

Abstract
Protein tyrosine phosphatase of liver regeneration-1 (Prl-1) is an immediate-early gene that is significantly induced during liver regeneration. Several in vitro studies have suggested that Prl-1 is important for the regulation of cell cycle progression. To evaluate its function in liver regeneration, we ablated the Prl-1 gene specifically in mouse hepatocytes using the Cre-loxP system. Prl-1 mutant mice (Prl-1loxP/loxP;AlfpCre) appeared normal and fertile. Liver size and metabolic function in Prl-1 mutants were comparable to controls, indicating that Prl-1 is dispensable for liver development, postnatal growth, and hepatocyte differentiation. Mutant mice demonstrated a delay in DNA synthesis after 70% partial hepatectomy, although ultimate liver mass restoration was not affected. At 40 h posthepatectomy, reduced protein levels of the cell cycle regulators cyclin E, cyclin A2, cyclin B1, and cyclin-dependent kinase 1 were observed in Prl-1 mutant liver. Investigation of the major signaling pathways involved in liver regeneration demonstrated that phosphorylation of protein kinase B (AKT) and signal transducer and activator of transcription (STAT) 3 were significantly reduced at 40 h posthepatectomy in Prl-1 mutants. Taken together, this study provides evidence that Prl-1 is required for proper timing of liver regeneration after partial hepatectomy. Prl-1 promotes G1/S progression via modulating expression of several cell cycle regulators through activation of the AKT and STAT3 signaling pathway.
Keywords: protein tyrosine phosphatase, hepatectomy
the liver retains the capacity to rapidly respond to changes in mass in adulthood in both humans and animals. The ability to restore its functional mass is essential in the response to clinical situations such as surgical removal of part of the organ, for instance, after tumor resection or living-related liver transplantation. Liver regeneration can be modeled by the partial hepatectomy paradigm in rodents, in which 70% of the liver is removed and liver mass is restored within 10–14 days (18). Within 30 min of surgery, growth factor and cytokine stimulation lead to the activation of preexisting transcription factors and activation of a genetic program that stimulates normally quiescent hepatocytes and nonparenchymal liver cells to reenter the cell cycle (11, 21, 24, 33). The genes that are either increased from basal levels of expression or are induced de novo encode proteins involved in maintaining homeostasis and stimulating cells to reenter the cell cycle and proliferate. There are at least two distinct phases of liver regeneration: a priming phase in which hepatocytes are induced to reenter the cell cycle, followed by a second phase in which hepatocytes respond to growth factors and progress through the G1 stage of the cell cycle (11). Among the genes induced massively following partial hepatectomy is “Phosphatase of regenerating liver 1” (Prl-1) (7), also known as PTP4a1. Expression of Prl-1 parallels hepatocyte proliferation during liver regeneration (8).
The Prl enzymes constitute a unique subfamily of protein tyrosine phosphatases and play an important role during cell development and tissue regeneration (5). Prl-1 expression is elevated in multiple rapidly proliferating cell and cancer types, including mitogen-induced hepatoma, NIH 3T3 cells, and a number of human tumor cell lines, suggesting that Prl-1 is involved in cell growth and tumorigenesis (7, 8, 10, 27, 31, 34). Short-hairpin RNA (shRNA)-mediated suppression of both Prl-1 and Prl-2 significantly inhibited colony formation of pancreatic cancer cell lines (31). Overexpression of Prl-1 in 3T3 cells resulted in rapid growth rates (7). Overexpression of Prl-1 and Prl-2 in the epithelial cell line D27 resulted in shorter doubling times relative to control cell lines (4). In addition, overexpression of Prl-1 and Prl-2 in these cells promoted tumor formation upon injection in nude mice (4).
Although Prl-1 expression is significantly induced after partial hepatectomy, its function during liver regeneration in vivo remains unknown. Given that Prl-1 is overexpressed in multiple tumor cell lines, and that suppression or mutation of Prl-1 leads to delayed cell growth in vitro, we hypothesized that PRL-1 contributes to hepatocyte proliferation during liver regeneration, possibly via modulating the activity of key cell cycle regulators or signaling pathways such as the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathway. To address this question directly, we derived a conditional Prl-1 allele and ablated the gene in hepatocytes. Hepatocyte-specific deletion of Prl-1 caused delayed liver regeneration due to altered activation of several proproliferative factors.
MATERIALS AND METHODS
Mice.
The Prl-1loxP targeting vector was constructed using standard subcloning techniques. The Prl-1loxP targeting vector introduces two loxP sites flanking exons 2–4 into the Prl-1 gene. To allow for selection of stably transfected ES cell clones, the Prl-1loxP targeting vector contained a neomycin resistance/thymidine kinase (neo-tk) cassette flanked by two FRT sites. The neo-tk cassette was subsequently deleted in the targeted ES clones by transient transfection with a Flp recombinase expression vector. Correctly targeted ES clones were screened by Southern blot analysis (data not shown) and injected into C57BL/6J mouse blastocysts, which were transferred to pseudopregnant NMRI females, and chimeric offspring were identified by the presence of agouti fur. Chimeric males were mated to C57BL/6 females to obtain ES cell-derived offspring, which were analyzed by genotyping of tail DNA to identify Prl-1loxP/+ heterozygous mice. Prl-1loxP/+ heterozygous mice were bred inter se to obtain Prl-1loxP/loxP homozygous mice. The α-fetoprotein Alfp-Cre mouse (38) was mated with Prl-1loxP/loxP homozygous mice to obtain mice with hepatocyte-specific deletion of Prl-1 (Prl-1loxP/loxP;Alfp-Cre mice, referred to below as “mutants”).
