

Abstract
Osteoarthritis (OA) is associated with increased mechanical damage to joint cartilage. We have previously found that extracellular superoxide dismutase (ECSOD) is decreased in OA joint fluid and cartilage, suggesting oxidant damage may play a role in OA. We explored the effect of forced running as a surrogate for mechanical damage in a transgenic mouse with reduced ECSOD tissue binding. Transgenic mice heterozygous (Het) for the human ECSOD R213G polymorphism and 129-SvEv (wild-type, WT) mice were exposed to forced running on a treadmill for 45 min/day, 5 days/wk, over 8 wk. At the end of the running protocol, knee joint tissue was obtained for histology, immunohistochemistry, and protein analysis. Sedentary Het and WT mice were maintained for comparison. Whole tibias were studied for bone morphometry, finite element analysis, and mechanical testing. Forced running improved joint histology in WT mice. However, when ECSOD levels were reduced, this beneficial effect with running was lost. Het ECSOD runner mice had significantly worse histology scores compared with WT runner mice. Runner mice for both strains had increased bone strength in response to the running protocol, while Het mice showed evidence of a less robust bone structure in both runners and untrained mice. Reduced levels of ECSOD in cartilage produced joint damage when joints were stressed by forced running. The bone tissues responded to increased loading with hypertrophy, regardless of mouse strain. We conclude that ECSOD plays an important role in protecting cartilage from damage caused by mechanical loading.
Keywords: extracellular superoxide dismutase, osteoarthritis, bone, cartilage, running
osteoarthritis (OA) is associated with abnormal mechanical loading on the cartilage surface, particularly in mal-aligned or unstable joints, or in posttraumatic arthritis after intra-articular fractures. In epidemiology studies, heavy laborers and individuals with high body mass index have increased rates of OA (1, 34). Excessive mechanical loading on cartilage and other tissues can generate reactive oxygen species and cartilage damage (16, 33), while antioxidants are protective (3).
We have previously shown that human OA cartilage and joint fluid have reduced levels of a key catalytic antioxidant, extracellular superoxide dismutase (ECSOD), and show evidence of oxidant damage (25). It is unknown whether this deficiency is a primary event in the pathogenesis of OA or a later consequence of the disease. Since OA is a disease process that occurs over a prolonged time period, the effect of excessive mechanical loading on cartilage with reduced antioxidant protection (i.e., loss of ECSOD) provides a clinically relevant model for studying OA.
A naturally occurring human single-nucleotide polymorphism (SNP) in ECSOD occurs at amino acid 213, where glycine is substituted for arginine (23). This substitution is in the heparin binding domain of ECSOD and causes the protein to lose its affinity for the negatively charged extracellular matrix (ECM). This leads to decreased levels of ECSOD in tissue and increased levels in plasma, resulting in a diminished ability of ECSOD to scavenge superoxide in tissue (11). This mutation has been associated with cardiovascular disease (12) but has not been studied in OA. A transgenic mouse model of this human mutation has been recently developed (9). We considered forced treadmill running as a surrogate for excessive mechanical loading given that strenuous running can cause both structural (32) and oxidative damage (2). We postulated that mice with the R213G SNP would be at increased risk of joint damage when exposed to forced treadmill running compared with wild-type mice with normal levels of cartilage ECSOD.
MATERIALS AND METHODS
Animals.
A knock-in mouse for the R213G variant of ECSOD was generated on a 129 SvEv background and characterized by Hartney et al. (9). The 129 SvEv (WT) mice were used as controls for mice heterozygous for the R213G variant of ECSOD (Het), and both strains were used in the following experiments. Animals were housed 5 per cage in standard mouse cages in the animal facility at National Jewish Health (NJH) where they were exposed to a 12:12-h light-dark cycle and fed and provided water ad libitum. The experimental protocol was reviewed and approved by the NJH Institutional Animal Care and Use Committee.
Training.
Male mice, 4–6 mo of age, were randomized to either the treadmill running (n = 21 per strain) or unstressed control groups (n = 12 per strain) over three experimental cohorts. Run training consisted of daily exercise on a rodent treadmill (Exer 3/6 Treadmill, Columbus Instruments, Columbus, OH). This regimen was adapted from a previous study showing strenuous exercise caused chondrocyte changes (2). The introductory week began with animals spending 10 min in the treadmill environment without the belt moving, followed by running for 10 min at a 10 m/min pace. On days 2–5, animals were exercised for 15 min at 12 m/min, 25 min at 13 m/min, 35 min at 14 m/min, and 45 min at 15 m/min. If animals refused to run, they came into contact with a metal grid that delivered a mild electrical shock (0–1.5 mA; 1–3 Hz) to motivate them to continue running. For the next 8 wk, animals ran for 45 min/day at 15 m/min, 5 days a week, as modified from Baur and colleagues (2). Although no physiological parameters were measured during exercise, extrapolations from previous studies would suggest that this running intensity would be between 50 and 70% of the maximum oxygen uptake (V̇o2max) for these mice (7, 27, 29). The Monday following the last running session, which occurred on a Friday, animals were euthanized with pentobarbital sodium (Fatal-Plus, Vortech Pharmaceuticals, Dearborn, MI). Both knee joints were dissected around the joint capsule, which included small portions of the distal femur and proximal tibia. The right knee was placed in 4% paraformaldehyde (PFA) in 1 M phosphate-buffered saline solution (PBS, pH 7.4), and the left knee was frozen in liquid nitrogen for protein or RNA extraction. Tibias were dissected and frozen in liquid nitrogen. Frozen tissues were stored at −80°C until further analyses.
