 2G).
TACE/ADAM17 mediates the spontaneous and accelerated shedding of KIM-1.
Next, we proceeded to characterize the sheddase responsible for KIM-1 proteolysis in RCC cells expressing endogenous KIM-1 (15, 84). Based on the inhibitory effect of hyroxamate-based metalloprotease inhibitors on KIM-1 shedding (7, 36, 39) and the knowledge that ADAM17 or TACE mediates PMA-induced shedding of various transmembrane proteins (22, 80, 85), we hypothesized that TACE/ADAM17 may be responsible for regulating KIM-1 shedding. ADAM (a disintegrin and metalloprotease) proteins are a family of zinc-dependent, membrane-anchored metalloproteases that are implicated in ectodomain shedding of several growth factors (30). TACE belongs to the ADAM family of metalloproteases (3) and is responsible for cleavage of several important surface proteins, including TNF-α (56, 59), transforming growth factor (TGF)-α (59), Notch1 receptor (12), and the type I TGF-β receptor (50). We first tested our hypothesis by pretreating 769-P cells with a chemical inhibitor of TACE (TAPI-0). While GM6001 inhibits both matrix metalloproteases and some members of the ADAM family of proteases, TAPI-0 is a more specific inhibitor of ADAM17/TACE (2, 54, 57). We found pretreatment of 769-P cells with TAPI-0 [10 μM], but not vehicle control (DMSO), effectively inhibited H2O2-induced (1 mM) KIM-1 shedding (Fig. 3A). TAPI-0 [10 μM], but not vehicle control (DMSO), also effectively inhibited both PV (50 μM)- and PMA-induced (1 μM) KIM-1 shedding (Fig. 3, B and C). In keeping with Zhang et al. (84), GM6001 effectively blocked KIM-1 shedding triggered by H2O2, PMA, and PV (Fig. 3, A–C). Although these data were highly suggestive that TACE is the KIM-1 sheddase, it was not conclusive because of the possible off-target effects of chemical inhibitors. Hence, we employed a shRNA-mediated knockdown strategy to inhibit TACE expression in 786-O RCC cells expressing endogenous KIM-1 (Gunaratnam L, unpublished observations). For this, we used previously generated 786-O cell clones stably transfected with pSilencer vector (Life Technologies, NY) or pSilencer encoding shRNA targeting TACE (28). Western blot analysis of the cell lysates with anti-TACE antibodies confirmed effective knockdown of TACE expression in our clone expressing TACE shRNA (shTACE) but not the vector (pSilencer) (Fig. 3D). We then analyzed the conditioned media of both clones (pSilencer and shTACE) for soluble KIM-1 both at baseline and after stimulation with PMA (1 μM) using Western blotting. We found that both PMA-unstimulated and PMA-stimulated KIM-1 cleavage were markedly reduced in the conditioned media of 786-O cells transfected with shRNA targeting TACE compared with the control cells (Fig. 3E). The acceleration of KIM-1 shedding via TACE is not entirely surprising given that PMA activates TACE via p38α MAP (80). Next, to determine the mechanism of H2O2-induced KIM-1 shedding is due to upregulation of TACE on cells, we examined cell-surface expression of TACE in 769-P cells exposed to H2O2 using PMA as a control and flow cytometry. Data presented in Fig. 3F suggest that H2O2 but not PMA upregulated cell-surface TACE expression in 769-P cells. The lack of effect of PMA on TACE expression is in keeping with previous findings (80). Together, these results indicated that TACE is responsible for both the constitutive and induced cleavage of endogenous KIM-1 in these cells.
Fig. 3.
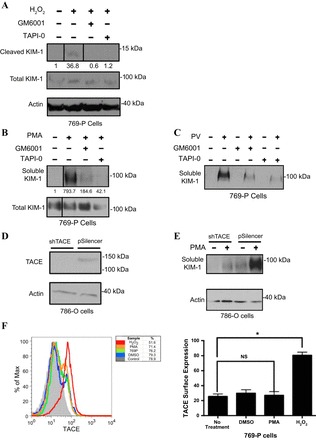
Constitutive and accelerated KIM-1 shedding is inhibited by TNF-α-converting enzyme (TACE) inhibitors and short hairpin (sh) RNA-mediated knockdown of TACE. A: confluent monolayers of 769-P renal cell carcinoma (RCC) cells were first pretreated with DMSO (control), GM6001 (1 μM), or TAPI-0 (10 μM) for 30 min following exposure to H2O (1 μl) or H2O2 (1 mM) for 30 min. Shed KIM-1 was detected in media with AKG antibody. Cleaved and total KIM-1 was detected in total cellular lysate with 195 antibody. B and C: experiment in A was repeated with PMA (1 μM) for 1 h and PV (50 μM) for 30 min. A and B: soluble KIM-1 relative to total KIM-1 was quantified by densitometry. Lines between the bands represent any lanes that were rearranged from the original blot. Cleaved KIM-1 was detected using a 15% gel, while total KIM-1 and actin were detected using a 10% gel. D: cell lysates of 786-O RCC cells stably expressing a vector encoding shRNA targeting TACE (shTACE) or the empty vector alone (pSilencer) were analyzed by Western blotting with anti-TACE antibody. A band corresponding to TACE is shown. E: confluent monolayers shTACE and pSilencer cells were preincubated with PMA for 30 min, and KIM-1 shedding was assessed by Western blot analysis. Actin was used as a loading control. The Western blot data are representative of the results obtained from 3 independent experiments. F: 769-P cells were incubated with H2O (1 μl), H2O2 (1 mM), DMSO, or PMA (1 μM) for 30 and 60 min, respectively, before detection of surface TACE expression by flow cytometry using a TACE antibody. Surface TACE expression is displayed in the form of a histogram against the % maximum mean fluorescent intensity and as a graph representative of data from 3 independent experiments. *P < 0.05.
Accelerated shedding of KIM-1 inhibits efferocytosis.