Animal procedures.
Seventy percent partial hepatectomy was performed on 12- to 14-wk-old male mice. Mice were anesthetized with isofluorane, and the upper left lobe, the upper right lobe, and lower right lobe were removed, resulting in resection of 70% of liver tissue (17). For bromodeoxyuridine (BrdU) labeling, the BrdU-labeling reagent (Zymed Laboratories, South San Francisco, CA) was injected subcutaneously 1 h before death at a dose of 1 ml reagent/100 g body wt. Livers were collected at different time points for histology, RNA isolation, and protein preparation. Liver sections were stained for BrdU or phosphorylated histone 3 (PH3), and three random areas of each liver were imaged. BrdU- or PH3-positive cells and the total cell number per image were counted manually in blinded fashion. For the 40-h time point, the percentage of BrdU- or PH3-positive cells was calculated separately for both central vein and portal triad regions. All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
RT-PCR and quantitative real-time PCR.
The liver was dissected and total RNA extracted in TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. RNA was reverse transcribed using 1 μg oligo(dT) primer, Super-Script II Reverse Transcriptase, and accompanying reagents (Invitrogen). The RT-PCR reaction mixture contained cDNA, 0.25 mM dNTP, 1× PCR buffer, 0.2 μl of Taq DNA polymerase (Invitrogen), and 0.25 μM forward (ggctgtatgattaggccacaa) and reverse (caccatggatggtctgagagt) primers for amplification of the Prl-1 cDNA. For quantitative real-time PCR, the reaction mixture was prepared using SYBR GreenER qPCR SuperMix Universal (Invitrogen). Mitogen primers are as follows: epithelial growth factor (EGF) (cacaactgcggtgagaatgc; aaatggtggccatcttcccc), hepatocyte growth factor (HGF) (atttacggctggggctacac; acatccacgaccaggaacaa) transforming growth factor (TGF) (tcccaacatccctttcctctc, agggaccaacgtcaccatcta), amphiregulin (Areg) (cagtgcacctttggaaacgata; acaactgggcatctggaacc), fibroblast growth factor (FGF) 1 (tggggacaatgtctgtcagc; gtgactgaccgttgagggtt), FGF2 (ggctgctggcttctaagtgt; ttctgtccaggtcccgtttt), interleukin (IL)-6 (gacaaagccagagtccttcaga; aacgcactaggtttgccgag), tumor necrosis factor (TNF) (agaccctcacactcagatca; acaaggtacaacccatcggc), platelet-derived growth factor (PDGF) (gtactgaatttcgccgccac; ctcagcccctacggagtcta), vascular endothelial growth factor (VEGF) (taaatcctggagcgttcactgt; ttccggtgagaggtctggtt), stem cell factor (SCF) (ctggaagaagaaacagtcaagtct, aggtcacgggtagcaagaac), angiopoietin1 (Angpt1) (atgttcgaaaactcatcttgggt; ttgctacacacatgttgggatt), and angiopoietin2 (Angpt2) (gagtccaactacaggattcacct; ccggaccccttccagtagta). Reactions were performed on a Mx4000 Multiplex Quantitative PCR System (Stratagene). All reactions were performed in triplicate with reference dye normalization, and median threshold cycle values were used for analysis.
Immunohistochemistry.
Livers were fixed in 4% paraformaldehyde overnight at 4°C, washed with 1× PBS and 70% ethanol, embedded in paraffin, and cut to 6-μm sections. Slides were subjected to microwave antigen retrieval by pressure cooker in 10 mM citric acid buffer (pH 6.0). Slides were quenched in 2.25% hydrogen peroxide at room temperature for 20 min. Slides were blocked with avidin D blocking reagent (Vector) and biotin blocking reagent (Vector) for 15 min each at room temperature, with a quick rinse of 1× PBS in between. All slides were blocked with protein-blocking reagent (Immunotech) for 20 min at room temperature. The primary antibodies against BrdU (B2850-01; U.S. Biological) or PH3 (06–570; Upstate Biotech) were diluted in 1× PBS 1,000- or 5,000-fold, respectively. Slides were incubated in primary antibody overnight at 4°C, washed in 1× PBS, and then incubated with horseradish peroxidase (HRP)-conjugated anti-sheep antibodies (1:200; Vector Laboratories) or anti-rabbit antibodies (1:200; Vector Laboratories) for 30 min at 37°C. Slides were rinsed with 1× PBS and incubated with HRP-conjugated ABC reagent (Vector Elite kit) for 30 min at 37°C. After being washed, slides were developed using a DAB substrate kit (Vector Laboratories) and counterstained with hematoxylin.
Western blot analysis.