Joint histology.
Following fixation, knee joints were decalcified for 1 wk (Calci-Clear Rapid, National Diagnostics, Atlanta, GA), dehydrated, processed with a clearing agent (Histo-Clear, National Diagnostics), and embedded in paraffin. Coronal sections were made (5 μm) through the anterior tibiofemoral compartment. Slides were then deparaffinized, rehydrated, and stained with a Safranin O staining kit (no. 8348, ScienCell Research Laboratories, Carlsbad, CA) and counterstained with Fast green to determine joint morphology. Sections were viewed using light microscopy, and color images were captured (10× magnification) using a camera and computer software program (Image-Pro Plus, Media Cybernetics, Bethesda, MD), with settings consistent between capture sessions. Sections stained with Safranin O were scored for the progression of OA according to a 0 to 6 subjective scoring system, adapted from Glasson and colleagues (8) and Pritzker and colleagues (24). The medial and lateral articular surfaces of the tibia and femur were scored by two readers blinded to group, and a blinded consensus read was performed to resolve any differences and achieve final consensus scores. A final, summed score was calculated for each animal. Each of the surfaces and the sum were analyzed using the Wilcoxon rank sums nonparametric test (JMP 10.0, SAS Institute, Cary, NC). Additionally, articular cartilage area, thickness, and subchondral bone thickness were also evaluated (18), and these scores were similarly analyzed.
Additional sections were used to detect DNA fragmentation using an In Situ Cell Death Detection Kit (TUNEL; Roche Applied Science, Indianapolis, IN) according to the instruction manual. Sections were subsequently mounted with Vectashield mounting medium for fluorescence with DAPI (Vector Laboratories, Burlingame, CA). TUNEL staining in the articular cartilage of each section was quantified using Image-Pro Plus and Adobe Photoshop CS5. First, articular cartilage area was determined for each section. The total number of chondrocytes was subsequently enumerated using a DAPI fluorescent filter, while cells that were TUNEL-positive were enumerated using a FITC filter. The sections were scored by two readers blinded to group, and a blinded review was performed to resolve any differences and achieve a final consensus score. These data were expressed as a ratio of positively stained cells to total cells enumerated.
Immunostaining of joint tissue for ECSOD and nitrotyrosine.
Tissue sections were deparaffinized, hydrated, boiled in 0.1 M sodium citrate, washed in PBS, and incubated with 3% H2O2 for 5 min to quench endogenous peroxidase activity. Slides were then washed in PBS and blocked with BSA in PBS for 30 min at 37°C. Primary antibody was applied and incubated overnight at 4°C (polyclonal rabbit antibody against mouse ECSOD, 1:1,000, generously provided by Dr. Tim Oury at the Univ. of Pittsburgh; polyclonal rabbit antibody against multispecies nitrotyrosine, 1:100, no. 06-284, EMD Millipore, Billerica, MA). Negative control sections were incubated in PBS overnight, without the primary antibody. The slides were stained using a Vectastain Elite ABC Kit, Rabbit IgG (Vector Laboratories) according to manufacturer's instructions, incubated with diaminobenzidine (DAB), and then counterstained with hematoxylin.
Joint and bone homogenization.
Frozen knee joints, including synovium and meniscus, and tibias, including bone marrow, were pulverized under liquid nitrogen using a tissue gun, placed in lysis buffer [5 mM EDTA, 50 mM Tris, 150 mM NaCl, 1% Nonidet P-40, Complete Mini Protease Inhibitor (Roche)], sonicated, and rotated overnight at 4°C. Homogenates were centrifuged at 12,000 rpm for 10 min, and supernatants were retained. Protein concentrations of the homogenates were determined using the Bradford method (4).
Western blots.
Western blotting was used to measure ECSOD, MnSOD, and CuZnSOD protein levels within joint homogenates. Equal amounts of protein were loaded on 12% Tris-HCl polyacrylamide gels, separated by one-dimensional electrophoresis, and electrophoretically transferred to polyvinylidene fluoride membranes. Nonspecific binding was blocked by incubating membranes in 5% nonfat milk in TBS-T (50 mM Tris, 150 mM NaCl, 0.1% Tween 20) overnight at 4°C. Membranes were incubated with primary antibody (polyclonal rabbit antibody against mouse ECSOD, 1:1,000; polyclonal rabbit antibody against multispecies nitrotyrosine, 1:1,000, no. 06-284, EMD Millipore; polyclonal rabbit anti-CuZnSOD, 1:1,000, no. ADI-SOD-100, Enzo Life Sciences; polyclonal rabbit anti-MnSOD, 1:1,000, no. ADI-SOD-110, Enzo Life Sciences, Farmingdale, NY) for 1 h in 1% nonfat milk in TBS-T, washed three times in TBS-T, and incubated with secondary antibody (goat-anti-rabbit conjugated to horseradish peroxidase, 1:10,000 in TBS-T). Blots were developed using ECL Plus Kit (GE Healthcare Biosciences, Pittsburgh, PA) and visualized via Molecular Dynamics Storm Imager (GE Healthcare). Densitometry was performed on visible bands using Image-Pro Plus Software (Media Cybernetics), which were normalized to the amount of loaded protein. To quantify loaded protein, all membranes were subsequently stained using a Ponceau S solution. Briefly, membranes were immersed in methanol, incubated in a 1% vol/vol Ponceau S and acetic acid solution for 5 min, and then destained using distilled water until bands were visible.