Ectodomain shedding has been shown to regulate the activities of numerous substrate proteins (22, 39, 60, 67, 68). We hypothesized that the proteolytic cleavage of KIM-1 might serve to regulate its function as a phagocytic receptor on PTECs. We postulated that the generation of the soluble form of membrane-bound KIM-1 could competitively inhibit phagocytic receptor signaling and engulfment of apoptotic cells by intact receptors on the cell surface (26, 59, 75). Alternatively, KIM-1 shedding could reduce the number of surface receptors available for phagocytosis. To test our hypothesis, we explored how excess KIM-1 shedding might affect KIM-1-dependent efferocytosis in LLC-PK1 PTECs. Here, the phagocytic uptake of fluorescently (CFSE) labeled apoptotic cells was quantified using flow cytometry as described in materials and methods. Importantly, LLC-PK1 cells lacking KIM-1 expression (pcDNA-PK1) inherently do not mediate efferocytosis (Fig. 4A). Untreated KIM-1-PK1 cells, on the other hand, were far more efficient at engulfing apoptotic cells compared with PCDNA-PK1 cells. The efficiency of apoptotic cell uptake by KIM-1-PK1 cells was significantly inhibited when the cells were pretreated with either 1 mM H2O2, 50 μM PV, or 1 μM PMA, which was previously shown to accelerate KIM-1 shedding (Fig. 4, B–D). It is pertinent to note that we did not notice any significant change in the viability of the KIM-1-PK1 cells upon addition of H2O2, PV, or PMA for the duration of the experiment (data not shown). The decreased ability of KIM-1-PK1 cells to engulf apoptotic cells could not be explained solely by reduced surface expression of (uncleaved) KIM-1 receptors as a result of accelerated shedding with PMA or PV (Fig. 4E). Although both H2O2 and PV inhibited phagocytosis, it remained possible that this effect was independent of its effect on KIM-1 shedding (27, 74, 83, 84). We demonstrate that the inhibitory effect of both H2O2 and PV on efferocytosis was rescued by blocking KIM-1 shedding with TAPI-0 pretreatment of cells (Fig. 4, F and G).
Fig. 4.
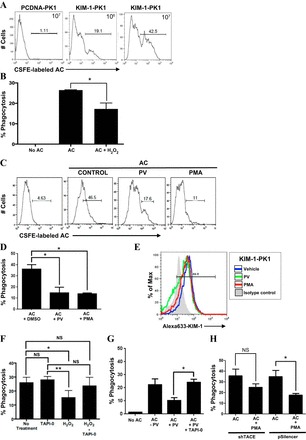
Accelerated KIM-1 shedding inhibits efferocytosis in KIM-1-expressing PTECs. A: PCDNA-PK1 or KIM-1-PK1 cells were incubated with 106 or 107 CFSE-labeled apoptotic cells(AC) in 6-well plates for 90 min, and the phagocytic uptake of apoptotic cells was analyzed by flow cytometry. Data re depicted in the form of a representative single-parameter (CFSE-labeled AC) histogram determined from 3 independent experiments. B: KIM-1-PK1 cells were untreated or pretreated with H2O (control) or H2O2 (1 mM) for 30 min in 6-well plates followed by incubation with 107 CFSE-labeled apoptotic cells for 90 min before measurement of phagocytic uptake of apoptotic cells by flow cytometry. C: KIM-1-PK1 cells were pretreated with DMSO (control), PV (50 μM) for 30 min, or PMA (1 μM) for 1 h in 6-well plates followed by incubation with 107 CFSE-labeled apoptotic cells for 90 min. Samples were analyzed by flow cytometry as in A. D: data from 3 independent experiments in C were graphed to show percent phagocytosis with error bars representing SD. E: change in surface KIM-1- expression in KIM-1-PK1 cells after addition of DMSO (vehicle), PMA, and PV as determined by flow cytometry using AKG primary antibody and Alexa Fluoro 633-conjugated anti-mouse IgG secondary antibody. F: 769-P cells were first pretreated with TAPI-0 (10 μM) for 30 min and then with H2O2 (1 mM) for 30 min before incubation with 106 CFSE-labeled apoptotic cells for an additional 90 min. Percent phagocytosis was determined as described in A from 3 independent experiments. G: KIM-1-PK1 cells were first pretreated with TAPI-0 (10 μM) for 30 min and then with DMSO (−PV) or PV (+PV) for another 30 min before incubation with 106 CFSE-labeled apoptotic cells for an additional 90 min. Percent phagocytosis was determined as described in A. H: shTACE and pSilencer cells were pretreated with and without PMA (1 μM) for 1 h followed by incubation with apoptotic cells for 90 min. Flow cytometry analysis was done as in A. *P < 0.05.
Given the data in Fig. 3 showing that TACE mediates the accelerated shedding of KIM-1 in 786-O cells, we next determined whether TACE knockdown would also rescue the inhibitory effect of PMA on KIM-1-mediated efferocytosis. For this, we once again used the previously established 786-O clones with stable knockdown TACE by shRNA (28). Silencing TACE expression in 786-O cells prevented the PMA-induced inhibition of apoptotic cell phagocytosis (Fig. 4H). We attribute the incomplete rescue of phagocytosis ability to the incomplete knockdown of TACE in 786-O cells or TACE-independent effects of PMA on efferocytosis. Together, these data suggest accelerating KIM-1 shedding can impede efferocytosis efficiency in cells expressing either endogenous (786-O) or recombinant KIM-1 (KIM-1-PK1). Although our data also suggest that this is mediated by TACE-dependent shedding of KIM-1, it still remains possible that other metalloproteases might also regulate efferocytosis in these cells (31).
Soluble KIM-1 serves as a decoy receptor to inhibit KIM-1-dependent efferocytosis.
