

Abstract
Methods for isolating elastin from fat, collagen, and muscle, commonly used in the design of artificial elastin based biomaterials, rely on exposing tissue to harsh pH levels and temperatures that usually denature many proteins. At present, a quantitative measurement of the modifications to elastin following isolation from other extracellular matrix constituents has not been reported. Using magic angle spinning 13C NMR spectroscopy and relaxation methodologies, we have measured the modification in structure and dynamics following three known purification protocols. Our experimental data reveal that the 13C spectra of the hydrated samples appear remarkably similar across the various purification methods. Subtle differences in the half maximum widths were observed in the backbone carbonyl suggesting possible structural heterogeneity across the different methods of purification. Additionally, small differences in the relative signal intensities were observed between purified samples. Lyophilizing the samples results in a reduction of backbone motion and reveals additional differences across the purification methods studied. These differences were most notable in the alanine motifs indicating possible changes in cross-linking or structural rigidity. The measured correlation times of glycine and proline moieties are observed to also vary considerably across the different purification methods, which may be related to peptide bond cleavage. Lastly, the relative concentration of desmosine cross-links in the samples quantified by MALDI mass spectrometry is reported.
Keywords: Elastin, Elastin Purification, 13C MAS NMR, MALDI mass spectrometry
Introduction
Elastin, the principle protein component of the elastic fiber found in vertebrate tissues, is a biopolymer that exhibits remarkable mechanical characteristics. Numerous examples of imbalance or degradation of elastin are associated to pathological disorders, such as aortic stenosis and Williams syndrome, in which the elastin gene is mutated1 or cutis laxa, in which connective tissues have shown a disruption of elastin2. The interplay between elastin degradation and disease3, in addition to efforts to design elastin-based biomaterials4,5, has for a long time been the stimulus of studies to understand the structure and source of elasticity of this biopolymer. For example, many efforts to design artificial skin rely on elastin as a mechanical scaffold6. Obtaining detailed structural information of this protein has posed a challenge, as elastin does not crystallize and is largely insoluble making X-ray and solution state NMR methods inapplicable. Tropoelastin, the soluble precursor of elastin, is a 60–70kDa monomer which is composed of hydrophobic domains rich in alanine, glycine, proline and valine, and cross-linking domains comprised of desmosine or isodesmosine7,8. Together with fibrillin-microfibrils, elastin comprises the elastic fiber and contributes to the elastomeric characteristics and longevity of many tissues in vertebrates.
The elasticity of elastin is currently believed to be driven by an entropic force that is mediated by conformational and/or dynamical changes9. Early models described elastin as a two phase system, composed of globular molecules that carry hydrophobic groups packed in the interior and hydrophilic groups on the surface10. Andersen et al. suggested the existence of both hydrophobic and hydrophilic groups on the surface of the macromolecule, adding an additional view to the interpretation of the entropic force that drives elasticity11. In particular, the underlying physics behind the entropic force is related to oscillations of the protein backbone but also the interactions between the hydrophobic domains and solvent molecules at the surface. The latter has been studied by Torchia and coworkers where experiments were carried out on purified 13C labeled elastin using nuclear magnetic resonance. Their measured relaxation times, line widths and correlation times showed that under physiological conditions all valine residues undergo rapid motion; on the other hand alanine and lysine residues exhibited both slow and fast motions12. Investigations by Partridge et. al. revealed that certain treatments of elastin may result in a product soluble in water that consists of two components with rather different physical characteristics13. Partridge also suggested that elastin may be composed of molecules in tetrahedral packing14. On the other hand, in earlier work, Ramachandran suggested a triple helical model for elastin similar to that of collagen15.
More recently, studies have aimed to further understand the source of elasticity in elastin by considering short repeating peptides comprising tropoelastin. For example the repeating motif (VPGVG)n has been given much attention; in human elastin this pentapeptide repeats nine times16. Urry and coworkers studied this polypeptide extensively and proposed a simple model, termed the librational entropy mechanism, for the elasticity of elastin17. According to this model elastin is composed of consecutive type II β-turns that make up an ‘idealized β-spiral’ and assuming fixed ends, the conformation of the macromolecule allows for librational motion. Upon mechanical deformation the libration of the peptide results in a change in entropy giving rise to an entropic force that mediates the elastic character. Tamburro et al. suggested an alternate model, referred to as the sliding β-turn, consisting of mobile conformations18. In this model the conformation of the polypeptide changes under deformation and results in reduction of entropy which gives rise to elastimicrofibrillarcity. An intriguing characteristic of the pentapeptide (VPGVG)n is that it undergoes coacervation (also termed the inverse temperature transition) over the temperature range of 20°C and 40°C, similar to elastin9,19–21. In practice, the inverse temperature transition may be experimentally controlled by the length or arrangement of the repeating motif22,23 and salt concentration24–26. The biomechanically tunable characteristics of elastin-like peptides of VPGVG motifs has led to many intriguing applications, including drug delivery27,28.
A well known characteristic of elastin is that its mechanical characteristics appear to be strongly related to the degree of hydration29 and the polarity of the solvent30,31. Dehydrated elastin exhibits brittle characteristics and many experimental efforts have focused on giving insight into the source of this behavior. The notion that the polymer backbone, not the solvent, must bear the entropic force arising in mechanically strained elastin was argued in a classic work by Hoeve and Flory32. Molecular dynamics simulations have been used to study the entropic contribution of localized water33 or exposed hydrophobic groups by modeling short pentapeptides undergoing extension34. Experiments on hydrated bovine elastin under mechanical deformation have recently provided a direct measurement of dynamical changes of localized water and the protein backbone and have confirmed that the protein backbone reduces in entropy upon extension giving rise to a restoring entropic force35.
A distinguishing feature of elastin is its resistance to high temperature and pH that usually denature many proteins. Various purification schemes take advantage of these characteristics by heating the tissue in order to purify it from collagen, fat and smooth muscle. A detailed comparison of the purity of the products resulting from five purification schemes has been recently documented by Daamen et al. using amino acid analysis, sulphydryl quantification, and transmission electron microscopy36. Their results showed that the purification methods may also result in a product that includes traces of the microfibrillar component of the elastic fiber. The present work reports on the structural and dynamical modifications of elastin upon purification using high field 13C solid state NMR methodology. Both hydrated and lyophilized elastin samples were studied using techniques that allow for the determination of the structural features in addition to dynamical characteristics measured over the time scale of microseconds to milliseconds. Remarkably, the overall structural features revealed upon purification do not seem to vary significantly between samples, however, the purification methods we studied appear to slightly affect the dynamics and the heterogeneity of structures revealed on the NMR time scale. The process of lyophilization quenches the proteins’ dynamics and the different purification schemes appear to add an additional contribution to this change. In particular, proline rich moieties appear more rigid after purifying the tissue whereas alanine moieties are more mobile. Lastly, based on our measured correlation times, the dynamics of the glycine and proline rich motifs across the samples were observed to vary slightly across the samples studied.
Materials and Methods
Sample Preparation
Both purified and unpurified bovine nuchal ligament elastin samples were purchased from Elastin Products Company, LLC (Owensville, MO). Elastin Products Company, LLC purified the tissue using the following three methods which are summarized in reference37.
Sample 1: Autoclaving38
The tissue was washed and autoclaved in 20 volumes of distilled water for 45 minutes at atmospheric pressure. This procedure is repeated sequentially until the sample contains no further protein trace.
Sample 2: Hot alkali39
Minced tissue was suspended in 0.1 N NaOH and mixed. The product is placed in boiling water for 45 minutes and stirred. The sample was the stored at room temperature and washed with cold 0.1N NaOH followed by centrifugation.