Whole cell lysates were prepared from livers after partial hepatectomy, and 50 μg of protein were resolved by SDS-PAGE (4–12% gel; Invitrogen). Resolved proteins were transferred to PVDF (Invitrogen) membranes, and cyclin A2 (ab7956, 1:200; Abcam), cyclin B1 (H-433, sc-752, 1:200; Santa Cruz Biotechnology), cyclin D1 (RM-9104-S, 1:150; Thermo Scientific), cyclin E (sc-247; Santa Cruz), cyclin-dependent kinase 1 (CDK1, 9112, 1:1,000; Cell Signaling), phosphorylated (p) CDK1 (Tyr15, 9111, 1:1,000; Cell Signaling), pan-AKT (9272, 1:1,000; Cell Signaling), p-AKT (Ser473, 9271, 1:1,000; Cell Signaling), glycogen synthase kinase (GSK) 3-β (9315, 1:1,000; Cell Signaling), p-GSK3-β (Ser9, 9336, 1:1,000; Cell Signaling), Erk1/2 (4695, 1:1,000; Cell Signaling), p-Erk1/2 (9101, 1:1,000; Cell Signaling), signal transducer and activator of transcription (STAT) 3 (9132, 1:1,000; Cell Signaling), and p-STAT3 (Tyr705, 9131, 1:1,000; Cell Signaling) immunoreactivity was detected using the antibodies specified. Immunoreactivity was visualized by use of ECL (manufacturer's protocol). For loading control, we employed an antibody against β-actin (4970, 1:1,000; Cell Signaling). Bands were quantified by densitometry.
RESULTS
Deletion of Prl-1 does not affect liver development and growth.
Previous studies have suggested that Prl-1 might play a role in hepatocyte proliferation, since Prl-1 expression is significantly induced during liver development and regeneration following partial hepatectomy (8). The function of Prl-1 in the liver, however, has never been investigated in vivo. To address this question directly, we derived mice deficient for Prl-1 in hepatocytes using the Cre-loxP system as shown in Fig. 1A. Exons 2–4 of the Prl-1 gene were flanked with loxP sites, which results in a null allele after Cre-mediated recombination. Prl-1loxP/loxP mice were bred with Alfp-Cre transgenic mice, which have been shown previously to efficiently target loxP-flanked loci in fetal hepatoblasts (38). Efficient deletion of Prl-1 in the liver was demonstrated by RT-PCR (Fig. 1B) and DNA sequencing of the deleted allele (data not shown). The RT-PCR product of the wild-type Prl-1 transcript has a size of 1,524 bp, and deletion of exons 2–4 of the Prl-1 gene generates a smaller transcript of 507 bp (Fig. 1B). Quantitative RT-PCR data confirm the absence of Prl-1 mRNA expression in Prl-1 mutant liver (data not shown).
Fig. 1.
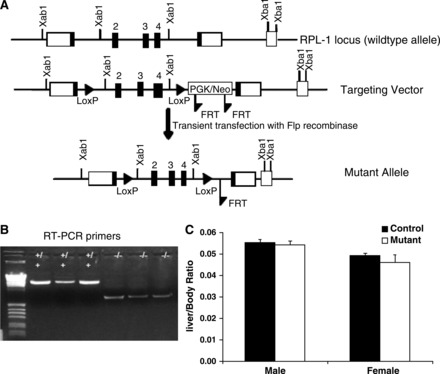
Derivation of mice with hepatcoyte-specific deletion of protein tyrosine phosphatase of liver regeneration-1 (Prl-1). A: scheme for derivation of a conditional Prl-1 null allele. Two loxP sites were inserted to flank exons 2 through 4 of the Prl-1 gene by homologous recombination in mouse embryonic stem cells. The FRT-flanked neomycin resistance cassette was removed by transient transfection with a Flp-recombinase expression plasmid. B: for gene ablation in hepatocytes, Prl-1loxP mice were bred to AlfpCre mice to obtain Prl-1loxP/loxP;AlfpCre mice. The deletion of Prl-1 in the liver was confirmed by RT-PCR. +/+, Control mice, the length of the wild-type Prl-1 transcript is 1,524 bp; −/−, mutant (Prl-1loxP/loxP;AlfpCre) mice, the length of the smaller transcript is 507 bp. C: the liver and body weight of 5-wk-old male and female mice were measured to calculate the liver-to-body weight ratio (n = 5).
To assess whether deletion of Prl-1 affects liver development or postnatal growth, we determined the liver-to-body ratio in control and mutant mice. For both male and female mice, the absence of Prl-1 did not affect liver size in adult mice significantly (Fig. 1C). Next, we measured various metabolic parameters to investigate whether liver function was altered by Prl-1 deletion (Table 1). Blood serum tests indicated that the levels of these metabolites and liver enzymes were comparable between control and mutant mice for both male and female mice. Alanine aminotransferase (ALT) values were slightly higher in mutant female mice compared with control values but fell within the normal ALT reference range (24–140 U/l). These data demonstrate that Prl-1 is dispensable for liver development, postnatal growth, and normal liver homeostasis.
Table 1.