We hypothesized that soluble KIM-1 in the conditioned media resulting from accelerated shedding might serve as a decoy receptor for PtdSer exposed on apoptotic cells to competitively block efferocytosis in our model. To test this, we measured the uptake of fluorescently (CFSE) labeled apoptotic cells by KIM-1-PK1 cells where we replaced culture medium with conditioned media derived from 769-P cells that were cultured at confluence for over three days. 769-P cells spontaneously shed KIM-1 and their conditioned media accumulated large amounts of soluble KIM-1 by day 3 (data not shown). To enhance the binding of soluble KIM-1 to apoptotic cells, we first preincubated apoptotic cells with conditioned media from 769-P cells for 30 min at 4°C before transferring the apoptotic cell-conditioned media mixture to the KIM-1-PK1 cells. Fresh medium was used to dilute the conditioned media to generate 0, 25, and 100% fractions of conditioned media that were preincubated with apoptotic cells, after which equal volumes of the apoptotic cell-conditioned media mixture were transferred to monolayers of KIM-1-PK1 cells plated in six-well plates. Figure 5A shows the percent phagocytosis of the treated cells as determined by flow cytometry. We observed a significant reduction in the phagocytic uptake when undiluted (100%) conditioned media from 769-P cells was used. To confirm that it was soluble KIM-1 contained in conditioned media and not other mediators that inhibited efferocytosis by KIM-1-PK1 cells, we employed a previously established (35) tetracycline-repressive KIM-1 expression system in MDCKII canine renal tubular cells. This cell line was generated by stably expressing the human KIM-1 transgene under the control of a tetracycline-dependent repressor system (KIM-1-Tet-off MDCKII). Withdrawal of tetracycline in the medium drives human KIM-1 expression that results in the efficient and spontaneous shedding of soluble KIM-1 into the conditioned media (labeled +sKIM-1 in Fig. 5B). Apoptotic cells were once again preincubated with conditioned media for 30 min at 4°C before transference of the apoptotic cell-conditioned media mixture to monolayers of KIM-1-PK1 cells and performance of phagocytosis assays. Efferocytosis efficiency was significantly retarded in the presence of conditioned media containing soluble KIM-1 (+sKIM-1) compared with conditioned media not containing soluble KIM-1 (−sKIM-1) (Fig. 5, B and C). To further confirm the inhibitory role of soluble KIM-1 in efferocytosis, we preincubated CFSE-labeled apoptotic cells with recombinant soluble KIM-1 (Sino Biological) consisting of the extracellular domain of human KIM-1 fused to the Fc region of human IgG1 at the N terminus (KIM-1-Fc). Nonspecific IgG was used as a negative control. CFSE-labeled apoptotic cells were preincubated with increasing concentrations of KIM-1-Fc (200, 400, or 800 ng/ml) or 800 ng/ml of human IgG diluted in standard culture medium (DMEM and 10% FBS) for 30 min at 4°C before transference of the solutions containing apoptotic cells-KIM-1-Fc or apoptotic cells-IgG complexes to monolayers of KIM-1-PK1 cells to allow for their phagocytic uptake (Fig. 5D). The phagocytic uptake of apoptotic cells was significantly inhibited in the presence of 800 ng/ml of KIM-1-Fc compared with the same concentration of IgG. It is important to note that the concentration of KIM-1-Fc required to block phagocytosis here was consistent with that required to competitively block PtdSer binding to KIM-1 in ELISA assays in previous studies (35). Together, these data suggested that soluble KIM-1 in the extracellular milieu resulting from accelerated shedding competitively inhibits efferocytosis by PTECs.
Fig. 5.
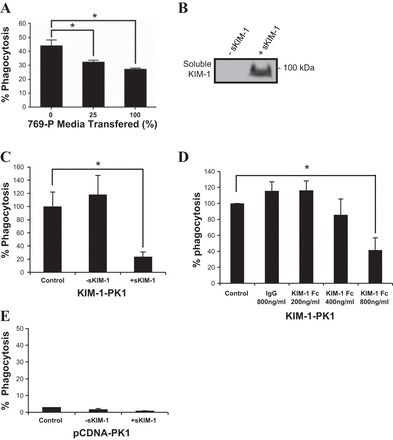
Soluble KIM-1 competitively inhibits apoptotic cell engulfment by KIM-1-expressing PTECs. A: CFSE-labeled apoptotic cells were incubated on ice for 30 min with increasing fractions of conditioned medium from 769-P cells cultured for 3 days. Thereafter, the conditioned medium containing the apoptotic cells was added to confluent monolayers of KIM-1-PK1 cells for 90 min before measurement of phagocytic uptake by flow cytometry. B: Western blot detecting soluble KIM-1 in the conditioned medium collected from KIM1-Tet-off MDCKII cells either treated with doxycycline (100 ng/ml) to inhibit KIM-1 expression (−sKIM-1) or no doxycycline for 5 days, permitting high-level expression of KIM-1 (+sKIM-1). C: CFSE-labeled apoptotic cells were incubated on ice for 30 min with conditioned medium from KIM1-Tet-off MDCKII cells either treated with doxycycline (−sKIM-1) or no doxycycline (+sKIM-1). Thereafter, the conditioned media containing the apoptotic cells were added to monolayers of KIM-1-PK1 cells for 90 min before measurement of phagocytic uptake by flow cytometry. D: relative phagocytic uptake of labeled apoptotic cells by KIM-1-PK1 cells incubated in the absence or presence of recombinant KIM-1-Fc protein (n = 3) or nonspecific IgG at the concentration indicated. C and D: results are expressed as percent relative phagocytosis of untreated control. A–D: fresh culture medium was used as the control. E: soluble KIM-1 does not confer on PTECs the ability to engulf apoptotic cells in the absence of KIM-1 expression. CFSE-labeled apoptotic cells were incubated on ice for 30 min with the conditioned (−sKIM-1 or +sKIM-1) medium from KIM1-Tet-off MDCKII cells, and thereafter the conditioned media containing the apoptotic cells were added to the PCDNA-PK1 cells for an additional 90 min before measurement of phagocytic uptake by flow cytometry. *P < 0.05.
Phagocytic cells can recognize eat-me signals like PtdSer on apoptotic cells either directly (e.g., KIM-1) (35, 44, 65) or indirectly via opsonin proteins such as Milk fat globule EGF factor 8 protein (via αvβ5-integrin) and growth arrest-specific factor-6 (via Mer), respectively (43, 53, 77). We speculated that soluble KIM-1 might serve as an opsonin (1, 23) to promote phagocytic uptake by tubular epithelial cells within the tubule that do not express KIM-1. To test this, we fed CFSE-labeled apoptotic cells that were preincubated with conditioned media from KIM-1-Tet-off MDCKII cells containing soluble KIM-1 (+sKIM-1) or no soluble KIM-1 (−sKIM-1) (as in Fig. 5C) to PCDNA-PK1 cells and measured their phagocytic uptake by flow cytometry. There was no significant increase in baseline efferocytosis by PCDNA-PK1 cells following the preincubation of apoptotic cells with soluble KIM-1 (Fig. 5E). These data suggested that soluble KIM-1 is unable to serve as an opsonin to promote efferocytosis by these PTECs. We argue that LLC-PK1 cells possess the signaling machinery required to mediate phagocytic signaling since transfection of KIM-1 alone into these cells converts them in avid phagocytes of apoptotic cells.
The juxtamembrane region containing aa 278–283 is vital for KIM-1-mediated phagocytosis of apoptotic cells by PTECs.