Sample 3: Starcher method40
0.05 M Na2HPO4 at 7.6 pH, 1% NaCl, 0.1%EDTA was used as a buffer to extract the tissue. The purification begins with suspending the tissue in the buffer for 72h. The product is washed twice with distilled water and lyophilzed. 200mg of the lyophilized product was suspended in 30ml of water and autoclaved for 45min at 25psi. The next step includes centrifugation and washing the tissue. The product is suspended in 30ml of 0.1M Tris buffer at a pH of 8.2, that contains 0.02 M CaCl2 and incubated with 4mg of trypsin at 37°C for 18h. The sample is centrifuged and the residue is washed and suspended in 10ml of 97% of formic acid. Cyanogen bromide (200mg) is added and the suspension shaken under a hood at room temperature for 5h. The sample is then centrifuged and the residue is washed twice with water and resuspended in 30ml of 0.05 M Tris buffer at a pH of 8.0 which contains 6 M urea and β-mercaptoethanol (0.5% v/v). The suspension is stirred overnight at room temperature, centrifuged, washed successively with the three washes each of ethanol and acetone, and dried in vacuum over P2O5.
The unpurified bovine nuchal ligament elastin sample was cleared of muscle and fat and dried by Elastin Products Company, LLC. All samples arrived having an average mesh size of 90μm (Samples 1 to 3) and 465μm (Unpurified) and were lyophilized for 72 hours. Experiments on hydrated samples were prepared as follows: the samples were suspended in distilled water for 48 hours and then centrifuged for 3 minutes to remove bulk water for packing into the NMR rotor. The water to protein concentration of all samples was determined to be approximately 40% by volume. Standard amino acid analysis was performed by J. Myron Crawford’s group at the W.M. Keck Biotechnology Resource Laboratory at Yale University using a Hitachi L8900 analyzer. Amino acid analysis on unpurified elastin was performed by New England Peptide (Cambridge, MA) using the same instrumentation.
Quantification of desmosine concentration
Elastin hydrolysis
2.1–2.2 mg of each sample was placed in 300μL of 6N HCL and 1μL of 0.5% w/w phenol solution in sealed glass tubes and flushed with argon gas. The samples were incubated at 110 °C for 96 hours. After incubation the solvent was frozen under liquid nitrogen and then lyophilized for 6–8 hours. The sample was then re-suspended in 50μL of solution of 95.5% 0.14M sodium acetate, 0.5% triethylamine, 5% acetonitrile (v/v/v) at a pH of 7.5.
Quantification with labeled desmosine
Resuspended samples were mixed in 1 : 1 (v/v) ratios with standard d4-desmosine (Toronto Research Chemicals, Toronto, Canada) at a final concentration of 0.250 mg/mL. The mass spectrometric peak intensity ratio in MS2 mode of desmosine to d4-desmosine was used to quantify the relative amount of desmosine in each sample.
MALDI-MS quantitative analysis
MALDI-MS2 experiments were performed using a Thermo LTQ XL ion trap mass spectrometer (Thermo Scientific, Waltham, MA, USA) equipped with a vacuum MALDI source. α-Cyano-4-hydroxycinnamic acid (CHCA) was purchased from Sigma-Aldrich (St. Louis, Missouri, USA) and used without additional purification. Solid CHCA was then mixed with a solution of 0.1% trifluoroacetic acid, 70% acetonitrile and 29.9% HPLC grade water until saturated, and used as a matrix solution. 1μL of the sample was added to 10 μL of solution containing standard desmosine and CHCA matrix solution. The solution of CHCA matrix and standard was prepared in the ratio 1:9 (1μL standard in 9μL of CHCA matrix). Finally, the solution was stirred and 1μL of the solution was spotted on the MALDI plate and allowed to dry prior to mass spectrometric analysis.
MALDI-MS2 analysis was conducted using a laser energy of 2.6 μJ, for 150 scans with the AGC set to 10, 000 ions. Precursor ions of unlabeled desmosine (m/z 526.3) and d4 labeled desmosine standard (m/z 530.3) were selected in a single isolation window (m/z 529 ± 6), and fragmented with a normalized collision energy = 35, activation Q = 0.25, activation time = 30 ms. The most intense product ions, at 397 m/z for unlabeled desmosine and 401 m/z for d4-desmosine, were used for relative quantification.
NMR experimental parameters
All the experiments were performed on a Bruker 750MHz system externally referenced to adamantane (TMS scale). The temperature in all experiments was set to regulated to within ±0.5°C. The three pulse sequences used in the experiments are shown in Figure S1 in the supporting material. In all experiments proton TPPM decoupling was implemented at 100kHz41. The 13C π/2 pulse was 5μs and the rotor spinning frequency was 10 kHz ± 5Hz. The sample temperature at 10kHz magic angle spinning was measured with lead nitrate and offset to account for additional heating effects. In the cross polarization experiments the contact time was set to 3ms and the recycle delay was set to 3s. For the T1ρ experiments the two spin locking fields used were 50kHz and 25kHz and the spin lock time interval was incremented from 50μs to 10ms. In the T1ρ experiments a π pulse was used to eliminate ring down effects. The sample was packed in a 3.2mm rotor and sealed using compression style caps that prevented water loss. The extent of water loss throughout all the studies on the hydrated samples was measured to be less than 1%. The 37°C spectra were acquired by accumulating 10k scans and the 75°C temperature spectra were acquired by accumulating 28k scans. Gaussian multiplication with a broadening factor of 30Hz was used and the analysis was performed using MATLAB and matNMR42. The dwell time in all the experiments was 8.8μs and total number of points acquired was 17k.
Results and Discussion
Amino acid and MALDI mass spectrometry studies of elastin
Amino acid analysis was performed on all the samples in order to analyze their purity. For each sample of HCl was used for hydrolysis in vacuum at 110°C for 48h. After drying off the acid, each aliquot was dissolved and serially diluted to load onto the amino acid analyzer. Table 1 highlights the results of the amino acid analysis. The results show that elastin is largely composed of glycine, proline, alanine and valine; more than 84% of the residues in a single molecule belong to these four amino acids. Additionally, in less concentration, these measurements reveal that elastin also contains isoleucine, leucine, phenylalanine, and glutamine. Additional traces of amino acids appearing in unpurified bovine ligament elastin, such as histidine, may be evidence of either residual collagen or smooth muscle. Note that the presence of these amino acids also skews the percentage contribution of other tabulated values. Our findings for purified samples are in agreement with the theoretical values computed from the cDNA of tropoelastin43 and with work reported elsewhere37.
Table 1.
Amino acid analysis of three purified bovine nuchal ligament elastin samples (samples 1–3, described in the text), unpurified elastin and their relative desmosine concentration. The amino acid measurements are reported as “residues per 1000 residues (res/1000)” for ease of comparison with other published results37. The theoretical values, shown in column 6, correspond to the amino acids computed from cDNA of bovine tropoelastin43. The relative desmosine concentration represents the ratio of desmosine to d4-desmosine, normalized by the unpurified sample (starting product). Note that the concentration of lysine in the theoretical value (column 6) is higher than that in samples 1–3 and the unpurified sample, as cross-links consist of lysine.