RNA extraction and real-time RT-PCR.
Real-time RT-PCR was used to measure the mRNA expression of the ECSOD gene. Briefly, RNA was extracted from crushed, isolated knee joints using RNeasy Mini Kit (QIAGEN, Valencia, TX). RNA concentration was verified using spectrophotometry. Reverse transcription was performed using Taqman RNA-to-Ct 1-Step kit with Taqman probes (Applied Biosystems, Foster City, CA). 18S was used as the housekeeping gene. The threshold cycle was recorded for each sample to reflect the mRNA expression levels. Results were analyzed using the −2ΔΔCT method as described, calculating fold gene expression change of treatment over untreated condition (30).
Bone morphometry.
Bone structural characteristics were assessed ex vivo by microcomputed tomography (μCT) (Siemens Inveon, Erlangen, Germany) of whole tibia scans using a resolution of 21 μm. Scans were imported into ImageJ, a public domain Java-based image processing program developed at the National Institutes of Health. The BoneJ plugin was used to determine cross-sectional and polar moments of inertia (Imax, Imin, Ip) and section moduli (Zmax, Zmin, Zp) of the principal axes, cross-sectional area (CSA), and cortical thickness at a slice just proximal to the junction of the tibia and fibula (22).
Bone mechanical testing.
After imaging, tibial fracture strength and stiffness were determined by performing fracture tests in compression using a material testing system (Insight 30; MTS Systems, Eden Prairie, MN). Load and displacement data were collected using TestWorks software and a 500 N MTS load cell. The ends of each bone were fixed in blocks of urethane, and loading occurred along the longitudinal axis. Force was applied with a preload of 1 N at a strain rate of 0.005/s until bone failure. Failure load (N) was defined as the maximum load on the load-displacement curve. Stiffness (N/mm) was defined as the slope of the load-displacement curve in the linear region.
Bone finite element modeling.
Models of the murine tibia were developed from computed tomography data (voxel size = 0.02 mm3). Segmentation and meshing were performed in ScanIP (Simpleware, Exeter, UK) to create three-dimensional models. To reproduce the mechanical testing conditions, blocks were incorporated into the model to imitate the urethane used for fixing the bones. The urethane was assigned an elastic modulus of 6,832 MPa and a Poisson's ratio of 0.3 (manufacturer's specifications). Models were then imported into Abaqus (Dassault Systemes, Waltham, MA) for finite element analysis. The bone was assigned an elastic modulus of 18,000 MPa and a Poison's ratio of 0.3. The failure load determined during testing for each sample was divided by the area of the block to obtain the pressure applied in the z-direction. Peak von Mises stress at the time of failure was computed by sampling 30 elements in the region containing the highest stress. To compare the stress levels experienced in each bone under a single, consistent load, a standard force of 100 N was also applied across the top of the urethane block. For this loading condition, an average von Mises stress was computed at the cross-section at the middiaphysis, just proximal to the junction of the tibia and fibula.
Statistics.
Data were pooled from all three experiments. Densitometric analysis of visible bands on ECSOD Western blots were analyzed using a one-way ANOVA followed by Tukey HSD post hoc tests. TUNEL staining was analyzed for the WT and Het runners using a two-tailed, unpaired t-test. Joint damage, articular cartilage, and subchondral bone parameters (area, thickness) were analyzed for the WT and Het runners using Wilcoxon nonparametric tests (JMP 10.0, SAS Institute). Bone data were analyzed using SAS 9.3 (SAS Institute, Cary, NC), and two-way ANOVAs were used to test for strain-by-exercise interaction effects, and for main effects of strain and exercise. In the case of a significant interaction term, Tukey-Kramer HSD tests were used to make pairwise comparisons. A P value <0.05 was considered significant. Values are presented as means ± SE.
RESULTS
Animal weights.
At the start of the experiment the average age and weight of the animals were 5.3 ± 1.1 mo and 33.4 ± 5.1 g, respectively. The percent changes in weight from the start to the end of the experiment were +0.6 ± 1.1% (WT Sed), −3.1 ± 1.5% (WT Run), +2.9 ± 1.5% (Het Sed), and −2.0 ± 1.5% (Het Run). The end weights of the WT Run and Het Run animals were significantly lower than the end weights of their unstressed counterparts (P < 0.01) but were not different from each other.
SOD isoenzymes: localization of proteins and gene expression.