Liver function tests in 5-wk-old mice
| Summary of Serum Biochemistry |
||||
|---|---|---|---|---|
| Controls | Mutants | P value* | Ref. range | |
| Female cohort | ||||
| n | 6 | 4 | ||
| AST, U/l | 60.83 ± 2.3 | 64.5 ± 8.4 | 0.625 | 72–288 |
| ALT, U/l | 26.5 ± 1.4 | 34.75 ± 1.9 | 0.008# | 24–140 |
| CPK, U/l | 300.8 ± 94.2 | 460.3 ± 180.6 | 0.414 | 0–800 |
| LDH, U/l | 295.8 ± 28.6 | 231.8 ± 36.7 | 0.202 | 200–600 |
| Amylase, U/l | 1,366 ± 45.6 | 1,473 ± 46.9 | 0.155 | 602–2,311 |
| BUN, mg/dl | 31.8 ± 0.86 | 24 ± 2.35 | 0.011 | 9–38 |
| Creat, mg/dl | 0.4 ± 0.026 | 0.425 ± 0.025 | 0.527 | 0.2–0.7 |
| T Bili, mg/dl | 0.3 ± 0.04 | 0.275 ± 0.03 | 0.684 | 0–0.9 |
| Uric, mg/dl | 1.9 ± 0.24 | 1.975 ± 0.66 | 0.904 | 2.2–4.6 |
| Albumin, g/dl | 3.033 ± 0.1 | 2.825 ± 0.1 | 0.206 | 2.6–4.6 |
| TP, g/dl | 4.5 ± 0.15 | 4.375 ± 0.11 | 0.573 | 4.0–6.2 |
| Male cohort | ||||
| n | 4 | 5 | ||
| AST, U/l | 49.25 ± 1.9 | 61 ± 7.1 | 0.195 | 72–288 |
| ALT, U/l | 27.75 ± 4.2 | 34.6 ± 4.9 | 0.339 | 24–140 |
| CPK, U/l | 316.8 ± 43.3 | 323.8 ± 139.2 | 0.967 | 0–800 |
| LDH, U/l | 344.3 ± 55.7 | 316.6 ± 50.8 | 0.725 | 200–600 |
| Amylase, U/l | 1,339 ± 162.1 | 1,537 ± 134.8 | 0.374 | 602–2,311 |
| BUN, mg/dl | 29.25 ± 1.7 | 27.6 ± 1.0 | 0.413 | 9–38 |
| Creat, mg/dl | 0.4 ± 2.5E-05 | 0.42 ± 0.04 | 0.651 | 0.2–0.7 |
| T Bili, mg/dl | 0.25 ± 0.06 | 0.22 ± 0.02 | 0.638 | 0–0.9 |
| Uric, mg/dl | 1.65 ± 0.23 | 2.02 ± 0.46 | 0.527 | 2.2–4.6 |
| Albumin, g/dl | 2.875 ± 0.05 | 3.1 ± 0.13 | 0.176 | 2.6–4.6 |
| TP, g/dl | 4.5 ± 0.03 | 4.8 ± 0.2 | 0.267 | 4.0–6.2 |
Sera of 5-wk-old mice were collected, and various metabolic parameters, including aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatine phosphokinase (CPK), lactate dehydrogenase (LDH), amylase, urea nitrogen (BUN), creatinine (Creat), total bilirubin (T Bili), uric acid (Uric), albumin, and total protein (TP) were determined. The levels of these metabolites were comparable between control and protein tyrosine phosphatase of liver regeneration-1 mutant mice.
P value comparing control and mutant biochemistry values using the Student's t-test.
Statistical significance, P < 0.01.
Prl-1 mutants exhibit delayed cell cycle progression during liver regeneration.
Expression of Prl-1 is nearly undetectable in the adult quiescent liver; however, it is markedly induced in rats 16 h posthepatectomy (8), indicating a potential role for Prl-1 in liver regeneration. We determined Prl-1 mRNA expression in mice and found it peaked 40–48 h posthepatectomy (Fig. 2E). To determine whether deletion of Prl-1 affects liver regeneration, we performed 70% partial hepatectomy in adult male control and mutant mice (12–14 wk old). BrdU was given to the mice 1 h before death, livers were collected, and sections were stained for BrdU at 0, 24 (1 day), 40, 48 (2 days), 72 (3 days), and 168 (7 days) h posthepatectomy (Fig. 2). Prl-1 mRNA was confirmed to be absent before and posthepatectomy in Prl-1 mutant liver, and Prl-2 mRNA expression remained unchanged between control and Prl-1 mutant liver (data not shown). Quantification of BrdU-positive hepatocytes during the posthepatectomy time course showed that controls had the highest proportion of cells in S phase at 40 h, whereas this posthepatectomy peak of BrdU incorporation was blunted in Prl-1 mutants (Fig. 2, A and B). Wnt signaling has been shown to determine metabolic zonation by regulating pericentral gene expression and proliferation in response to liver injury (2, 29, 30, 32). To investigate if the zonation of replication is altered in Prl-1 mutants, the percent of proliferation was quantified at the central vein and portal triad regions of the liver. Prl-1 mutants showed a significant reduction in the hepatocyte proliferation rate in both central vein (30%) and portal triad (34%) regions at 40 h posthepatectomy (Fig. 2C). Mutant hepatocytes exhibited higher labeling indexes at 48 and 72 h posthepatectomy than controls (Fig. 2B). This delayed proliferative response did not affect liver mass restoration, as indicated by the liver-to-body weight ratio (Fig. 2D).
Fig. 2.