| res/1000 | |||||
|---|---|---|---|---|---|
|
| |||||
| Sample 1 | Sample 2 | Sample 3 | Unpurified | Theory* | |
| Asx | 5.6 | 4.6 | 4.1 | 4.2 | 4 |
| Thr | 6.9 | 5.5 | 6.8 | 1.0 | 11 |
| Ser | 7.1 | 6.4 | 7.3 | 0.0 | 10 |
| Glx | 14.5 | 14.9 | 14.0 | 19.7 | 14 |
| Pro | 147.5 | 149.2 | 153.2 | 121.0 | 119 |
| Gly | 335.9 | 332.6 | 329.7 | 325.3 | 319 |
| Ala | 238.3 | 237.6 | 238.4 | 229.1 | 211 |
| Val | 121.8 | 125.3 | 123.7 | 125.5 | 126 |
| Cys | - | - | - | - | - |
| Met | 0.0 | 0.0 | 0.0 | 0.0 | - |
| IIe | 21.9 | 22.2 | 22.2 | 21.1 | 25 |
| Leu | 53.7 | 54.6 | 53.9 | 62.7 | 60 |
| Tyr | 6.9 | 7.3 | 7.2 | 17.4 | 10 |
| Phe | 33.2 | 34.3 | 34.6 | 38.4 | 29 |
| His | 0.0 | 0.0 | 0.0 | 10.4 | 0 |
| Lys | 2.4 | 1.9 | 1.9 | 10.9 | 53 |
| Arg | 4.3 | 3.6 | 3.0 | 13.3 | 7 |
|
| |||||
| Relative desmosine concentration | 1.00±0.10 | 1.09±0.12 | 0.86±0.10 | 1.00±0.14 | - |
To compare these three purification methods for tissue samples, MS/MS experiments were performed to quantify the amount of desmosine in the samples treated with and without the purification steps. The internal standard, d4-desmosine, was added prior to the MS/MS experiments where the precursor ions for desmosine and d4-desmosine were fragmented into their product ions (m/z 397 and 401, respectively), shown in Figure 1. In addition, Table 1 includes the relative concentration of desmosine in each sample studied in this work. The numbers reported are shown as the ratio of desmosine to d4-desmosine, normalized to the unpurified sample. Note that this metric is thus sensitive to traces of residual fat and collagen. The results of Table 1 indicate that the relative desmosine concentration in the samples are very similar, with similar overlap in error bars; sample 2 (hot alkali protocol) appears to have slightly higher desmosine concentration with respect to all the other samples. The small differences between sample 2 and all the other samples may be attributed to traces of fat and collagen, or differences in the extent of cross-linking across the samples. As the starting tissue was the same in all three isolation schemes, the amount of cross-linking is presumably similar. Thus, these results would indicate that sample 2 may contains slightly less residual fat and collagen compared to the other isolation schemes studied. However, this protocol is known to cause significant peptide-bond cleavage37, which we discuss later in the manuscript.
Figure 1.

Mass spectrometric spectra of hydrated samples treated with various purification methods: A) autoclaving (sample 1), B) hot alkali (sample 2), C) Starcher (sample 3), and D) unpurified. The relative amount of desmosine in the samples was measure by monitoring peak intensity of MS2 fragmentation of unlabeled desmosine (m/z 397) to d4-desmosine (m/z 401).
Dynamical and structural characteristics of hydrated elastin
Magic angle spinning 13C-NMR was used to investigate the structural modifications of elastin following purification. Two standard pulse sequences were used in the experiments; a direct polarization experiment which allows for the observation of both highly mobile and restricted spins and a cross polarization experiment which allows for the transfer of magnetization from protons to carbon spins via the carbon-proton dipole-dipole interaction. A signal resulting from cross-polarization implies a non-zero dipolar interaction between a 13C nucleus and 1H nuclei. Fast isotropic motion, however, results in suppression of the heteronuclear dipolar interaction, whereas slow or anisotropic motion would allow for magnetization to transfer between 13C and 1H nuclear spin baths44.
Figure 2 highlights the direct polarization (DP) and cross polarization (CP) 13C NMR spectra of hydrated bovine nuchal ligament elastin purified by the autoclaving method at 37°C. When completely hydrated, elastin is known to exhibit high mobility resulting in a low 1H-13C cross polarization signal, as noted elsewhere45,46. A detailed discussion of the chemical shift and secondary structure assignments is provided in the supporting material, and summarized in Table 2.
Figure 2.

a,b) Direct polarization (DP) 13C NMR spectrum of hydrated bovine nuchal ligament elastin purified by the autoclaving method (Sample 1) at 37°C. The aliphatic and carbonyl regions are presented with the same scale as the spectra acquired at 75°C (Figure 5) for ease of comparison. c,d) Cross Polarization (CP) 13C NMR spectrum of hydrated bovine nuchal ligament elastin purified by the autoclaving method (Sample 1) at 37°C. When completely hydrated, elastin exhibits high mobility resulting in a low 1H-13C cross polarization signal, as noted elsewhere45,46.
Table 2.
Tabulated results of bovine ligament elastin chemical shifts observed in this work at 37°C. Additional peaks that were resolvable at 75°C are also shown and indicated with *. As discussed in the text, the chemical shifts are identical across the samples (all three purification schemes, as well as the unpurified starting tissue) within our experimental uncertainty so one table is reported. A † indicates that the value shown was recomputed for the TMS scale53. The error bar reported in our measured chemical shift (column 2) corresponds to the half-width at half maximum of the corresponding lines.
| Observed Chemical Shift [ppm] | Assignment | Random Coil | α-helix | β-strand | Ref. | |
|---|---|---|---|---|---|---|
| 13Cα | (53.3 ± 1.3) | Ala | 51.14 ± 1.05 | 53.13 ± 1.05 | 49.83 ± 1.48 | 47 † |
| (48.65 ± 1.5) | 50.97 ± 1.94 | 53.16 ± 0.94 | 49.16 ± 1.28 | 48 † | ||
| (57.5 ± 0.7)* | Phe | 56.28 ± 2.02 | 59.11 ± 1.90 | 54.95 ± 1.59 | 47 † | |
| 55.24 ± 1.98 | 59.04 ± 1.63 | 54.63 ± 1.31 | 48 † | |||
| (43.4 ± 3.2) | Gly | 43.81 ± 1.05 | 45.21 ± 1.10 | 43.52 ± 1.17 | 47 † | |
| 43.64 ± 1.17 | 45.32 ± 0.90 | 43.38 ± 1.20 | 48 † | |||
| 42.7 | 43.0 | - | 49 | |||
| 43.1 | - | 43.2 – 44.3 | 50 | |||
| (61.4 ± 0.6)* | Ile | 59.33 ± 1.90 | 62.87 ± 1.74 | 58.35 ± 1.57 | 47 † | |
| 58.94 ± 2.08 | 62.98 ± 1.66 | 58.30 ± 1.51 | 48 † | |||
| (53.3 ± 1.4)* | Leu | 53.22 ± 1.70 | 55.82 ± 1.23 | 52.38 ± 1.31 | 47 † | |
| 53.15 ± 1.79 | 55.84 ± 0.98 | 52.24 ± 1.19 | 48 † | |||
| (60.4 ± 3.5) | Pro | 61.77 ± 1.26 | 63.79 ± 1.08 | 60.94 ± 1.03 | 47 † | |
| 61.83 ± 1.26 | 63.82 ± 1.01 | 61.09 ± 1.22 | 48 † | |||
| 61.65 | - | - | 51 † | |||
| (60.4 ± 3.5) | Val | 60.36 ± 2.16 | 64.46 ± 1.55 | 59.13 ± 1.64 | 47 † | |
| 60.10 ± 2.25 | 64.26 ± 1.39 | 59.02 ± 1.59 | 48 † | |||
| 60.38 | - | - | 51 † | |||
| 13Cβ | (17.7 – 22.0 ± 0.7) | Ala | 17.36 ± 1.26 | 16.56 ± 0.88 | 19.44 ± 2.05 | 47 † |
| 17.33 ± 1.27 | 16.57 ± 1.08 | 20.02 ± 1.77 | 48 † | |||
| 17.86 | - | - | 51 † | |||
| - | - | 19.9 | 52 | |||
| (37.3 ± 2.1) | Ile | 36.95 ± 1.69 | 35.90 ± 1.15 | 38.16 ± 1.98 | 47 † | |
| 36.56 ± 1.06 | 35.89 ± 1.08 | 38.39 ± 1.85 | 48 † | |||
| 37.25 | - | - | 51 † | |||
| (31.0 ± 1.8) | Pro | 31.94 ± 0.95 | 31.46 ± 0.95 | 32.27 ± 1.20 | 47 † | |
| 30.17 ± 0.96 | 29.38 ± 0.84 | 30.75 ± 0.93 | 48 † | |||
| (31.0 ± 1.8) | Val | 31.01 ± 1.37 | 29.79 ± 0.72 | 32.21 ± 1.61 | 47 † | |
| 30.98 ± 1.76 | 29.71 ± 1.74 | 32.11 ± 1.79 | 48 † | |||
| CO | (174.0 – 177.9 ± 2.1)* | Ala | 175.97 ± 1.57 | 177.70 ± 1.32 | 174.39 ± 1.51 | 47 † |
| 175.69 ± 1.45 | 177.88 ± 1.39 | 173.60 ± 1.61 | 48 † | |||