So far, we have provided experimental evidence to suggest that soluble KIM-1 resulting from accelerated shedding inhibits KIM-1-mediated efferocytosis by PTECs. We have, however, not addressed how baseline shedding affects this process. To address this, we employed a mutant form of KIM-1 containing a six-amino acid deletion (aa 278–283) that disrupts the helical polypeptide structure in the juxtamembrane region predicted to contain the putative cleavage site of KIM-1 shedding (84). When expressed in COS-7 cells, this mutant was shown to be resistant to both constitutive and PV-accelerated KIM-1. We therefore generated stable LLC-PK1 cells that expressed equivalent amounts either the wild-type (KIM-1-PK1) or the cleavage-mutant (Δ278–283-PK1) proteins as determined by Western blotting (Fig. 6A). In keeping with the data published by Zhang et al. (84), we found that Δ278–283-PK1 generated neither the 90-kDa (soluble KIM-1) nor the 14-kDa (cleaved membrane-bound KIM-1) fragments when stimulated with PMA (Fig. 6A). Next, we studied the cell-surface expression of KIM-1 by generating (10 μM) a single-cell suspension of KIM-1-PK1 and Δ278–283-PK1 cells incubated with anti-KIM-1 antibody (AKG) (7) raised against the extracellular (mucin) domain of KIM-1. The cell-surface expression of KIM-1 was comparable between Δ278–283-PK1 and KIM-1-PK1 cells (Fig. 6B) However, we detected a dramatic decrease in the phagocytosis efficiency of Δ278–283-PK1 compared with KIM-1-PK1 cells (Fig. 6C). This was not explained by a difference in the binding of apoptotic cells to the surface of Δ278–283-PK1 or KIM-1-PK1 cells (Fig. 6, D and E). The binding of fluorescently (pHRODO, Life Technologies) labeled apoptotic cells to KIM-1-PK1 and Δ278–283-PK1 cells was done at 4°C to block phagocytosis (that requires ATP) and visualized by fluorescence microscopy as previously described (35). The data presented so far suggest that PTECs expressing the deletion mutant of KIM-1 are significantly impaired in their ability to mediate efferocytosis compared with those expressing the wild-type protein. However, these data do not exclude the possibility that the observed defect in phagocytic ability is due to a structural effect resulting from deleting aa 278–283 rather than a defect in KIM-1 cleavage. We thus measured phagocytic uptake of apoptotic cells after pretreating KIM-1-PK1 with GM6001 to block KIM-1 shedding. Surprisingly, there was no significant difference in phagocytosis when baseline KIM-1 shedding was blocked (Fig. 6F). Together, the above data suggested that even though baseline cleavage is not required for KIM-1-mediated phagocytosis, the juxtamembrane region containing aa 278–283 is required for KIM-1-mediated phagocytosis of apoptotic cells. One potential limitation of our data is that TACE may be affecting KIM-1-dependent efferocytosis independently of its effect on KIM-1 shedding. Since Δ278–283-PK1 cells do not shed KIM-1 but are still able to engulf apoptotic cells, we used these cells to show that PMA-mediated inhibition of phagocytosis of apoptotic cells is due to KIM-1 shedding. We thus compared the relative decrease in the phagocytic efficiency between KIM-1-PK1 and Δ278–283-PK1 cells upon exposure to PMA. While PMA significantly inhibited the engulfment of apoptotic cells by KIM-1-PK1 cells, it had no significant effect on Δ278–283-PK1 engulfment of apoptotic cells (Fig. 6G).
Fig. 6.
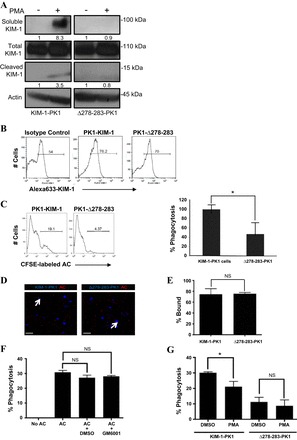
Phagocytic uptake of apoptotic cells is impaired in PTECs expressing a cleavage-mutant of KIM-1. A: LLC-PK1 cells stably expressing wild-type (KIM-1-PK1) or a cleavage-mutant of KIM-1 (Δ278–283-PK1) were treated with PMA (1 μM) or vehicle control (DMSO) for 30 min. Soluble and cleaved KIM-1 were detected from media and cell lysate samples, respectively. Actin was used as a loading control. Soluble and cleaved KIM-1 relative to total KIM-1 was quantified by densitometry. B: surface expression of KIM-1 in KIM-1-PK1 and Δ278–283-PK1 cells was evaluated by flow cytometry using both AKG or control antibody and Alexa Fluoro 633-conjugated secondary without permeabilization. C: KIM-1-PK1 and Δ278–283-PK1 cells were incubated with 106 CFSE-labeled apoptotic cells for 90 min, and phagocytosis was analyzed by flow cytometry. Data are depicted in the form of a representative single-parameter histogram (left) and a graph summarizing the relative (%) phagocytosis determined from 3 independent experiments (right). D: binding of pHrodo-labeled apoptotic cells(AC) to confluent monolayers of KIM-1-PK1 and Δ278–283-PK1 cells at 4°C as observed by confocal microscopy. Images were captured at 400× magnification, and the scale bar represents 10 μm. E: percent bound apoptotic cells relative to the total number of nuclei determined from 3 independent experiments in D. F: KIM-1-PK1 cells were pretreated with DMSO (control) or GM6001 (1 μM) for 30 min followed by incubation with 107 CFSE-labeled apoptotic cells (AC) for 90 min. Flow cytometric analysis was used to determine the percent phagocytosis from 3 independent experiments. G: KIM-1-PK1 and Δ278–283-PK1 cells were pretreated with DMSO (control) or PMA (1 μM) for 1 h in 6-well plates followed by incubation with 107 CFSE-labeled apoptotic cells for 90 min. Percent phagocytosis was determined by flow cytometry from 3 independent experiments. *P < 0.05.
DISCUSSION
KIM-1 undergoes membrane-proximal cleavage by a metalloproteinase, resulting in shedding of a soluble form of KIM-1 into the kidney tubular lumen and subsequently the urine (7, 64). Using the same culture models described in this study, Zhang et al. (84) previously demonstrated that KIM-1 is shed constitutively into the culture medium of cell lines expressing either endogenous or recombinant KIM-1 and is enhanced dramatically by PV, a potent inhibitor of protein tyrosine phosphatases (84). They found that ERK-MAPK and p38-MAPK regulate spontaneous KIM-1 shedding and accelerated shedding of KIM-1, respectively (84). The functional significance of KIM-1 ectodomain shedding, however, was not addressed.