The amount of ECSOD protein in the knee joint in each group of animals was determined via Western blot and densitometric analysis of visible bands (Fig. 1, A and B). There was significantly more ECSOD in the whole joint of WT mice compared with Het mice (P < 0.01). Robust ECSOD immunostaining was present in the articular cartilage and subchondral bone of unstressed WT animals (Fig. 1C), while ECSOD was minimally present in unstressed Het animals (Fig. 1D). Eight weeks of running did not cause significant changes in joint ECSOD protein levels in WT (Fig. 1E) or Het animals (Fig. 1F). The pattern of ECSOD gene expression mirrored the pattern of ECSOD protein levels, although there were no significant differences between groups (Fig. 1G). There was no difference between groups in the amount of CuZnSOD or MnSOD protein present in knee joint homogenates (Fig. 1H). There were no differences in nitrotyrosine levels between groups when analyzed via immunoblot or immunostaining (data not shown).
Fig. 1.
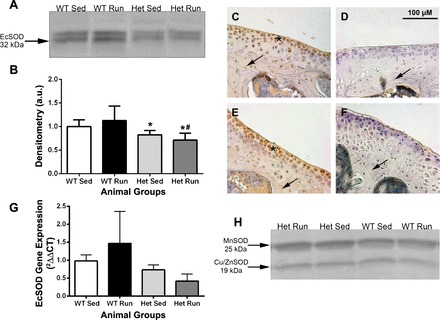
Extracellular superoxide dismutase (ECSOD) localization in the joint tissues. A: ECSOD Western blot from mouse knee joint homogenates. Equal protein loading was verified with Ponceau S staining of blots. B: densitometric analysis of visible bands on ECSOD Western blot (*P < 0.05: Het Sed vs. WT Run, and Het Run vs. WT Sed; #P < 0.01: Het Run vs. WT Run). ECSOD immunostaining of sections counterstained with hematoxylin through the tibiofemoral joint compartment in WT Sed (C, n = 8), Het Sed (D, n = 8), WT Run (E, n = 6), and Het Run (F, n = 6) animals. ECSOD-positive staining in cartilage is visualized by the brown color and indicated by the asterisks in C and E. Arrows indicate bone tissue with (C and E) and without (D and F) ECSOD-positive immunostaining. G: ECSOD gene expression in knee joint homogenates. H: MnSOD and Cu,ZnSOD Western blots did not show differences between any groups. WT, wild-type; Het, heterozygous for the R213G polymorphism; Sed, sedentary, home-cage untrained; Run, mice exposed to forced running.
No changes in chondrocyte cell death (TUNEL staining).
Following 8 wk of forced running, there was no difference in the number of TUNEL-positive chondrocytes in articular cartilage (Fig. 2B) in Het mice compared with WT animals (Fig. 2A).
Fig. 2.

Chondrocyte cell death in WT vs. Het runners. Tissue sections through the tibiofemoral joint compartment stained using a cell death detection (TUNEL) kit in WT Run (A), Het Run (B), positive control (C), and negative control (D) sections. Cell nuclei are visualized by the punctate, fluorescent blue staining (DAPI), while TUNEL-positive cells are visualized by the punctate, fluorescent green staining, indicated by the arrows in A and B. E: the number of TUNEL-positive cells was enumerated and expressed as a ratio of the total number of cells per section (WT, n = 11; Het, n = 13).
Increased joint damage in Het animals following running.
There were worse overall histology scores in the Het mice compared with WT mice [2.6 (2.2) vs. 1.0 (0.7), P = 0.036] after 8 wk of forced running. The medial tibia surface showed the most striking damage (P = 0.001), but lateral tibias also had significantly worse scores (P = 0.04, Figs. 3 and 4). There were no significant differences in the two femoral surfaces. No differences between groups were observed for articular cartilage area and thickness or subchondral bone thickness. WT sedentary mice had significantly more joint damage than WT mice exposed to running (P = 0.049).
Fig. 3.
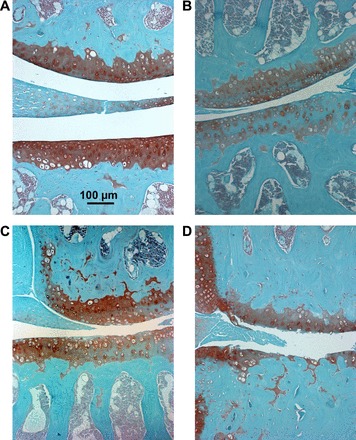
Morphological changes in cartilage in WT vs. Het runners. A: histological sections through the medial tibiofemoral joint compartment in WT runner with a score of 0 for each surface. B: Het runner with a score of 0.5 for each surface in the lateral tibiofemoral joint. C: medial tibiofemoral joint with score of 2 for each surface in a Het runner. D: Het runner with a score of 4 in the lateral femur and 5 for the lateral tibial. Sections were stained with Safranin O and counterstained with fast green and images were captured at 10× magnification.
Fig. 4.