| (171.8 – 174.7 ± 2.8)* | Gly | 172.19 ± 1.42 | 173.81 ± 1.23 | 170.85 ± 1.58 | 47 † | |
| 172.60 ± 1.80 | 174.61 ± 1.50 | 171.31 ± 2.59 | 48 † | |||
| (174.0 – 177.9 ± 2.8)* | Pro | 175.19 ± 1.34 | 176.64 ± 1.45 | 174.48 ± 1.40 | 47 † | |
| 175.21 ± 1.72 | 176.64 ± 1.53 | 174.71 ± 1.50 | 48 † | |||
| (174.0 – 174.7 ± 1.4)* | Val | 173.96 ± 1.47 | 175.95 ± 1.38 | 173.10 ± 1.39 | 47 † | |
| 174.06 ± 1.63 | 176.05 ± 1.49 | 172.96 ± 1.36 | 48 † |
Structural and dynamical differences across purified elastins
Subtle differences were observed in the 13C NMR spectra acquired at 37°C of hydrated elastin purified by the three methods. Figture 3.a shows the direct polarization spectra of the three purified samples and the unpurified bovine nuchal ligament. The resolution of the direct polarization spectra is approximately 1–3ppm and within our experimental uncertainty the spectra appear remarkably similar in terms of the chemical shifts and relative signal intensities. However, the line width of the backbone carbonyl peak at approximately 172.0ppm seems to vary slightly across the different purification schemes and in comparison with the unpurified sample. Specifically, the backbone carbonyl of the unpurified sample appears more narrow compared with that of sample 1 indicating structural differences between the two. Again, as discussed in the Supplementary Materials, there is a signature of a splitting in the carbonyl peak, but our resolution is not sufficient for a more quantitative analysis. The signals observed in the aliphatic region of all hydrated samples (Figure 3.a) are well resolved with no observed differences in terms of the chemical shifts, relative signal intensities and line widths. It should be noted that an enhanced signal intensity at approximately 70ppm was observed in the unpurified sample and was assigned to threonine-Cβ.
Figure 3.

a) Direct polarization (DP) 13C NMR spectra of hydrated (a) and lyophilized (b) bovine nuchal ligament elastin samples at 37°C. For each set of figures (a, or b) the spectra shown are as follows: A) sample 1 purified by the autoclaving method38, B) sample 3 purified by the Starcher method40, C) sample 2 purified by the alkaline extraction method39 and D) unpurified elastin. As discussed in the text, the spectra appear remarkably similar in terms of the chemical shifts, however a signal enhancement of the valine or proline-Cβ at approximately 30.0ppm was observed in D-2. Note that differences shown in Table 1 in regards to amino acid concentration may not be reflected in a simple difference in signal intensity in the NMR spectra due differences in mass of the sample packed into the rotor or potential structural heterogeneity across the samples. The reader should note the different scales used in the spectra, as they were all scaled such that the standard deviation of the noise was set to unity.
Upon lyophilization of the samples, the dynamics of the ensemble seem to change dramatically in comparison with the hydrated samples. Dynamical changes between lyophilized and hydrated elastin were reported previously by Kumashiro and coworkers where they showed that the temperature and hydration levels seem to affect the underlying interactions and dynamics45. Additionally, previous studies on a small elastin peptide showed a decrease in the relaxation times, T1ρ, with increasing the hydration level of the protein54. The hydration dependence is also directly reflected in the 13C NMR spectra acquired by cross polarizing the carbon spins with the proton bath (Figure 4). One of the key characteristics of these NMR spectra is the broad lines of approximately 7ppm line width, which do not allow for an accurate determination of secondary structure. A second key observation is that these spectra appear remarkably similar in terms of chemical shifts which suggests that the structure is not affected upon purification.
Figure 4.

Cross polarization (CP) 13C NMR spectra of lyophilized bovine nuchal ligament elastin samples at 37°C. A) sample 1 purified by the autoclaving method38, B) sample 3 purified by the Starcher method40, C) sample 2 purified by the alkaline extraction method39 and D) unpurifed elastin. As discussed in the text, a signal enhancement which corresponds to alanine Cα at approximately 53ppm, of the unpurified sample (D) was observed.
In the spectra of the unpurified sample shown in Figure 4, an enhanced signal intensity is observed at approximately 53.0ppm in comparison with the purified samples. Although the spectra in Figure 4 are quite broad, this peak was assigned to alanine-Cα and according to the literature the observed chemical shift points to α-helical structure47,48. In previous NMR studies of elastin, the chemical shift of the alanine-Cα was observed at approximately 52.0ppm to 53.0ppm but different structures have been reported. Kumashiro et al. reported that the alanine-Cα signal at 53.0ppm was more α-helical which in agreement with our observation55. On the other hand, Witterbort et al. reported that the alanine-Cα chemical shift points to random coil/β-sheet like structure51. The signal enhancement of the alanine motifs in the unpurified sample suggests that the Cα spins experience a more rigid environment compared with that in the purified elastin and thus cross polarization is more efficient. Additionally, across the purified samples shown in Figure 4, the signal intensity of the alanine-Cα seems to vary; in sample 2 (hot alkali) the peak is no longer observed. Therefore, a portion of the alanine-Cα carbons seem to become more mobile upon purification with the alkaline extraction technique. This behavior may be due to peptide-bond cleavage which occurs with the hot alkali method, as pointed out by R. Mecham37.
A more quantitative interpretation of the signal enhancement of the alanine residues in Figure 4 may be given by the amino acid analysis data and by normalizing the spectra to account for differences in mass (data not shown). Table 1 shows a higher concentration of alanine residues in purified elastin of approximately 3.8% compared to unpurified elastin. As mentioned earlier the presence of histidine, glutamine and arginine might skew the calculated concentration of alanine in the unpurified sample. While the values tabulated in table 1 would suggest that the signal intensity of alanine be lower in the unpurified sample the integral of the peak at 53ppm (corresponding to alanine-Cα) of Figure 4D.2 is markedly larger than that of all purified elastin spectra by approximately 18%. The differences in the signal intensity of the alanine motifs between the purified and unpurified spectra may be due to dynamical changes but also due to residual amino acids from smooth muscle or collagen. For example, the glutamine-Cα signal arising from β-strand like motifs have a chemical shift of (54.95 ± 1.59)ppm47 (TMS) which might contribute to the observed signal in the unpurified sample. However, this signal enhancement was not evident in the spectra obtained at 37° or 75° from elastin in the hydrated state (see below). Therefore, a more likely explanation for the differences in the spectra shown in Figure 4 is that the alanine motifs in the unpurified sample are structurally more rigid than that of any of the purified samples, which results in a higher cross polarization enhancement.