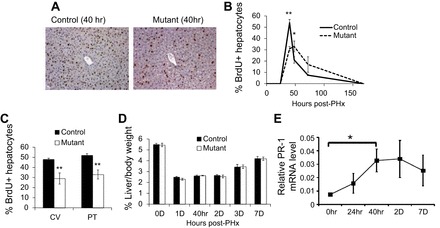
Delayed S phase entry in Prl-1 mutant mice following partial hepatectomy. Partial hepatectomy (70%) was performed on 12- to 14-wk-old male mice (n = 5). Bromodeoxyuridine (BrdU) was injected sc 1 h before mice were killed, and liver sections were collected. A: BrdU staining of control and mutant mice 40 h posthepatectomy. There are fewer BrdU+ cells in the mutant mice. B: time course of BrdU+ cells in the liver of control and mutant mice posthepatectomy. Mutant mice show a blunted and delayed peak of proliferation. C: quantification of BrdU+ cells at 40 h posthepatectomy in central vein (CV) and portal triad (PT) regions (n = 5). D: the ratio of liver-to-body weight was not significantly different for control mice vs. Prl-1 mutants during the posthepatectomy time course (n = 4, male). E: quantitative RT-PCR analysis shows that Prl-1 mRNA expression peaks at 40 h posthepatectomy. Relative expression shown normalized to β-actin mRNA levels. Data are means ± SE. *P < 0.05 and **P < 0.01.
Next, we analyzed the number of hepatocytes in mitosis by immunohistochemical staining of liver sections for PH3 (Fig. 3A). Quantification of PH3-positive cells showed a 69% decrease of mitotically active hepatocytes in Prl-1 mutants at 40 h posthepatectomy (Fig. 3B). Taken together, these data demonstrate that Prl-1 is important for the timing of cell cycle progression during liver regeneration. Similar effects on the time course, but not on ultimate restoration of liver mass, have been observed in previous mouse mutants for key regulators, indicative of the multiple redundant pathways that ensure adequate liver mass in vertebrates (see discussion).
Fig. 3.
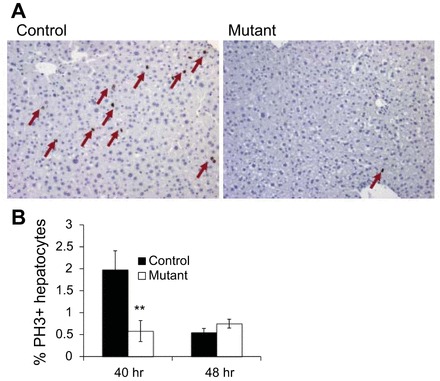
Blunted liver regeneration in Prl-1 male mutant mice detected by phosphorylated histone 3 (PH3) staining. A: liver sections were collected at 40 h posthepatectomy and stained for PH3, a marker of M phase. The number of PH3-positive cells was significantly reduced in Prl-1 mutants. B: quantification of PH3-positive cells at 40 h posthepatectomy (n = 5). Data are means ± SE. **P < 0.01 vs. controls.
Prl-1 regulates the protein levels of cyclin E, B1, A2, and p-CDK1 at 40 h posthepatectomy.
We investigated the molecular mechanism responsible for the delay in hepatocyte cell cycle progression in Prl-1 mutant mice detailed above. Protein levels of key cell cycle regulators were measured by Western blot analysis in livers 40 h posthepatectomy, the time point with the largest impact of Prl-1 deficiency. We observed reduced protein levels of the cell cycle regulators cyclin E, cyclin B1, and cyclin A2 in Prl-1 mutants (Fig. 4, A–E). Unphosphorylated and p-CDK1 protein levels were also significantly reduced at 40 h posthepatectomy (Fig. 4, A and F, and data not shown). The decrease of the G1/S cyclin E likely contributes to the delayed cell cycle progression in mutant hepatocytes detected by BrdU incorporation. The lower levels of the G2/M cyclins A2 and B1 reflect the delayed cell cycle progression in Prl-1 mutant mice.
Fig. 4.
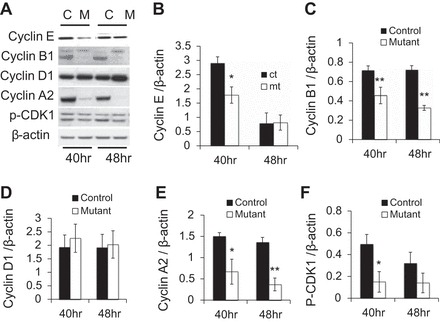
Cell cycle regulators are reduced in Prl-1 mutant mice during liver regeneration. Whole cell lysate was prepared from livers collected 40 and 48 h posthepatectomy for both controls (C) and Prl-1 mutants (M) and subjected to Western blot analysis. A: representative blots for cyclin E, cyclin B1, cyclin D1, cyclin A2, phosphorylated (p) cyclin-dependent kinase 1 (CDK1), unphosphorylated CDK1 (the band above the p-CDK1 band), and β-actin as loading control. Protein levels were quantified by densitometry for 4 different blots for cyclin E (B), cyclin B1 (C), cyclin D1 (D), cyclin A2 (E), and p-CDK1 (F). *P < 0.05 and **P < 0.01.