The primary objective of this work was to explore the functional significance of KIM-1 shedding with respect to its ability to transform PTECs into phagocytes for clearance of apoptotic cells. By utilizing various in vitro cell culture models that either express endogenous or recombinant KIM-1, we provide evidence suggesting that KIM-1 shedding is accelerated by H2O2-mediated cellular injury or exposure to apoptotic cells, both of which mimic the tubular microenvironment during severe AKI (10, 11, 18) and PMA. We also show that efferocytosis by PTECs is competitively inhibited by excess soluble KIM-1 resulting from accelerated shedding. Furthermore, we demonstrate that both baseline and accelerated KIM-1 shedding are mediated by TACE/ADAM17. Finally, we report that spontaneous or baseline KIM-1 shedding does not affect KIM-1-dependent efferocytosis by PTECs. These results suggest that soluble KIM-1 has different effects at different concentrations.
TACE is a membrane-anchored, Zn-dependent metalloprotease and was the first member of the ADAM family to be characterized (8, 56, 59). It functions as a membrane sheddase for numerous transmembrane proteins, including the precursors of TNF-α, TGF-α, several other cytokines, as well as the receptors for TNF-α and neuregulin (ErbB4) (30). The implication of TACE in KIM-1 cleavage is not surprising. We have long speculated that a metalloprotease is involved because shedding is highly sensitive to inhibition by broad-spectrum metalloprotease inhibitors (7) and is activated by MAPK-mediated activation (81, 84). Our findings, however, do not exclude the possibility that other metalloproteases might also mediate KIM-1 shedding under different conditions (31 49). Whether apoptotic cell-mediated acceleration of KIM-1 shedding is also mediated by TACE remains to be tested. Our results surprisingly showed that H2O2 can upregulate cell- surface expression of TACE. The mechanism of the H2O2-mediated increase in TACE expression is unclear. We also cannot exclude the possibility that H2O2 also increases TACE activity. Furthermore, we cannot rule out the possibility that mechanisms other than ROS generation might be involved in H2O2-induced KIM-1 shedding. In addition, it would also be interesting to ascertain whether H2O2 and apoptotic cells trigger accelerated KIM-1 cleavage through p38-MAPK activation of TACE in PTECs, given that TACE activity is enhanced during ischemia-reperfusion injury in vivo (70).
Membrane-bound receptors can be converted into soluble receptors using alternative mRNA splicing to remove the transmembrane region, or through proteolytic cleavage and release of the receptor ectodomain. In this report, we have shown that accelerated shedding and excess soluble KIM-1 in the conditioned medium competitively inhibits efferocytosis. The KIM-1 ectodomain-Fc fusion protein has been shown previously to effectively block the binding of PtdSer-containing liposomes to the surface of KIM-1-PK1 cells (35). Akin to the phagocytosis receptor Mer, when present in excess amounts, soluble KIM-1 molecules likely serve as decoy PtdSer receptors to inhibit PTEC-mediated engulfment of apoptotic cells (67). Importantly, we did not find any evidence suggesting that soluble KIM-1 can serve as a PtdSer opsonin like MFG-E8 and Gas6 to promote efferocytosis by PTECs that do not express KIM-1 (78). However, our data do not exclude the possibility that shed KIM-1 might have a nonphagocytic role during AKI or in cancers of the kidney (15). Nevertheless, the fact that pretreatment with TAPI-0 rescued the inhibitory effect of H2O2 and PV on KIM-1-mediated efferocytosis suggests that the reduction in phagocytosis is independent of other potential effects of these agents on cells and is dependent on KIM-1 shedding.
Our studies also revealed that LLC-PK1 cells expressing Δ278–283 mutant KIM-1 were impaired in their uptake of apoptotic cells compared with those expressing the wild-type protein. This difference was not explained by reduced binding of the mutant KIM-1 to apoptotic cells or inefficient surface expression of the receptor on PTECs. Moreover, the inability of Δ278–283 KIM-1 to undergo shedding did not explain the ineffective phagocytosis by LLC-PK1 cells expressing this mutant. Thus the juxtamembrane region (aa 278–283) of KIM-1 may be required for phagocytosis independently of its requirement for shedding or binding to apoptotic cells. We postulate that this region might be important for cross talk with other cell-surface proteins that might be involved in phagocytosis. Nonetheless, while efferocytosis of KIM-1-PK1 cells was inhibited by PMA treatment, the phagocytic capacity of Δ278–283-PK1 was unaffected by PMA treatment. This suggests that PMA-mediated inhibition of phagocytosis is due to accelerated KIM-1 shedding because Δ278–283-PK1 are defective at KIM-1 shedding.
What is the physiological significance of our findings given that KIM-1 is expressed on the apical surface of PTECs? It is unlikely that soluble KIM-1 will accumulate in the tubular lumen when glomerular filtration is maintained in each nephron. However, it is plausible that excess soluble KIM-1 might accumulate within the tubular lumen when the tubules are obstructed, as occurs during severe AKI (29, 61, 69). Perhaps inhibition of phagocytosis by excess soluble KIM-1 triggered by accumulation of apoptotic cells in the tubule, under conditions of severe injury when blood flow is minimal, may represent an energetically favorable process to reduce ATP consumption. The increase in shedding in response to excess apoptotic cells could be a potential mechanism to limit the engulfment of additional apoptotic cells by PTECs. Our data suggest that baseline KIM-1 shedding is accelerated by conditions that mimic severe AKI and that accelerated shedding inhibits KIM-1-mediated uptake of apoptotic cells by PTECs. Taken together, we infer that the increased inflammation and inefficient repair that is associated with severe AKI (6, 82) might be in part due to inefficient efferocytosis during severe AKI. This notion is supported by the fact that primary PTECs isolated from KIM-1 knockout mice (76) are virtually incapable of engulfing apoptotic cells compared with wild type mice (Gunaratnam L, unpublished observations). Furthermore, mice expressing a mucin domain deletion, mutant KIM-1, rendering it phagocytosis defective (79), have been reported to exhibit severe inflammation and tissue injury after being subjected to renal ischemia-reperfusion injury (82a).
In summary, the experimental evidence described in this report suggests that the baseline ectodomain cleavage of KIM-1 observed in PTECs is accelerated by severe cellular injury or PMA. While baseline KIM-1 cleavage is not necessary for the phagocytic uptake of apoptotic cells by PTECs, excess shedding can competitively inhibit the process by generating soluble decoy (KIM-1) receptors (see hypothetical model, Fig. 7). Given that clearance of apoptotic cells is central to maintaining tissue homeostasis and avoiding unwanted inflammation (14, 55), it might be interesting to speculate that excess KIM-1 proteolysis might occur during severe AKI and limit renal recovery. Future studies will be required to formally test this notion in vivo.
Fig. 7.