Subtle differences in the spectra were observed in the methyl region between 16.0ppm and 22.0ppm. Specifically, the peak at 16.1ppm that was assigned to the isoleucine-Cγ, seems to be more resolvable in the unpurified sample in comparison with the purified elastin samples (Figure 4-D). It should be noted here that previous studies on lyophilized elastin reported this peak as alanine-Cβ 55 or valine-Cγ 56, whereas other studies on hydrated elastin appear to be in agreement with our assignment51. The resolution enhancement indicates that upon purification isoleucine motifs would seem to experience a more mobile environment. Slightly more in the downfield region, the peaks at approximately 18.0ppm and 24.0ppm were assigned to alanine/valine and leucine residues respectively. The peak at 29.5ppm arises from the proline-Cβ and the small shoulder at roughly 32.0ppm was assigned to the valine-Cβ. The glycine-Cα signal was observed at 42.3ppm in all the samples with an approximate 3ppm line width. The peaks at 47.6ppm and 59.3ppm were assigned to proline-Cδ and valine-Cα which is in agreement with previous NMR studies on elastin56. Additionally, the small shoulder at approximately 55ppm may be a signature of the leucine-Cα carbons. According to references47,48 the Cα of leucine has chemical shift in the region between 55ppm to 59ppm (TMS) but our spectral resolution does not allow for a reliable structural assignment.
Figure 3.b shows the direct polarization spectra of the lyophilized samples studied in this work. The spectra look remarkably similar in terms of chemical shifts, however some subtle differences should be noted in the signal intensities. The peak at approximately 30.0ppm was assigned to proline-Cβ; however the valine-Cβ signal has almost identical chemical shift as noted earlier. A larger signal at 30.0ppm was observed before purifying the tissue (Figure 3.b-D4). However, upon purification the relative signal intensity is reduced and the peak is broader (Figure 3.b-A through Figure 3.b-C). This observation indicates that upon purification some of the proline-Cβ or valine-Cβ seem to exhibit increased structural heterogeneity. Additionally, the peak at 22.7ppm was assigned to alanine-Cβ, which is well resolved in the purified sample and seems to be suppressed by overlapping peaks upon purification (Figure 3.b-D4).
High temperature 13C-NMR spectra of hydrated elastin
Previous 13C MAS NMR studies of elastin performed by Wittebort et al. showed that there is an enhancement in the spectral resolution by performing experiments at 75°C51. We have adopted the same approach to investigate structural variations in the purified samples. Figure 5 shows the 13C-NMR spectrum of hydrated bovine ligament elastin purified by the autoclaving method (Sample 1) at 75°C. One of the key observations is the enhancement of the resolution with 0.3ppm line width, in both the aliphatic region and the backbone carbonyl in comparison with the 37°C spectrum (Figure 2). This enhancement allows for a more accurate chemical shift and structural assignment. Narrower lines may point to higher structural homogeneity and increased mobility at high temperature. Another important finding is that within our experimental uncertainty, both 37°C and 75°C spectra appear remarkably similar in terms of the observed chemical shifts. Experimentally, we found that the spectra of the purified and unpurified samples appeared identical in terms of chemical shifts and relative signal intensities; for that reason only one spectrum is shown in Figure 5.
Figure 5.

Normalized 13C NMR direct polarization spectrum of hydrated bovine nuchal ligament elastin (sample 1) at 75°C. As discussed in the text, all the spectra from purified elastin were identical in terms of the measured chemical shifts and relative signal intensities, thus only one spectrum is shown. a) The backbone carbonyl is shown and a characteristic splitting is observed(structural assignments provided and discussed in the text). b) The aliphatic region is shown; our spectral resolution is approximately 0.3ppm and allows for a more accurate chemical shift and secondary structure assignment compared with data acquired at 37°C.
Comparing the 37°C (Fig. 2) and 75°C data (Fig. 5) we observed subtle differences in the relative signal intensities of the valine-Cγ;/alanine-Cβ signals (at approximately 18.8ppm), leucine-Cδ at 23.5ppm and isoleucine-Cγ signals (at 16.7ppm). These differences may be attributed to overlapping peaks in the 37°C spectra.
The backbone carbonyl has characteristic splitting at 171.8ppm and 174.0ppm. The peak at 171.8ppm was assigned to glycine residues in β-strand like structure, which is in agreement with previous studies52; however, glycine residues in random coil motifs appear very close to the observed chemical shift47. The peak at 174.0ppm points to glycine in α-helical secondary structure, proline in random coil and/or β-strand, alanine in β-strand and valine in random coil and/or β-strand conformations. The small shoulder at approximately 174.7ppm points to either glycine in α-helical structure, alanine in random coil and/or β-strand, valine in random coil and/or β-strand and proline in random coil and/or β-strand. Finally, the peak at 177.9ppm could be evidence of alanine or proline residues in β-strand like structures. In addition, glycine and valine residues may also contribute to this peak.
In the aliphatic region we observe more β-strand and random coil like structures, similar to the 37°C spectra. Specifically, starting with the upfield region we observe the isoleucine Cδ and Cγ at 11.4ppm and 16.0ppm respectively. The peaks at 17.8ppm, 18.5ppm and 19.6ppm may be alanine and valine motifs in β-strand or random coil structures, which is in agreement with the observations at 37°C. According to Pometun et al. the alanine-Cβ at approximately 19.0ppm points to a random coil structure51. Additionally, the peaks at 22.1ppm and 23.5ppm were assigned to alanine-Cβ or leucine-Cδ respectively. The signal at 22.1ppm would imply an alanine β-strand structure. Proline-Cγ and valine-Cβ are observed at 25.3ppm, 29.9ppm and 30.9ppm; these chemical shifts point to either β-strand, random coil or α-helical secondary structures.
Isoleucine-Cβ was assigned to the peak at approximately 37.2ppm and this chemical shift could point to either β-strand, random coil or α-helical structures. The peak at 40.9ppm was assigned to isoleucine-Cβ which implies a β-strand secondary structure47. It should be noted that at 37°C isoleucine exhibited β-strand and/or random coil conformations. Previous studies showed that isoleucine rich motifs might be in a more random coil secondary structure51. Additionally, leucine-Cβ contributes to the signal at 40.9ppm in either β-strand, random coil or α-helical structure. The peak at 43.7ppm was assigned to glycine-Cα, however as in the 37°C spectra, no assignment can be made regarding structure. The signals at 48.7ppm and 50.5ppm correspond to the alanine-Cα in random coil and/or β-strand structures; proline-Cδ also contributes to the signal at 50.5ppm. The peak 53.6ppm was assigned to the alanine-Cα and indicates an α-helical conformation in agreement with our observation at 37°C. In addition, leucine-Cα and phenylalanine-Cα contribute to the signal at 53.6ppm in random coil and β-strand structures.
The Cα signals of phenylalanine and isoleucine, two of the low abundance amino acids of elastin, appear at 57.6ppm and 61.5ppm respectively and indicate either α-helical, random coil and β-strand structures respectively. The peak at 60.4ppm was assigned to valine-Cα; this chemical shift points to β-strand and random coil secondary structure as discussed in the Supplementary Materials. In addition, proline-Cα in random coil or β-strand conformation appears to have a chemical shift of 60.4ppm; isoleucine in random coil structure appears to contribute to this signal. Lastly, the small peak at 70.7ppm was assigned to threonine.