Prl-1 mutants display attenuated activation of AKT and STAT3 at 40 h posthepatectomy.
After loss of liver mass following partial hepatectomy, or in response to liver injury due to carbon tetrachloride, ethanol, or acetaminophen exposure, several signaling networks are activated by cytokines and growth factors, including IL-6, TNF-α, hepatocyte growth factor, epidermal growth factor, and TGF-β (3, 12). To investigate whether the expression of the hepatic mitogens HGF, FGF1 and -2, and the EGFR ligands were changed due to Prl-1 loss, we assessed the levels of HGF, EGF, TGFα, Amphiregulin, FGF1, and FGF2 in the liver of Prl-1 mutants at 0, 24, and 40 h posthepatectomy. Although HGF mRNA levels were near-significantly reduced in Prl-1 mutant livers at 0 h (P = 0.05), all four mitogens increased to control levels by 24 h posthepatectomy (data not shown). Similarly, no significant difference in mRNA expression of the indirect hepatocyte mitogens TNF-α or IL-6, the hepatocyte-produced stellate cell mitogen PDGF, or the endothelial mitogens VEGF, SCF, Angpt1, or Angpt2 were detected between Prl-1 mutants and control liver at 0, 24, or 40 h posthepatectomy (data not shown).
Signaling networks such as the Ras/ERK/MAPK, PI3K/AKT, JAK/STAT3, and GSK3-β pathways cooperate in liver repair and lead to full regeneration of the organ. Therefore, we assessed the activation of these signaling pathways in the Prl-1-deficient regenerating liver. Prl-1 mutants exhibited lower levels of total and p-AKT at 40 h posthepatectomy, but neither total nor p-ATK levels were significantly different at 0 h (Fig. 5, A–C, and data not shown). In contrast, Erk1/2 and p-Erk1/2 expression was unchanged between control and mutant mice (Fig. 5A). Similarly, there were no differences in p-GSK3-β levels (Fig. 5A). In addition, total STAT3 and p-STAT3 levels were moderately but significantly lower at 40 h, but not 48 h, posthepatectomy in Prl-1 mutants (Fig. 5, A, D, and E). These data indicate that hepatocyte-specific deletion of Prl-1 altered AKT and STAT3 signaling pathways during liver regeneration.
Fig. 5.
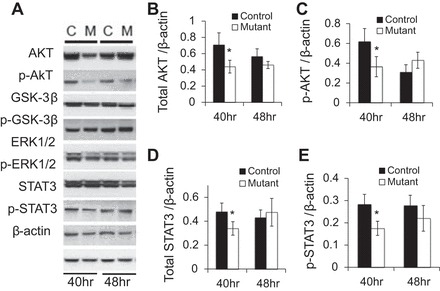
Decreased activation of protein kinase B (AKT) and signal transducer and activator of transcription (STAT) 3 40 h posthepatectomy in Prl-1 mutants. Whole cell lysate was prepared from livers collected at 40 and 48 h posthepatectomy for controls and Prl-1 mutants. Western blots were performed to detect the levels of the proteins indicated (A). Protein levels were quantified by densitometry for four different blots for total Akt (B), p-Akt (C), STAT3 (D), and p-STAT3 (E). *P < 0.05.
DISCUSSION
During liver regeneration after 70% partial hepatectomy, quiescent hepatocytes enter the cell cycle and undergo one or two rounds of replication to restore liver mass. This process is regulated by cytokines, growth factors, and the signaling pathways activated by these factors (3, 12). The protein tyrosine phosphatase Prl-1 contributes to cell proliferation in cultured cell lines, and its expression is induced dramatically in the regenerating liver (1, 7, 8, 13, 31, 34, 35). Here, we demonstrate that Prl-1 is important for the timing of hepatocyte cell cycle entry in vivo. Consistent with this observation, shRNA-mediated suppression of Prl-1 causes decreased proliferation in human lung cancer cells in vitro (1), and overexpression of Prl-1 promotes cell growth and accelerated entry into S phase in NIH 3T3 cells and D27 hamster pancreatic ductal epithelial cells (7, 35).
Although Prl-1 deletion caused a delay in cell cycle progression during liver regeneration, liver mass was nevertheless restored by the 7th day posthepatectomy. There are two possible explanations for this observation. First, Prl-2 is ubiquitously expressed in many tissues, including the liver (9, 37), and may compensate for the loss of Prl-1 function in hepatocytes. Thus, pancreatic cell lines showed decreased cell growth only when treated with both Prl-1 and Prl-2 small-interfering RNA (31), indicating that these two proteins have redundant roles in this context. However, we saw no compensatory upregulation of Prl-2 expression in Prl-1 mutants compared with controls throughout the posthepatectomy time course (data not shown). Second, regaining liver mass despite delayed entry into S phase is a common phenomenon in animal models that are defective in liver regeneration (16, 22, 25, 28). Maintaining functional liver mass is so essential for life that vertebrates have evolved multiple redundant pathways to guarantee this process (23).