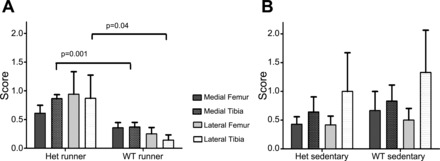
Histological grades of runners and sedentary animals. Sections were scored by 2 readers blinded to both group assignment and genetic background and resolved to a consensus score for each of the four joint surfaces using the method of Glasson et al. (8). A mean score for each of the four joint surfaces was calculated for the WT runners and Het runners (A). There were significant differences between WT (n = 8) and Het (n = 13) runners in both the medial (P = 0.001) and lateral (P = 0.04) tibial surfaces. A mean score for each joint surface was also calculated for the sedentary mice (B). There were no significant differences between WT and Het Sedentary animals by joint surface.
Bone adaptations.
Running increased (P < 0.05) the resistance to bending (Imax, Imin, Zmax, Zmin) and torsion (Zp) at the cross-sectional slice just proximal to the junction of the tibia and fibula in both mouse strains (Table 1). Failure loads from mechanical testing were higher, but not significantly, in the running groups compared with controls (P = 0.13, main effect of running). The simulated peak von Mises stresses were not different when failure load (StressFail) was used in the finite element model. However, Het mice had higher von Mises stresses (P < 0.05) at the same slice where bone morphometry was measured when a standardized load (100 N) (StressStd) was used. The higher bone stress in response to standardized load suggests that Het mice had less favorable overall bone structure.
Table 1.
Structural and mechanical characteristics of the tibiae after 8 wk of intervention
| Het-Run (n = 21) | Het-Sed (n = 12) | WT-Run (n = 15) | WT-Sed (n = 11) | |
|---|---|---|---|---|
| Midshaft bone morphology | ||||
| CSA, mm2 | 0.817 ± 0.024 | 0.745 ± 0.014 | 0.778 ± 0.010 | 0.751 ± 0.023 |
| Cort ThickMax, mm | 0.372 ± 0.009 | 0.359 ± 0.008 | 0.368 ± 0.006 | 0.359 ± 0.012 |
| Imax†, mm4 | 0.123 ± 0.007 | 0.094 ± 0.005 | 0.112 ± 0.004 | 0.098 ± 0.007 |
| Zmax*, mm3 | 0.161 ± 0.007 | 0.138 ± 0.005 | 0.154 ± 0.004 | 0.142 ± 0.008 |
| Imin*, mm4 | 0.074 ± 0.004 | 0.066 ± 0.004 | 0.077 ± 0.002 | 0.066 ± 0.005 |
| Zmin*, mm3 | 0.131 ± 0.005 | 0.120 ± 0.005 | 0.133 ± 0.003 | 0.119 ± 0.006 |
| Ip, mm4 | 0.093 ± 0.011 | 0.086 ± 0.012 | 0.105 ± 0.008 | 0.092 ± 0.013 |
| Zp*, mm3 | 0.254 ± 0.010 | 0.232 ± 0.010 | 0.258 ± 0.005 | 0.233 ± 0.012 |
| Mechanical testing | ||||
| Failure load, N | 65.07 ± 5.34 | 55.89 ± 6.07 | 66.4 ± 5.0 | 57.05 ± 7.84 |
| Stiffness, N/mm | 139.6 ± 24.9 | 122.7 ± 20.3 | 130.1 ± 21.2 | 139.9 ± 25.2 |
| Finite element analysis | ||||
| StressFail, MPa | 136.0 ± 8.4 | 120.4 ± 15.1 | 126.2 ± 8.2 | 115.6 ± 16.2 |
| StressStd‡, MPa | 114.8 ± 3.6 | 110.5 ± 4.6 | 97.6 ± 3.4 | 107.4 ± 6.5 |
Values are means ± SE.
WT, wild-type; Het, heterozygous for the R213G polymorphism; Sed, sedentary mice, home-cage untrained; Run, mice exposed to forced running;CSA, cross-sectional area; Cort ThickMax, maximum cortical thickness; Imax, maximum area moment of inertia; Zmax, maximum section modulus; Imin, minimum area moment of inertia; Zmin, minimum section modulus; Ip, polar moment of inertia; Zp, polar section modulus; StressFail, von Mises stress at failure load; StressStd, von Mises stress at standard load.
P ≤ 0.01,
P < 0.05, main effect of exercise;
P ≤ 0.05, main effect of mouse strain.
DISCUSSION
Oxidative damage to cartilage has been identified as a factor in osteoarthritis (5, 17). We have previously found that extracellular superoxide dismutase (ECSOD) is highly expressed in human cartilage and decreased in OA cartilage and joint fluid, with footprints of oxidative damage (nitrotyrosine) present in extracellular matrix of cartilage (6, 25, 26). The R213G mutation studied here for joint and bone effects has been shown previously to have reduced vascular tissue levels of ECSOD and an association with increased risk of ischemic heart disease (12).
Mice heterozygous for the R213G mutation in our study had reduced levels of ECSOD in their joint tissue and when exposed to the stress of forced running demonstrated significantly more joint damage compared with WT runner mice. These Het mice did not have any observable physical differences or changes in weight compared with WT animals; thus the degree of mechanical loading on the knee joint was similar between the two runner groups. Sedentary mice from both Het and WT backgrounds had significantly higher body weights by the end of the study, did not show a difference in joint histology based on genetic background, but did have greater joint damage than WT runner mice.