13C-1H Rotational correlation times
A measurement of the dynamics of the system can be obtained by considering the average rotational correlation times of the effective carbon-proton internuclear vectors, obtained by NMR relaxation methods. Previous studies for describing the molecular motions of elastin and short elastin peptides included measurements of a single longitudinal relaxation time for each spectroscopically resolvable moiety12,57, following Solomon’s equations58. Their findings indicate that molecular motions of the backbone carbonyl carbons of elastin are in the ns time scale. However, Urry et al. showed that it is ambiguous to determine the correlation times from a single relaxation time and that this ambiguity arises from the degeneracy of τc with respect to T1 due to the shape of the spectral density function (eq. S6)59. Large proteins, such as elastin are known to exhibit a distribution of dynamics on different time scales. The collective dynamical behavior of elastin and small elastin peptides have been studied by acoustic and dielectric methods that probed frequencies from 0.1 to 100kHz and 0.1 to 100MHz, respectively9,60,61. However, these motions are different than what is studied by 13C NMR, which is sensitive to local 13C-1H nuclei internuclear fluctuations. The 13C T1ρ relaxation time is sensitive to motions spanning the dynamic range of μs to ms62 and we have provided an overview of the salient equations used in the Supplementary materials. Without knowledge of the spectral density functions
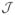 (ω) it is not possible to know how many correlation times govern the relaxation behavior of a given 13C-1H fluctuation. we have therefore restricted the discussion below under the assumptions of an average single correlation time derived from two T1ρ measurements.
(ω) it is not possible to know how many correlation times govern the relaxation behavior of a given 13C-1H fluctuation. we have therefore restricted the discussion below under the assumptions of an average single correlation time derived from two T1ρ measurements.
Table 3 summarizes the rotating frame relaxation times T1ρ at two different fields and the correlation times of the spectroscopically resolvable moieties of the hydrated elastin samples at 37°C (example T1ρ relaxation curves of proline-Cβ are shown in the Supplementary Materials, Figure S2).
Table 3.
Tabulated results of T1ρ relaxation times at two different fields and correlation times of the spectroscopically resolved moieties of hydrated elastin samples at 37°C.
| Sample 1 | Sample 2 | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| ppm | T1ρ [ms] (50kHz) | T1ρ [ms] (25kHz) | τc[μs] | ppm | T1ρ [ms] (50kHz) | T1ρ [ms] (25kHz) | τc[μs] |
| 18.8 | 5.90±0.18 | 3.83±0.10 | 3.27±0.05 | 18.8 | 9.60±0.54 | 6.50±0.23 | 3.02±0.25 |
| 19.5 | 5.62±0.12 | 4.05±0.08 | 2.65±0.02 | 19.5 | 8.88±0.35 | 6.08±0.15 | 2.95±0.18 |
| 22.0 | 3.11±0.13 | 1.95±0.06 | 3.49±0.13 | 22.0 | 3.23±0.31 | 2.76±0.11 | 1.62±0.73 |
| 23.4 | 2.51±0.08 | 1.58±0.04 | 3.46±0.08 | 23.4 | 2.91±0.15 | 2.61±0.09 | 1.30±0.24 |
| 25.2 | 1.05±0.02 | 0.71±0.01 | 3.03±0.06 | 25.2 | 2.20±0.09 | 1.69±0.05 | 2.27±0.13 |
| 30.9 | 1.12±0.02 | 0.53±0.01 | 5.35±0.01 | 30.9 | 1.74±0.05 | 1.24±0.02 | 2.71±0.15 |
| 43.3 | 0.60±0.04 | 0.27±0.01 | 5.71±0.44 | 43.3 | 1.01±0.03 | 0.32±0.02 | 8.65±0.63 |
| 60.3 | 0.76±0.06 | 0.23±0.03 | 9.11±1.05 | 60.3 | 1.92±0.02 | - | - |
| 172.1 | 1.14±0.02 | 0.70±0.02 | 3.62±0.14 | 172.1 | 1.94±0.14 | 0.96±0.04 | 5.03±0.42 |
|
| |||||||
| Sample 3 | Unpurified | ||||||
|
| |||||||
| 18.8 | 6.12±0.22 | 3.92±0.10 | 3.36±0.13 | 18.1 | 4.44±0.14 | 3.23±0.08 | 2.59±0.08 |
| 19.5 | 6.46±0.20 | 4.34±0.09 | 3.07±0.13 | 18.8 | 5.14±0.09 | 3.01±0.05 | 3.91±0.01 |
| 22.0 | 2.21±0.11 | 1.52±0.05 | 2.92±0.20 | 21.3 | 3.30±0.19 | 2.07±0.05 | 3.48±0.41 |
| 23.4 | 2.24±0.11 | 1.21±0.03 | 4.44±0.32 | 22.7 | 3.16±0.16 | 1.71±0.05 | 4.43±0.28 |
| 25.2 | 0.86±0.03 | 0.63±0.01 | 2.55±0.23 | 24.5 | 1.54±0.06 | 1.18±0.02 | 2.28±0.26 |
| 30.9 | 0.97±0.02 | 0.55±0.01 | 4.12±0.03 | 30.5 | 1.36±0.05 | 0.76±0.02 | 4.21±0.14 |
| 43.3 | 0.51±0.03 | 0.34±0.01 | 3.11±0.35 | 42.9 | 1.05±0.07 | 0.58±0.02 | 4.29±0.42 |
| 60.3 | 0.59±0.13 | 0.32±0.05 | - | 61.7 | 1.53±0.13 | 1.27±0.07 | 1.80±0.37 |
| 172.1 | 1.24±0.10 | 1.13±0.06 | - | 172.1 | 2.80±0.11 | 2.17±0.13 | 2.22±0.24 |
All the measured correlation times appear to be in the μs time scale. The spectral density function
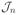 (ω) contains the term (ωτc)2 in the denominator which for the rotor frequency is of the order of ~ 0.05 ≪ 1 (computed by substituting the average correlation time < τc >= 3.63μs from Table 3 and the rotor frequency ω = 2π * 104 rad). Therefore the interference of the molecular dynamics and the sample spinning has negligible effects on R2. Referring to Table 3, small differences in the correlation times were observed across the samples. Specifically, the peak at approximately 23.5ppm of sample 2 that was assigned to leucine-Cδ appears to experience a more mobile environment in comparison with the other samples. Additionally, the correlation times of the proline-Cβ at 31.0ppm seem to vary slightly across the samples from 2.7μs of sample 2 to 5.3μs of sample 1, indicating less mobility. The glycine-Cα however seems to be more rigid in sample 2, with an average correlation time of 8.6μs, compared to sample 3 which has a correlation time of 3.1μs. Based on our correlation time measurements, the backbone carbonyl of the unpurified sample appears slightly more mobile in comparison with sample 1 and sample 2.
(ω) contains the term (ωτc)2 in the denominator which for the rotor frequency is of the order of ~ 0.05 ≪ 1 (computed by substituting the average correlation time < τc >= 3.63μs from Table 3 and the rotor frequency ω = 2π * 104 rad). Therefore the interference of the molecular dynamics and the sample spinning has negligible effects on R2. Referring to Table 3, small differences in the correlation times were observed across the samples. Specifically, the peak at approximately 23.5ppm of sample 2 that was assigned to leucine-Cδ appears to experience a more mobile environment in comparison with the other samples. Additionally, the correlation times of the proline-Cβ at 31.0ppm seem to vary slightly across the samples from 2.7μs of sample 2 to 5.3μs of sample 1, indicating less mobility. The glycine-Cα however seems to be more rigid in sample 2, with an average correlation time of 8.6μs, compared to sample 3 which has a correlation time of 3.1μs. Based on our correlation time measurements, the backbone carbonyl of the unpurified sample appears slightly more mobile in comparison with sample 1 and sample 2.
The correlation times of the alanine-Cβ (22.0ppm) of sample 2 seem to be shorter than any other of the samples by a factor of two. These differences may be due to peptide bond cleavage which may result from the hot alkali treatment, discussed earlier. The correlation times of the valine-Cα which was observed at approximately 60.4ppm were different between the unpurified sample and sample 1; the valine-Cα appears more rigid in sample 1 as the correlation time is more than eight times larger in comparison with the unpurified sample. It should be noted here that the correlation times of the valine-Cα of sample 2 and 3 could not be determined because of relatively large error bars in the T1ρ measurements. However, the estimated values are of the order of μs.