In parallel with the delay of liver regeneration in Prl-1 mutants, we also observed a lack of upregulation of cyclin E, cyclin A2, cyclin B1, and CDK1 at 40 h posthepatectomy, the peak of DNA synthesis during liver regeneration in mice. The failure to induce cyclin E to the full extent likely contributes to the G1/S delay in Prl-1 mutant mice detected by BrdU incorporation. In the liver, STAT3 is activated by the IL-6 family cytokines and IL-22, and the IL-6/STAT3 signaling pathway plays a critical role in liver regeneration (6, 15, 19, 22, 25). STAT3 has been suggested to regulate the G1/S transition during liver regeneration through the upregulation of G1/S cyclins, including cyclin E (14, 22). We found STAT3 expression attenuated 40 h posthepatectomy in Prl-1 mutants, which correlated with decreased cyclin E and cyclin A2 expression and delayed cell cycle progression. Ultimately, liver mass was restored by the 7th day posthepatectomy in Prl-1 mutants. Similarly, hepatocyte-specific deletion of STAT3 causes reduced DNA synthesis at 40 h posthepatectomy; however, liver mass is restored normally in this model 1 wk posthepatectomy due to intact MAPK activation and upregulation of STAT1 (22, 25).
AKT signaling is known for its roles in cell survival and cell cycle regulation in the regenerating liver (20). During liver regeneration, AKT is rapidly activated and promotes cell survival and cell proliferation (19). Mice transgenic for a constitutively active AKT mutant increase liver size by three- to fourfold (26). Furthermore, a recent study demonstrated that micro-RNA-21 accelerates hepatocyte proliferation via activating the PI3K/AKT pathway and elevating cyclin E levels (36). Consistent with the role of AKT in liver regeneration, we observed reduced activation of AKT in Prl-1 mutant livers at 40 h posthepatectomy, which might contribute to delayed liver regeneration in Prl-1 mutants. Future studies will be needed to elucidate the precise molecular links between Prl-1 and its effect on AKT, STAT3, and liver regeneration.
GRANTS
These studies were facilitated by Molecular Pathology and Imaging Core and the Transgenic Mouse Core of the Penn Center for Molecular Studies in Digestive and Liver Disease (P30-DK-50306). The study was supported by the PhRMA Foundation Postdoctoral Fellowship to D. Z. Ye, and National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-049210 and DK-053839 to K. H. Kaestner and R01-DK-044237 to R. Taub.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.J., D.Z.Y., Z.L., and Y.P. performed experiments; Y.J., D.Z.Y., and K.H.K. analyzed data; Y.J., D.Z.Y., L.E.G., and K.H.K. interpreted results of experiments; Y.J. and D.Z.Y. prepared figures; Y.J., D.Z.Y., and K.H.K. drafted manuscript; M.T.-B., L.E.G., and K.H.K. edited and revised manuscript; M.T.-B. and K.H.K. approved final version of manuscript; R.T. and K.H.K. conception and design of research.
ACKNOWLEDGMENTS
We thank Sophia Hammani, Beth Helmbrecht, Karrie Brondell, and Tia Bernard-Banks for help in managing the mouse colony.
Current addresses: Y. Peng, Discovery Biology, Centaurus Biopharma Co., Ltd. Beijing 100195, China; L. E. Greenbaum, Janssen R&D, Spring House, PA 19477.
REFERENCES
- 1.Achiwa H, Lazo JS. PRL-1 tyrosine phosphatase regulates c-Src levels, adherence, and invasion in human lung cancer cells. Cancer Res 67: 643–650, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, Vasseur-Cognet M, Kuo CJ, Kahn A, Perret C, Colnot S. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell 10: 759–770, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Bohm F, Kohler UA, Speicher T, Werner S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol Med 2: 294–305, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cates CA, Michael RL, Stayrook KR, Harvey KA, Burke YD, Randall SK, Crowell PL, Crowell DN. Prenylation of oncogenic human PTP(CAAX) protein tyrosine phosphatases. Cancer Lett 110: 49–55, 1996. [DOI] [PubMed] [Google Scholar]
- 5.Chiarugi P, Buricchi F. Protein tyrosine phosphorylation and reversible oxidation: two cross-talking posttranslation modifications. Antioxidants Redox Signal 9: 1–24, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 274: 1379–1383, 1996. [DOI] [PubMed] [Google Scholar]
- 7.Diamond RH, Cressman DE, Laz TM, Abrams CS, Taub R. PRL-1, a unique nuclear protein tyrosine phosphatase, affects cell growth. Mol Cell Biol 14: 3752–3762, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diamond RH, Peters C, Jung SP, Greenbaum LE, Haber BA, Silberg DG, Traber PG, Taub R. Expression of PRL-1 nuclear PTPase is associated with proliferation in liver but with differentiation in intestine. Am J Physiol Gastrointest Liver Physiol 271: G121–G129, 1996. [DOI] [PubMed] [Google Scholar]
- 9.Dumaual CM, Sandusky GE, Crowell PL, Randall SK. Cellular localization of PRL-1 and PRL-2 gene expression in normal adult human tissues. J Histochem Cytochem 54: 1401–1412, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dumaual CM, Sandusky GE, Soo HW, Werner SR, Crowell PL, Randall SK. Tissue-specific alterations of PRL-1 and PRL-2 expression in cancer. Am J Trans Res 4: 83–101, 2012. [PMC free article] [PubMed] [Google Scholar]
- 11.Fausto N. Liver regeneration. J Hepatol 32, Suppl 1: 19–31, 2000. [DOI] [PubMed] [Google Scholar]