Our data suggest that in WT mice forced running is associated with reduced joint damage and lower body weight compared with the sedentary state. However, when ECSOD levels are reduced, the benefits of running are lost. The combination of reduced levels of local antioxidant protection and the stress of increased mechanical loading from running resulted in more joint damage, akin to the effect of being sedentary. Other authors have previously found that a sedentary state increased the risk of OA. Hubbard-Turner et al. (10) found that C57Bl/6J mice exposed to running were protected from OA and had thicker joint spaces compared with sedentary mice up to 12 mo of age. Lapvetelainen and colleagues (13) found that mice at risk of OA from a collagen mutation were protected from arthritis by running.
The Het animals displayed a significant reduction in ECSOD protein in the tibiofemoral joint compartment and subchondral bone. The residual ECSOD appearing on the Western blots was likely derived from the single copy of the wild-type ECSOD gene present in these animals, and small amounts of ECSOD present in the synovial fluid or the small portions of the femur and tibia extracted with the joint capsule. Previous studies have shown an impact of exercise on ECSOD protein or mRNA including upregulation in skeletal muscle and aorta following a single bout of running in mice (6) and increases in plasma levels after acute exercise in endurance-trained humans (20). We did not observe a statistical change in ECSOD mRNA or protein levels due to training.
We evaluated CuZnSOD and MnSOD levels in the R213G mice for compensatory changes, and we observed no difference in either CuZnSOD or MnSOD protein levels associated with animal strain or training regimen. Others have shown that both CuZnSOD and MnSOD are important antioxidant enzymes in protecting joints from osteoarthritis (28, 31). Each of the SOD isoforms has a unique role in oxidant scavenging (SOD1 cytosolic and intracellular, SOD2 mitochondrial, ECSOD or SOD3 predominantly secreted and extracellular). The differences between studies assessing the impact of modification of antioxidant protection may be attributed to this specificity as well as different strains of animals used and/or different training protocols and time points analyzed.
Previous work has shown that forced exercise can cause oxidative stress, chondrocyte death, and cartilage damage depending on the model and the time points used for analysis (14, 15, 21, 32). After 8 wk of training, we found no difference in nitrotyrosine levels or the number of chondrocytes undergoing cell death between the Het and WT trained mice although structural damage was identified. Baur and colleagues (2) used a similar training method to ours and did not see any evidence of structural cartilage damage in a mouse heterozygous for a MnSOD deficiency following running. However, they did observe oxidative damage in the form of increased intracellular nitrotyrosine and 15-F2t-isoprostane in cartilage tissue of runners.
The differences in damage may be attributed to the differences in location of the antioxidant enzyme. MnSOD protects the mitochondria in chondrocytes from oxidative stress. Baur et al. (2) found that a reduction in MnSOD levels causes more oxidative stress and cell death of chondrocytes but no structural changes in the cartilage. ECSOD is a secreted protein that binds to collagen and proteoglycans providing oxidant scavenging in the extracellular matrix. We did not observe any significant changes in the chondrocytes in terms of cell death, or nitrotyrosine, but did see more damage to the cartilage in mice deficient in joint ECSOD. Our earlier work identified nitrotyrosine formation in the extracellular matrix of human cartilage with ECSOD deficiency (25). Failure of cartilage under mechanical stress may be a consequence of inadequate oxidant scavenging and oxidant damage to the structural molecules in the extracellular matrix. We also speculate that oxidative and structural damage to joint tissues may manifest at different times. Waiting longer to euthanize animals following a running regimen may allow these slow degradative processes to progress further, ultimately leading to more visible damage within the joint compartment.
We explored the role that ECSOD plays in bone tissue and in response to mechanical loading. Using a high scan resolution, we detected favorable changes in the distribution of the cortical bone tissue in response to training, which increases the resistance to mechanical failure by bending and torsion. We did not detect an effect of ECSOD deficiency on the structural bone adaptations to training. Additionally, the difference between Het and WT mice in bone stresses in response to a standardized load was not altered by exercise. However, the lower bone stress exhibited by WT tibias in response to the standardized load suggests that WT mice had a more favorable overall bone structure compared with Het animals. The volume and magnitude of loading in the training regimen was probably not sufficient to be considered pathological loading in bone (2). Thus it is likely the training protocol did not stimulate a strong enough oxidative stress response to cause excessive bone resorption in the face of reduced ECSOD but produced a strong enough loading stimulus to promote bone formation.
Mechanical loading may be able to stimulate intracellular antioxidant mechanisms and protect bone. MnSOD-deficient mice that ran on an inclined treadmill had lower levels of nitrotyrosine and 15-F2t-isoprostane within bone compared with MnSOD-deficient untrained controls, suggesting that other antioxidant enzymes were elevated to compensate for the loss of MnSOD (2). Other work suggests that skeletal unloading exacerbates oxidative stress within the cytoplasm; and that CuZnSOD-deficient mice experience greater reductions in bone formation and greater bone loss in response to unloading compared with wild-type mice and the effect was blocked by administration of vitamin C (19).