We note that the correlation times in hydrated bovine nuchal ligament elastin determined by a static (i.e. no sample spinning) measurement performed by us,46 and others12,57, appear smaller than the values reported in this study. In our previous work 46 the values reported were in units of s/cycle and need to be divided by 2π for comparison with the units here (s/rad). In unstrained nuchal ligament elastin the average aliphatic correlation time measured was 8.02×10−8s and for the carbonyl, measurements on nuchal ligament elastin in unstrained conditions the average correlation time was 1.29 ×10−7s.35 In a study by Torchia and coworkers the 13C NMR spin-lattice relaxation times of unstretched calf ligamentum nuchae in 0.15M NaCl were also measured without sample spinning. Approximately 80% of the backbone carbonyl carbons were shown to exhibit a correlation time of approximately 4 ×10−8s.57 Fleming, Sullivan, and Torchia also reported 13C-1H correlation times of purified labeled chick aorta, again without sample spinning. Their correlation times ranged from 6 to 15 ×10−8 s for all the valine labeled residues and 5 to 20 ×10−8 s for 75 % of the alanine and 60% of the lysine labeled resides.12 A likely cause for the differences in the correlation times measured in this work, and that of the previous studies, may have arisen from the fact that we measured the relaxation times in the rotating frame. The relaxation time in the rotating frame has a different dependence on the spectral density than the spin lattice relaxation time and is therefore sensitive to different timescales of motion.
Conclusions
The present work highlights the structural and dynamical modifications resulting from three methods to isolate elastin from fat, collagen and muscle studied by high field 13C NMR spectroscopy and relaxometry. Our results show that elastin maintains its structure upon purification, as the NMR chemical shifts are identical within our experimental uncertainty. However, small differences were observed in the dynamics and the relative signal intensities. The NMR spectra of the hydrated nuchal ligament elastin indicate high mobility of the backbone carbonyl at 37°C. By increasing the temperature to 75°C, the line width of the backbone carbonyl appears narrower allowing for some structural assignment. In the aliphatic region a comparison between the direct polarization and cross polarization experiments showed that the alanine motifs are comparatively more rigid than other residues. Additionally, glycine residues appear highly mobile resulting in narrower spectral lines in the direct polarization experiment and smaller signal intensity in the cross polarization experiment. The main feature of the cross polarization experiments on all lyophilized nuchal ligament elastins (purified & unpurified) was that the chemical shifts appear identical within our experimental uncertainty. The studies of the lyophilized samples revealed that upon purification the dynamical characteristics of the alanine motifs seem to be enhanced. Additionally, the direct polarization spectra of unpurified lyophilized elastin showed a signal at 30ppm which was assigned to proline-Cβ and/or valine-Cβ and is reduced upon purification indicating a possible structural heterogeneity in the proline or valine rich motifs or decrease in molecular motion. Across the samples, our results indicate that elastin largely maintains its structure upon purification, as well as the extent of cross linking, however the dynamics seem to be affected. Based on the line widths of the NMR spectra, the backbone appears structurally different and the backbone mobility also appears different based on the measured correlation times. The glycine rich motifs of elastin purified by alkaline extraction seem to be more rigid in comparison with elastin purified by the Starcher method37,40. Lastly, our measurements reveal that the hot alkali purification protocol results in a product with relatively less fat and collagen in comparison with the other purification schemes studied. However, this method for isolating elastin from other tissues constituents has been reported to cause significant peptide-bond cleavage and appears to alter the dynamics of alanine and glycine residues.
Supplementary Material
Highlights.
13C NMR methods were applied to study elastin structure/dynamics after purification.
13C-1H correlation times of Gly and Pro moieties vary across purification methods.
Desmosine cross-links in the samples were quantified by MALDI mass spectroscopy.
Differences in carbonyl line-widths suggest possible structural heterogeneity.
CP intensity of Ca-Ala appears greater before purification.
Acknowledgments
G.S. Boutis acknowledges support from award No. SC1GM086268-07 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health (NIH). The authors thank S. W. Morgan, M. C. Silverstein, B. Itin and M. Goger for useful discussions and help with the experiments. G. S. Boutis is a member of the New York Structural Biology Center. The data collected at NYSBC was made possible by a grant from NYSTAR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT. Human Molecular Genetics. 1997;6:1021–8. doi: 10.1093/hmg/6.7.1021. [DOI] [PubMed] [Google Scholar]
- 2.Urry DW. Arterial Mesenchyme and Arteriosclerosis. Springer; 1974. pp. 211–243. [PubMed] [Google Scholar]
- 3.Kielty CM. Expert Reviews in Molecular Medicine. 2006;8:1–23. doi: 10.1017/S146239940600007X. [DOI] [PubMed] [Google Scholar]
- 4.Sell SA, Wolfe PS, Garg K, McCool JM, Rodriguez IA, Bowlin GL. Polymers. 2010;2:522–553. [Google Scholar]
- 5.Wise SG, Mithieux SM, Weiss AS. Advances in Protein Chemistry and Structural Biology. 2009;78:1–24. doi: 10.1016/S1876-1623(08)78001-5. [DOI] [PubMed] [Google Scholar]
- 6.Lamme EN, De Vries H, van Veen H, Gabbiani G, Westerhof W, Middelkoop E. Journal of Histochemistry & Cytochemistry. 1996;44:1311–1322. doi: 10.1177/44.11.8918906. [DOI] [PubMed] [Google Scholar]
- 7.Debelle L, Alix AJ. Biochimie. 1999;81:981–94. doi: 10.1016/s0300-9084(99)00221-7. [DOI] [PubMed] [Google Scholar]
- 8.Debelle L, Tamburro AM. The International Journal of Biochemistry & Cell Biology. 1999;31:261–72. doi: 10.1016/s1357-2725(98)00098-3. [DOI] [PubMed] [Google Scholar]
- 9.Urry D, Parker T. Mechanics of Elastic Biomolecules. Springer; 2003. pp. 543–559. [Google Scholar]
- 10.Partridge SM. The Physiology and Biochemistry of Muscle as a Food. University of Wisconsin Press; 1966. [Google Scholar]
- 11.Weis-Fogh T, Andersen S. Nature. 1970;227:718–721. doi: 10.1038/227718a0. [DOI] [PubMed] [Google Scholar]
- 12.Fleming W, Sullivan C, Torchia D. Biopolymers. 1980;19:597–617. doi: 10.1002/bip.1980.360190311. [DOI] [PubMed] [Google Scholar]
- 13.Partridge S, Davis H, Adair G. Biochemical Journal. 1955;61:11. doi: 10.1042/bj0610011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Partridge SM. Symposium on Fibrous Proteins. Butterworths; 1968. [Google Scholar]