- 12.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology 43: S45–S53, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Fiordalisi JJ, Keller PJ, Cox AD. PRL tyrosine phosphatases regulate rho family GTPases to promote invasion and motility. Cancer Res 66: 3153–3161, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Fukada T, Ohtani T, Yoshida Y, Shirogane T, Nishida K, Nakajima K, Hibi M, Hirano T. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. Embo J 17: 6670–6677, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol 2: 92–100, 2005. [PubMed] [Google Scholar]
- 16.Greenbaum LE, Li W, Cressman DE, Peng Y, Ciliberto G, Poli V, Taub R. CCAAT enhancer- binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J Clin Invest 102: 996–1007, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greene AK, Puder M. Partial hepatectomy in the mouse: technique and perioperative management. J Invest Surg 16: 99–102, 2003. [PubMed] [Google Scholar]
- 18.Higgins GM, Anderson RM. Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol 12: 186–202, 1931. [Google Scholar]
- 19.Hong F, Nguyen VA, Shen X, Kunos G, Gao B. Rapid activation of protein kinase B/Akt has a key role in antiapoptotic signaling during liver regeneration. Biochem Biophys Res Commun 279: 974–979, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Jackson LN, Larson SD, Silva SR, Rychahou PG, Chen LA, Qiu S, Rajaraman S, Evers BM. PI3K/Akt activation is critical for early hepatic regeneration after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol 294: G1401–G1410, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koniaris LG, McKillop IH, Schwartz SI, Zimmers TA. Liver regeneration. J Am Coll Surg 197: 634–659, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Li W, Liang X, Kellendonk C, Poli V, Taub R. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem 277: 28411–28417, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Michalopoulos GK. Liver regeneration. J Cell Physiol 213: 286–300, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michalopoulos GK, DeFrances MC. Liver regeneration. Science 66: 60–66, 1997. [DOI] [PubMed] [Google Scholar]
- 25.Moh A, Iwamoto Y, Chai GX, Zhang SS, Kano A, Yang DD, Zhang W, Wang J, Jacoby JJ, Gao B, Flavell RA, Fu XY. Role of STAT3 in liver regeneration: survival, DNA synthesis, inflammatory reaction and liver mass recovery. Lab Invest 87: 1018–1028, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Mullany LK, Nelsen CJ, Hanse EA, Goggin MM, Anttila CK, Peterson M, Bitterman PB, Raghavan A, Crary GS, Albrecht JH. Akt-mediated liver growth promotes induction of cyclin E through a novel translational mechanism and a p21-mediated cell cycle arrest. J Biol Chem 282: 21244–21252, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Nakashima M, Lazo JS. Phosphatase of regenerating liver-1 promotes cell migration and invasion and regulates filamentous actin dynamics. J Pharmacol Exp Ther 334: 627–633, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roselli HT, Su M, Washington K, Kerins DM, Vaughan DE, Russell WE. Liver regeneration is transiently impaired in urokinase-deficient mice. Am J Physiol Gastrointest Liver Physiol 275: G1472–G1479, 1998. [DOI] [PubMed] [Google Scholar]
- 29.Sekine S, Gutierrez PJ, Lan BY, Feng S, Hebrok M. Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology 45: 361–368, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Sekine S, Lan BY, Bedolli M, Feng S, Hebrok M. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 43: 817–825, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Stephens B, Han H, Hostetter G, Demeure MJ, Von Hoff DD. Small interfering RNA-mediated knockdown of PRL phosphatases results in altered Akt phosphorylation and reduced clonogenicity of pancreatic cancer cells. Mol Cancer Ther 7: 202–210, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology 131: 1561–1572, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Taub R, Greenbaum LE. Transcriptional regulatory signals define cytokine-dependent and independent pathways in liver regeneration. Semin Liver Dis 19: 117–127, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Kirby CE, Herbst R. The tyrosine phosphatase PRL-1 localizes to the endoplasmic reticulum and the mitotic spindle and is required for normal mitosis. J Biol Chem 277: 46659–46668, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Werner SR, Lee PA, DeCamp MW, Crowell DN, Randall SK, Crowell PL. Enhanced cell cycle progression and down regulation of p21(Cip1/Waf1) by PRL tyrosine phosphatases. Cancer Lett 202: 201–211, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Yan-Nan B, Zhao-Yan Y, Li-Xi L, Jiang Y, Qing-Jie X, Yong Z. MicroRNA-21 accelerates hepatocyte proliferation in vitro via PI3K/Akt signaling by targeting PTEN. Biochem Biophys Res Commun 443: 802–807, 2014. [DOI] [PubMed] [Google Scholar]
- 37.Zeng Q, Hong W, Tan YH. Mouse PRL-2 and PRL-3, two potentially prenylated protein tyrosine phosphatases homologous to PRL-1. Biochem Biophys Res Commun 244: 421–427, 1998. [DOI] [PubMed] [Google Scholar]
- 38.Zhang L, Rubins NE, Ahima RS, Greenbaum LE, Kaestner KH. Foxa2 integrates the transcriptional response of the hepatocyte to fasting. Cell Metab 2: 141–148, 2005. [DOI] [PubMed] [Google Scholar]