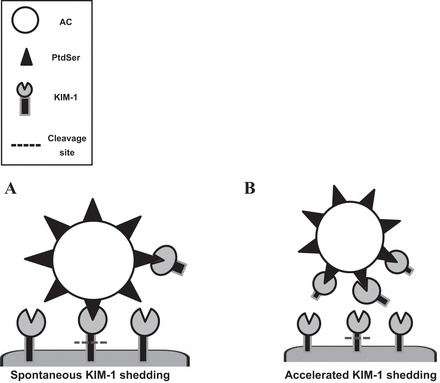
Hypothetical model showing the regulation of KIM-1-mediated efferocytosis by receptor shedding. A: spontaneous KIM-1 shedding does not affect the efficiency of KIM-1-dependent efferocytosis. B: competitive inhibition of efferocytosis by decoy (soluble KIM-1) receptors resulting from excess KIM-1 shedding due to exaggerated cellular injury (e.g., H2O2).
GRANTS
J. Yi was supported by the Summer Research Opportunities Program of the Schulich School of Medicine and Dentistry (Western University). L. Gunaratnam is the recipient of a New Investigator Award from the Kidney Research Scientist Core Education and Training (KRESCENT) program and Kidney Foundation of Canada, and supported by the Academic Medical Organization of Southern Ontario. This work was supported by operating grants from the Canadian Institutes of Health Research (HDK 232429 and 244945) and the Department of Medicine (Western University) awarded to L. Gunaratnam.
DISCLOSURES
J. V. Bonventre is a coinventor of KIM-1 patents that are assigned to Partners Healthcare and licensed by Partners to Johnson&Johnson, Sekisui, Biogen Idec, and a number of research reagent companies. J. V. Bonventre is a consultant for Sekisui.
AUTHOR CONTRIBUTIONS
Author contributions: R.G., J.Y., J.H., H.S., O.I., S.N., X.Z., and L.G. performed experiments; R.G. and L.G. analyzed data; R.G., J.Y., and L.G. interpreted results of experiments; R.G., J.Y., J.H., H.S., O.I., and L.G. prepared figures; R.G., J.Y., and L.G. drafted manuscript; R.G., J.V.B., and L.G. edited and revised manuscript; L.G. provided conception and design of research; L.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Rita Suri for help with editing the manuscript and Marie Sarabusky for technical expertise.
REFERENCES
- 1.Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, Shacter E. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol 4: 87–91, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Arribas J, Coodly L, Vollmer P, Kishimoto TK, Rose-John S, Massague J. Diverse cell surface protein ectodomains are shed by a system sensitive to metalloprotease inhibitors. J Biol Chem 271: 11376–11382, 1996. [DOI] [PubMed] [Google Scholar]
- 3.Arribas J, Esselens C. ADAM17 as a therapeutic target in multiple diseases. Curr Pharm Des 15: 2319–2335, 2009. [DOI] [PubMed] [Google Scholar]
- 4.Arroyo A, Modriansky M, Serinkan FB, Bello RI, Matsura T, Jiang J, Tyurin VA, Tyurina YY, Fadeel B, Kagan VE. NADPH oxidase-dependent oxidation and externalization of phosphatidylserine during apoptosis in Me2SO-differentiated HL-60 cells. Role in phagocytic clearance. J Biol Chem 277: 49965–49975, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Atabai K, Jame S, Azhar N, Kuo A, Lam M, McKleroy W, Dehart G, Rahman S, Xia DD, Melton AC, Wolters P, Emson CL, Turner SM, Werb Z, Sheppard D. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J Clin Invest 119: 3713–3722, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azuma H, Nadeau K, Takada M, Mackenzie HS, Tilney NL. Cellular and molecular predictors of chronic renal dysfunction after initial ischemia/reperfusion injury of a single kidney. Transplantation 64: 190–197, 1997. [DOI] [PubMed] [Google Scholar]
- 7.Bailly V, Zhang Z, Meier W, Cate R, Sanicola M, Bonventre JV. Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. J Biol Chem 277: 39739–39748, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385: 729–733, 1997. [DOI] [PubMed] [Google Scholar]
- 9.Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol 6: 32–43, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Bonegio R, Lieberthal W. Role of apoptosis in the pathogenesis of acute renal failure. Curr Opin Nephrol Hypertens 11: 301–308, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell 5: 207–216, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med 196: 135–140, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuadros T, Trilla E, Vila MR, de Torres I, Vilardell J, Messaoud NB, Salcedo M, Sarro E, Lopez-Hellin J, Blanco A, Mir C, Ramon y Cajal S, Itarte E, Morote J, Meseguer A. Hepatitis A virus cellular receptor 1/kidney injury molecule-1 is a susceptibility gene for clear cell renal cell carcinoma and hepatitis A virus cellular receptor/kidney injury molecule-1 ectodomain shedding a predictive biomarker of tumour progression. Eur J Cancer 49: 2034–2047, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Cui T, Miksa M, Wu R, Komura H, Zhou M, Dong W, Wang Z, Higuchi S, Chaung W, Blau SA, Marini CP, Ravikumar TS, Wang P. Milk fat globule epidermal growth factor 8 attenuates acute lung injury in mice after intestinal ischemia and reperfusion. Am J Respir Crit Care Med 181: 238–246, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cutrn JC, Perrelli MG, Cavalieri B, Peralta C, Rosell Catafau J, Poli G. Microvascular dysfunction induced by reperfusion injury and protective effect of ischemic preconditioning. Free Radic Biol Med 33: 1200–1208, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Daemen MA, van 't Veer C, Denecker G, Heemskerk VH, Wolfs TG, Clauss M, Vandenabeele P, Buurman WA. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest 104: 541–549, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demirjian S, Chertow GM, Zhang JH, O'Connor TZ, Vitale J, Paganini EP, Palevsky PM. Model to predict mortality in critically ill adults with acute kidney injury. Clin J Am Soc Nephrol 6: 2114–2120, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol 17: 1503–1520, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Driscoll WS, Vaisar T, Tang J, Wilson CL, Raines EW. Macrophage ADAM17 deficiency augments CD36-dependent apoptotic cell uptake and the linked anti-inflammatory phenotype. Circ Res 113: 52–61, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Etzerodt A, Maniecki MB, Moller K, Moller HJ, Moestrup SK. Tumor necrosis factor alpha-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J Leukoc Biol 88: 1201–1205, 2010. [DOI] [PubMed] [Google Scholar]
- 23.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101: 890–898, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 148: 2207–2216, 1992. [PubMed] [Google Scholar]