- 15.Ramachandran GN. Aspects of protein structure. International Symposium on Protein Structure and Crystallography; 1963; Madras, India. 1963. [Google Scholar]
- 16.He D, Chung M, Chan E, Alleyne T, Ha KC, Miao M, Stahl RJ, Keeley FW, Parkinson J. Matrix Biology. 2007;26:524–540. doi: 10.1016/j.matbio.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Urry D. Arterial Mesenchima and Arterioscleresis. Wiley Online Library; 1974. pp. 219–243. [Google Scholar]
- 18.Tamburro A, Guantieri V, Pandolfo L, Scopa A. Biopolymers. 1990;29:855–870. doi: 10.1002/bip.360290419. [DOI] [PubMed] [Google Scholar]
- 19.Huang J, Sun C, Mitchell O, Ng N, Wang ZN, Boutis GS. The Journal of Chemical Physics. 2012;136:1–27. doi: 10.1063/1.3685454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B, Alonso DO, Daggett V. Journal of Molecular Biology. 2001;305:581–92. doi: 10.1006/jmbi.2000.4306. [DOI] [PubMed] [Google Scholar]
- 21.Schreiner E, Nicolini C, Ludolph B, Ravindra R, Otte N, Kohlmeyer A, Rousseau R, Winter R, Marx D. Physical review letters. 2004;92:148101. doi: 10.1103/PhysRevLett.92.148101. [DOI] [PubMed] [Google Scholar]
- 22.Meyer DE, Chilkoti A. Biomacromolecules. 2004;5:846–851. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- 23.Martín L, Castro E, Ribeiro A, Alonso M, Rodríguez-Cabello JC. Biomacromolecules. 2012;13:293–298. doi: 10.1021/bm201436y. [DOI] [PubMed] [Google Scholar]
- 24.Serrano V, Liu W, Franzen S. Biophysical Journal. 2007;93:2429–2435. doi: 10.1529/biophysj.106.100594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luan CH, Parker TM, Prasad KU, Urry DW. Biopolymers. 1991;31:465–475. doi: 10.1002/bip.360310502. [DOI] [PubMed] [Google Scholar]
- 26.Reguera J, Urry DW, Parker TM, McPherson DT, Rodríguez-Cabello JC. Biomacromolecules. 2007;8:354–358. doi: 10.1021/bm060936l. [DOI] [PubMed] [Google Scholar]
- 27.MacEwan SR, Chilkoti A. Peptide Science. 2010;94:60–77. doi: 10.1002/bip.21327. [DOI] [PubMed] [Google Scholar]
- 28.Girotti A, Fernández-Colino A, López IM, Rodríguez-Cabello JC, Arias FJ. Biotechnology Journal. 2011;6:1174–1186. doi: 10.1002/biot.201100116. [DOI] [PubMed] [Google Scholar]
- 29.Lillie MA, Gosline JM. International Journal of Biological Macromolecules. 2002;30:119–27. doi: 10.1016/s0141-8130(02)00008-9. [DOI] [PubMed] [Google Scholar]
- 30.Lillie M, Gosline J. Biorheology. 1992;30:229–242. doi: 10.3233/bir-1993-303-408. [DOI] [PubMed] [Google Scholar]
- 31.Mistrali F, Volpin D, Garibaldo G, Ciferri A. The Journal of Physical Chemistry. 1971;75:142–150. doi: 10.1021/j100671a023. [DOI] [PubMed] [Google Scholar]
- 32.Hoeve C, Flory P. Biopolymers. 1974;13:677–686. doi: 10.1002/bip.1974.360130404. [DOI] [PubMed] [Google Scholar]
- 33.Li B, Alonso DO, Bennion BJ, Daggett V. Journal of the American Chemical Society. 2001;123:11991–11998. doi: 10.1021/ja010363e. [DOI] [PubMed] [Google Scholar]
- 34.Wasserman Z, Salemme F. Biopolymers. 1990;29:1613–1631. doi: 10.1002/bip.360291211. [DOI] [PubMed] [Google Scholar]
- 35.Sun C, Mitchell O, Huang J, Boutis GS. The Journal of Physical Chemistry B. 2011;115:13935–13942. doi: 10.1021/jp207607r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daamen WF, Hafmans T, Veerkamp JH, Kuppevelt THV. Biomaterials. 2001;22:1997–2005. doi: 10.1016/s0142-9612(00)00383-5. [DOI] [PubMed] [Google Scholar]
- 37.Mecham RP. Methods (San Diego, Calif) 2008;45:32–41. doi: 10.1016/j.ymeth.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Partridge S, Davis H, Adair S. Biochemical Journal. 1952:6. [Google Scholar]
- 39.Lowry O, Gilligan D, Katersky E. Journal of Biological Chemistry. 1941:139. [Google Scholar]
- 40.Starcher BC, Galione MJ. Analytical Biochemistry. 1976;74:441–447. doi: 10.1016/0003-2697(76)90224-4. [DOI] [PubMed] [Google Scholar]
- 41.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. The Journal of Chemical Physics. 1995;103:6951. [Google Scholar]
- 42.Van Beek JD. Journal of Magnetic Resonance. 2007;187:19–26. doi: 10.1016/j.jmr.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 43.Daamen WF, Hafmans T, Veerkamp JH, Kuppevelt THV. Tissue Engineering. 2005;11:1168–1176. doi: 10.1089/ten.2005.11.1168. [DOI] [PubMed] [Google Scholar]
- 44.Duer MJ. Introduction to Solid-State NMR Spectroscopy. Wiley-Blackwell; UK: 2005. [Google Scholar]
- 45.Perry A, Stypa MP, Tenn BK, Kumashiro KK. Biophysical Journal. 2002;82:1086–95. doi: 10.1016/S0006-3495(02)75468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun C, Mitchell O, Huang J, Boutis GS. The Journal of Physical Chemistry B. 2011;115:13935–42. doi: 10.1021/jp207607r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Neal S, Wishart DS. Journal of Biomolecular NMR. 2003;25:173–95. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Jardetzky O. Protein Science. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asakura T, Iwadate M, Demura M, Williamson MP. International Journal of Biological Macromolecules. 1999;24:167–71. doi: 10.1016/s0141-8130(98)00082-8. [DOI] [PubMed] [Google Scholar]
- 50.Jenkins JE, Creager MS, Lewis RV, Holland GP, Yarger JL. Biomacromolecules. 2010;11:192–200. doi: 10.1021/bm9010672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pometun MS, Chekmenev EY, Wittebort RJ. The Journal of Biological Chemistry. 2004;279:7982–7. doi: 10.1074/jbc.M310948200. [DOI] [PubMed] [Google Scholar]
- 52.Kumashiro KK, Kim MS, Kaczmarek SE, Sandberg LB, Boyd CD. Biopolymers. 2001;59:266–75. doi: 10.1002/1097-0282(20011005)59:4<266::AID-BIP1023>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 53.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. Journal of Biomolecular NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 54.Yao XL, Conticello VP, Hong M. Magnetic Resonance in Chemistry. 2004;42:267–75. doi: 10.1002/mrc.1330. [DOI] [PubMed] [Google Scholar]
- 55.Kumashiro KK, Ho JP, Niemczura WP, Keeley FW. The Journal of Biological Chemistry. 2006;281:23757–65. doi: 10.1074/jbc.M510833200. [DOI] [PubMed] [Google Scholar]
- 56.Lusceac SA, Vogel MR, Herbers CR. Biochimica et Biophysica acta. Proteins and Proteomics. 2010;1804:41–48. doi: 10.1016/j.bbapap.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 57.Torchia D, Piez K. Journal of Molecular Biology. 1973;76:419–424. doi: 10.1016/0022-2836(73)90514-7. [DOI] [PubMed] [Google Scholar]
- 58.Solomon I. Physical Review. 1955;99:559. [Google Scholar]
- 59.Urry DW, Trapane T, Iqbal M, Venkatachalam C, Prasad K. Biochemistry. 1985;24:5182–5189. doi: 10.1021/bi00340a034. [DOI] [PubMed] [Google Scholar]
- 60.Samouillan V, Lamure A, Lacabanne C. Chemical Physics. 2000;255:259–271. [Google Scholar]
- 61.Samouillan V, Dandurand J, Lacabanne C, Hornebeck W. Biomacromolecules. 2002;3:531–537. doi: 10.1021/bm015655m. [DOI] [PubMed] [Google Scholar]
- 62.VanderHart D, Garroway A. The Journal of Chemical Physics. 1979;71:2773–2787. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.