In conclusion, we have shown that a unique knock-in mouse model for the human R213G polymorphism in ECSOD has decreased levels of ECSOD in cartilage and bone compared with WT mice. Following 8 wk of forced running, Het animals had significantly more cartilage damage compared with WT runners. Interestingly, both Het and WT animals had beneficial bone responses to forced mechanical loading, although Het mice had less favorable overall bone structure. This animal strain may be used for future studies aimed at elucidating the detailed mechanisms of cartilage pathogenesis related to deficiencies in ECSOD, and understanding the role of oxidative stress in bone health.
GRANTS
Funding for these studies was provided by the Shramm Foundation of Denver, CO, and the Department of Defense USAMRC Proposal no. 07355003.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.M.P., J.D.C., W.M.K., R.P.B., R.E.O.-D., and E.A.R. conception and design of research; K.M.P., V.D.S., R.D.C., M.R.W., F.G., L.S.C., and D.A.G. performed experiments; K.M.P., V.D.S., R.D.C., M.R.W., S.C., R.E.O.-D., and E.A.R. analyzed data; K.M.P., V.D.S., R.D.C., R.E.O.-D., and E.A.R. interpreted results of experiments; K.M.P., M.R.W., and E.A.R. prepared figures; K.M.P. and E.A.R. drafted manuscript; K.M.P., V.D.S., R.D.C., M.R.W., F.G., L.S.C., D.A.G., J.D.C., W.M.K., R.P.B., R.E.O.-D., and E.A.R. edited and revised manuscript; K.M.P., V.D.S., R.D.C., M.R.W., F.G., L.S.C., D.A.G., J.D.C., W.M.K., R.P.B., R.E.O.-D., and E.A.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Tim Oury at the Univ. of Pittsburgh for generously providing the anti-mouse ECSOD primary antibody used in immunostaining and Western blots.
REFERENCES
- 1.Apold H, Meyer HE, Nordsletten L, Furnes O, Baste V, Flugsrud GB. Risk factors for knee replacement due to primary osteoarthritis, a population based, prospective cohort study of 315,495 individuals. BMC Musculoskelet Disord 15: 217, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baur A, Henkel J, Bloch W, Treiber N, Scharffetter-Kochanek K, Bruggemann GP, Niehoff A. Effect of exercise on bone and articular cartilage in heterozygous manganese superoxide dismutase (SOD2) deficient mice. Free Radic Res 45: 550–558, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Beecher BR, Martin JA, Pedersen DR, Heiner AD, Buckwalter JA. Antioxidants block cyclic loading induced chondrocyte death. Iowa Orthop J 27: 1–8, 2007. [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254, 1976. [DOI] [PubMed] [Google Scholar]
- 5.Del Carlo M, Schwartz D, Erickson EA, Loeser RF. Endogenous production of reactive oxygen species is required for stimulation of human articular chondrocyte matrix metalloproteinase production by fibronectin fragments. Free Radic Biol Med 42: 1350–1358, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest 105: 1631–1639, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Valles R, Gomez-Cabrera MC, Rodriguez-Manas L, Garcia-Garcia FJ, Diaz A, Noguera I, Olaso-Gonzalez G, Vina J. Life-long spontaneous exercise does not prolong lifespan but improves health span in mice. Longev Healthspan 2: 14, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 18, Suppl 3: S17–S23, 2010. [DOI] [PubMed] [Google Scholar]
- 9.Hartney JM, Stidham T, Goldstrohm DA, Oberley-Deegan RE, Weaver MR, Valnickova-Hansen Z, Scavenius C, Benninger RK, Leahy KF, Johnson R, Gally F, Kosmider B, Zimmermann AK, Enghild JJ, Nozik-Grayck E, Bowler RP. A common polymorphism in EC-SOD affects cardiopulmonary disease risk by altering protein distribution. Circ Cardiovasc Genet 7: 659–666, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hubbard-Turner T, Guderian S, Turner MJ. Lifelong physical activity and knee osteoarthritis development in mice. Int J Rheum Dis 2014March18. doi: 10.1111/1756-185X.12291 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 11.Iida S, Chu Y, Weiss RM, Kang YM, Faraci FM, Heistad DD. Vascular effects of a common gene variant of extracellular superoxide dismutase in heart failure. Am J Physiol Heart Circ Physiol 291: H914–H920, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, Jensen G, Nordestgaard BG. Genetically reduced antioxidative protection and increased ischemic heart disease risk: The Copenhagen City Heart Study. Circulation 109: 59–65, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Lapvetelainen T, Hyttinen M, Lindblom J, Langsjo TK, Sironen R, Li SW, Arita M, Prockop DJ, Puustjarvi K, Helminen HJ. More knee joint osteoarthritis (OA) in mice after inactivation of one allele of type II procollagen gene but less OA after lifelong voluntary wheel running exercise. Osteoarthritis Cartilage 9: 152–160, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Lapvetelainen T, Hyttinen MM, Saamanen AM, Langsjo T, Sahlman J, Felszeghy S, Vuorio E, Helminen HJ. Lifelong voluntary joint loading increases osteoarthritis in mice housing a deletion mutation in type II procollagen gene, and slightly also in non-transgenic mice. Ann Rheum Dis 61: 810–817, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lapvetelainen T, Nevalainen T, Parkkinen JJ, Arokoski J, Kiraly K, Hyttinen M, Halonen P, Helminen HJ. Lifelong moderate running training increases the incidence and severity of osteoarthritis in the knee joint of C57BL mice. Anat Rec 242: 159–165, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Li R, Chen B, Wang G, Yu B, Ren G, Ni G. Effects of mechanical strain on oxygen free radical system in bone marrow mesenchymal stem cells from children. Injury 42: 753–757, 2011. [DOI] [PubMed] [Google Scholar]