- 25.Famulski KS, de Freitas DG, Kreepala C, Chang J, Sellares J, Sis B, Einecke G, Mengel M, Reeve J, Halloran PF. Molecular phenotypes of acute kidney injury in kidney transplants. J Am Soc Nephrol 23: 948–958, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fanslow WC, Anderson DM, Grabstein KH, Clark EA, Cosman D, Armitage RJ. Soluble forms of CD40 inhibit biologic responses of human B cells. J Immunol 149: 655–660, 1992. [PubMed] [Google Scholar]
- 27.Fantus IG, Kadota S, Deragon G, Foster B, Posner BI. Pervanadate [peroxide(s) of vanadate] mimics insulin action in rat adipocytes via activation of the insulin receptor tyrosine kinase. Biochemistry 28: 8864–8871, 1989. [DOI] [PubMed] [Google Scholar]
- 28.Franovic A, Robert I, Smith K, Kurban G, Pause A, Gunaratnam L, Lee S. Multiple acquired renal carcinoma tumor capabilities abolished upon silencing of ADAM17. Cancer Res 66: 8083–8090, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Goligorsky MS, Lieberthal W, Racusen L, Simon EE. Integrin receptors in renal tubular epithelium: new insights into pathophysiology of acute renal failure. Am J Physiol Renal Fluid Electrolyte Physiol 264: F1–F8, 1993. [DOI] [PubMed] [Google Scholar]
- 30.Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol 45: 146–169, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo L, Takino T, Endo Y, Domoto T, Sato H. Shedding of kidney injury molecule-1 by membrane-type 1 matrix metalloproteinase. J Biochem 152: 425–432, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney injury molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 62: 237–244, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol 27: 244–250, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Hung CC, Ichimura T, Stevens JL, Bonventre JV. Protection of renal epithelial cells against oxidative injury by endoplasmic reticulum stress preconditioning is mediated by ERK1/2 activation. J Biol Chem 278: 29317–29326, 2003. [DOI] [PubMed] [Google Scholar]
- 35.Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 118: 1657–1668, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 273: 4135–4142, 1998. [DOI] [PubMed] [Google Scholar]
- 37.Ichimura T, Brooks CR, Bonventre JV. Kim-1/Tim-1 and immune cells: shifting sands. Kidney Int 81: 809–811, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ichimura T, Hung CC, Yang SA, Stevens JL, Bonventre JV. Kidney injury molecule-1: a tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am J Physiol Renal Physiol 286: F552–F563, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Jasuja RR, Mier JW. Differential effects of hydroxamate inhibitors on PMA and ligand-induced l-Selectin down-modulation: role of membrane proximal and cytoplasmic domains. Int J Immunopathol Pharmacol 13: 1–12, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, Ravichandran KS. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 493: 547–551, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaushal GP, Basnakian AG, Shah SV. Apoptotic pathways in ischemic acute renal failure. Kidney Int 66: 500–506, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239–257, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim S, Park SY, Kim SY, Bae DJ, Pyo JH, Hong M, Kim IS. Cross talk between engulfment receptors stabilin-2 and integrin alphavbeta5 orchestrates engulfment of phosphatidylserine-exposed erythrocytes. Mol Cell Biol 32: 2698–2708, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 27: 927–940, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuchroo VK, Meyers JH, Umetsu DT, DeKruyff RH. TIM family of genes in immunity and tolerance. Adv Immunol 91: 227–249, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol 282: C227–C241, 2002. [DOI] [PubMed] [Google Scholar]
- 47.Liangos O, Perianayagam MC, Vaidya VS, Han WK, Wald R, Tighiouart H, MacKinnon RW, Li L, Balakrishnan VS, Pereira BJ, Bonventre JV, Jaber BL. Urinary N-acetyl-beta-(D)-glucosaminidase activity and kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J Am Soc Nephrol 18: 904–912, 2007. [DOI] [PubMed] [Google Scholar]
- 48.Lieberthal W, Levine JS. Mechanisms of apoptosis and its potential role in renal tubular epithelial cell injury. Am J Physiol Renal Fluid Electrolyte Physiol 271: F477–F488, 1996. [DOI] [PubMed] [Google Scholar]
- 49.Lim AI, Chan LY, Lai KN, Tang SC, Chow CW, Lam MF, Leung JC. Distinct role of matrix metalloproteinase-3 in kidney injury molecule-1 shedding by kidney proximal tubular epithelial cells. Int J Biochem Cell Biol 44: 1040–1050, 2012. [DOI] [PubMed] [Google Scholar]
- 50.Liu C, Xu P, Lamouille S, Xu J, Derynck R. TACE-mediated ectodomain shedding of the type I TGF-beta receptor downregulates TGF-beta signaling. Mol Cell 35: 26–36, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matute-Bello G, Martin TR. Science review: apoptosis in acute lung injury. Crit Care 7: 355–358, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, Freeman GJ, Umetsu DT, DeKruyff RH. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat Immunol 2: 1109–1116, 2001. [DOI] [PubMed] [Google Scholar]
- 53.Miksa M, Amin D, Wu R, Ravikumar TS, Wang P. Fractalkine-induced MFG-E8 leads to enhanced apoptotic cell clearance by macrophages. Mol Med 13: 553–560, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mohler KM, Sleath PR, Fitzner JN, Cerretti DP, Alderson M, Kerwar SS, Torranee DS, Otten-Evans C, Greenstreet T, Weerawarna K, Kronheim SR, Petersen M, Gerhart M, Kozlosky CJ, March CJ, Black RA. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature 370: 218–220, 1994. [DOI] [PubMed] [Google Scholar]
- 55.Morimoto K, Janssen WJ, Fessler MB, McPhillips KA, Borges VM, Bowler RP, Xiao YQ, Kench JA, Henson PM, Vandivier RW. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol 176: 7657–7665, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Moss ML, Catherine Jin SL, Milla ME, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Warner J, Willard D, Becherer JD. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385: 733–736, 1997. [DOI] [PubMed] [Google Scholar]
- 57.Moss ML, Rasmussen FH. Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal Biochem 366: 144–148, 2007. [DOI] [PubMed] [Google Scholar]
- 58.Nissenson AR. Acute renal failure: definition and pathogenesis. Kidney Int Suppl 66: S7–S10, 1998. [PubMed] [Google Scholar]
- 59.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science 282: 1281–1284, 1998. [DOI] [PubMed] [Google Scholar]
- 60.Qu D, Wang Y, Esmon NL, Esmon CT. Regulated endothelial protein C receptor shedding is mediated by tumor necrosis factor-alpha converting enzyme/ADAM17. J Thromb Haemost 5: 395–402, 2007. [DOI] [PubMed] [Google Scholar]
- 61.Racusen LC, Fivush BA, Li YL, Slatnik I, Solez K. Dissociation of tubular cell detachment and tubular cell death in clinical and experimental “acute tubular necrosis.” Lab Invest 64: 546–556, 1991. [PubMed] [Google Scholar]
- 62.Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, Reiss K, Saftig P, Bianchi ME. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J 22: 3716–3727, 2008. [DOI] [PubMed] [Google Scholar]
- 62a.Ren Y, Savill J. Apoptosis: the importance of being eaten. Cell Death Differ 5: 563–568, 1998. [DOI] [PubMed] [Google Scholar]
- 63.Rovere P, Sabbadini MG, Vallinoto C, Fascio U, Zimmermann VS, Bondanza A, Ricciardi-Castagnoli P, Manfredi AA. Delayed clearance of apoptotic lymphoma cells allows cross-presentation of intracellular antigens by mature dendritic cells. J Leukoc Biol 66: 345–349, 1999. [DOI] [PubMed] [Google Scholar]
- 64.Sabbisetti VS, Ito K, Wang C, Yang L, Mefferd SC, Bonventre JV. Novel assays for detection of urinary KIM-1 in mouse models of kidney injury. Toxicol Sci 131: 13–25, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santiago C, Ballesteros A, Martinez-Munoz L, Mellado M, Kaplan GG, Freeman GJ, Casasnovas JM. Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity 27: 941–951, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Santiago C, Ballesteros A, Tami C, Martinez-Munoz L, Kaplan GG, Casasnovas JM. Structures of T Cell immunoglobulin mucin receptors 1 and 2 reveal mechanisms for regulation of immune responses by the TIM receptor family. Immunity 26: 299–310, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, Graham DK. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 109: 1026–1033, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol 32: 380–387, 2011. [DOI] [PubMed] [Google Scholar]
- 69.Singh P, Okusa MD. The role of tubuloglomerular feedback in the pathogenesis of acute kidney injury. Contrib Nephrol 174: 12–21, 2011. [DOI] [PubMed] [Google Scholar]
- 70.Tang ZY, Loss G, Carmody I, Cohen AJ. TIMP-3 ameliorates hepatic ischemia/reperfusion injury through inhibition of tumor necrosis factor-alpha-converting enzyme activity in rats. Transplantation 82: 1518–1523, 2006. [DOI] [PubMed] [Google Scholar]
- 70a.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science 296: 155–158, 2002. [DOI] [PubMed] [Google Scholar]
- 71.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 334: 1448–1460, 1996. [DOI] [PubMed] [Google Scholar]
- 72.Vaidya VS, Ozer JS, Dieterle F, Collings FB, Ramirez V, Troth S, Muniappa N, Thudium D, Gerhold D, Holder DJ, Bobadilla NA, Marrer E, Perentes E, Cordier A, Vonderscher J, Maurer G, Goering PL, Sistare FD, Bonventre JV. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol 28: 478–485, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaidya VS, Ramirez V, Ichimura T, Bobadilla NA, Bonventre JV. Urinary kidney injury molecule-1: a sensitive quantitative biomarker for early detection of kidney tubular injury. Am J Physiol Renal Physiol 290: F517–F529, 2006. [DOI] [PubMed] [Google Scholar]
- 73a.Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 129: 1673–1682, 2006. [DOI] [PubMed] [Google Scholar]
- 74.Vecchi M, Rudolph-Owen LA, Brown CL, Dempsey PJ, Carpenter G. Tyrosine phosphorylation and proteolysis. Pervanadate-induced, metalloprotease-dependent cleavage of the ErbB-4 receptor and amphiregulin. J Biol Chem 273: 20589–20595, 1998. [DOI] [PubMed] [Google Scholar]
- 75.Waetzig GH, Rosenstiel P, Arlt A, Till A, Brautigam K, Schafer H, Rose-John S, Seegert D, Schreiber S. Soluble tumor necrosis factor (TNF) receptor-1 induces apoptosis via reverse TNF signaling and autocrine transforming growth factor-beta1. FASEB J 19: 91–93, 2005. [DOI] [PubMed] [Google Scholar]
- 76.Wong SH, Barlow JL, Nabarro S, Fallon PG, McKenzie AN. Tim-1 is induced on germinal centre B cells through B-cell receptor signalling but is not essential for the germinal centre response. Immunology 131: 77–88, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci 118: 539–553, 2005. [DOI] [PubMed] [Google Scholar]
- 78.Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol 16: 189–197, 2006. [DOI] [PubMed] [Google Scholar]
- 79.Xiao S, Brooks CR, Zhu C, Wu C, Sweere JM, Petecka S, Yeste A, Quintana FJ, Ichimura T, Sobel RA, Bonventre JV, Kuchroo VK. Defect in regulatory B-cell function and development of systemic autoimmunity in T-cell Ig mucin 1 (Tim-1) mucin domain-mutant mice. Proc Natl Acad Sci USA 109: 12105–12110, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell 37: 551–566, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu P, Liu J, Sakaki-Yumoto M, Derynck R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci Signal 5: ra34, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82a.Yang L, Brooks CR, Xiao S, Sabbisetti V, Ichimura, Vijay K, Kuchroo JVB. Tubular kidney injury molecule-1 mediated phagocytosis downregulates the kidney's innate immune response after injury (Abstract). In: Kidney Week 2012. San Diego, CA: American Society of Nephrology, 2012, p. 51A. [Google Scholar]
- 83.Yu W, Beaudry S, Negoro H, Boucher I, Tran M, Kong T, Denker BM. H2O2 activates G protein, alpha 12 to disrupt the junctional complex and enhance ischemia reperfusion injury. Proc Natl Acad Sci USA 109: 6680–6685, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Z, Humphreys BD, Bonventre JV. Shedding of the urinary biomarker kidney injury molecule-1 (KIM-1) is regulated by MAP kinases and juxtamembrane region. J Am Soc Nephrol 18: 2704–2714, 2007. [DOI] [PubMed] [Google Scholar]
- 85.Zhang Z, Oliver P, Lancaster JR, Jr, Schwarzenberger PO, Joshi MS, Cork J, Kolls JK. Reactive oxygen species mediate tumor necrosis factor alpha-converting, enzyme-dependent ectodomain shedding induced by phorbol myristate acetate. FASEB J 15: 303–305, 2001. [DOI] [PubMed] [Google Scholar]
- 86.Zhang ZX, Wang S, Huang X, Min WP, Sun H, Liu W, Garcia B, Jevnikar AM. NK cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J Immunol 181: 7489–7498, 2008. [DOI] [PubMed] [Google Scholar]