- 17.Martin JA, Buckwalter JA. Post-traumatic osteoarthritis: the role of stress induced chondrocyte damage. Biorheology 43: 517–521, 2006. [PubMed] [Google Scholar]
- 18.McNulty MA, Loeser RF, Davey C, Callahan MF, Ferguson CM, Carlson CS. Histopathology of naturally occurring and surgically induced osteoarthritis in mice. Osteoarthritis Cartilage 20: 949–956, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morikawa D, Nojiri H, Saita Y, Kobayashi K, Watanabe K, Ozawa Y, Koike M, Asou Y, Takaku T, Kaneko K, Shimizu T. Cytoplasmic reactive oxygen species and SOD1 regulate bone mass during mechanical unloading. J Bone Miner Res 28: 2368–2380, 2013. [DOI] [PubMed] [Google Scholar]
- 20.Ookawara T, Haga S, Ha S, Oh-Ishi S, Toshinai K, Kizaki T, Ji LL, Suzuki K, Ohno H. Effects of endurance training on three superoxide dismutase isoenzymes in human plasma. Free Radic Res 37: 713–719, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Pap G, Eberhardt R, Sturmer I, Machner A, Schwarzberg H, Roessner A, Neumann W. Development of osteoarthritis in the knee joints of Wistar rats after strenuous running exercise in a running wheel by intracranial self-stimulation. Pathol Res Pract 194: 41–47, 1998. [DOI] [PubMed] [Google Scholar]
- 22.Papaioannou A, Joseph L, Ioannidis G, Berger C, Anastassiades T, Brown JP, Hanley DA, Hopman W, Josse RG, Kirkland S, Murray TM, Olszynski WP, Pickard L, Prior JC, Siminoski K, Adachi JD. Risk factors associated with incident clinical vertebral and nonvertebral fractures in postmenopausal women: the Canadian Multicentre Osteoporosis Study (CaMos). Osteoporos Int 16: 568–578, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Petersen SV, Olsen DA, Kenney JM, Oury TD, Valnickova Z, Thogersen IB, Crapo JD, Enghild JJ. The high concentration of Arg213→Gly extracellular superoxide dismutase (EC-SOD) in plasma is caused by a reduction of both heparin and collagen affinities. Biochem J 385: 427–432, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, Salter D, van den Berg WB. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage 14: 13–29, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Regan E, Flannelly J, Bowler R, Tran K, Nicks M, Carbone BD, Glueck D, Heijnen H, Mason R, Crapo J. Extracellular superoxide dismutase and oxidant damage in osteoarthritis. Arthritis Rheum 52: 3479–3491, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Regan EA, Bowler RP, Crapo JD. Joint fluid antioxidants are decreased in osteoarthritic joints compared to joints with macroscopically intact cartilage and subacute injury. Osteoarthritis Cartilage 16: 515–521, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Rezende EL, Chappell MA, Gomes FR, Malisch JL, Garland T Jr. Maximal metabolic rates during voluntary exercise, forced exercise, and cold exposure in house mice selectively bred for high wheel-running. J Exp Biol 208: 2447–2458, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martinez-Gomariz M, Fernandez M, Blanco FJ. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics 8: 172–189, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schefer V, Talan MI. Oxygen consumption in adult and AGED C57BL/6J mice during acute treadmill exercise of different intensity. Exp Gerontol 31: 387–392, 1996. [DOI] [PubMed] [Google Scholar]
- 30.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Scott JL, Gabrielides C, Davidson RK, Swingler TE, Clark IM, Wallis GA, Boot-Handford RP, Kirkwood TB, Taylor RW, Young DA. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann Rheum Dis 69: 1502–1510, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang T, Muneta T, Ju YJ, Nimura A, Miyazaki K, Masuda H, Mochizuki T, Sekiya I. Serum keratan sulfate transiently increases in the early stage of osteoarthritis during strenuous running of rats: protective effect of intraarticular hyaluronan injection. Arthritis Res Ther 10: R13, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomiyama T, Fukuda K, Yamazaki K, Hashimoto K, Ueda H, Mori S, Hamanishi C. Cyclic compression loaded on cartilage explants enhances the production of reactive oxygen species. J Rheumatol 34: 556–562, 2007. [PubMed] [Google Scholar]
- 34.Yucesoy B, Charles LE, Baker B, Burchfiel CM. Occupational and genetic risk factors for osteoarthritis: a review. Work 2013 Sep 4. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]