

Abstract
Background
A classic response to acute vasodilator testing (drop of > 10mmHg in mean pulmonary artery pressure [mPAP] to < 40mmHg) confers an excellent prognosis in patients with idiopathic pulmonary arterial hypertension (IPAH) and identifies candidates for treatment with calcium channel blockers (CCB). Little is known about vasodilator responsiveness (VR) in other types of PAH, or about outcomes in patients with a significant but non-classic decrease in mPAP. We hypothesized that VR occurs in non-idiopathic PAH and ‘non-classic’ VR portends a better prognosis than no VR in PAH.
Methods
Acute VR testing with nitric oxide was performed on 155 consecutive patients referred for PH evaluation. Non-classic response was defined as decrease in mPAP > 10mmHg to > 40mmHg with preserved cardiac output. Demographics and functional status were assessed at baseline and the first clinic visit after VR testing, and survival was followed over time.
Results
Twenty patients (13%) displayed classic VR. Among classic responders, 12 (60%) had IPAH and 8 (40%) had connective tissue disease-associated PAH (CTD-PAH); however, only responders with IPAH had improved survival compared with non-responders (p=0.02). Thirteen patients (8%) had a non-classic VR. Non-classic response was not associated with improved survival compared to non-responders (p=0.86). Acute change in mPAP or pulmonary vascular resistance in the entire cohort did not predict survival.
Conclusions
Classic acute VR occurs in CTD-PAH as well as IPAH; however, only IPAH patients have improved outcomes. A significant but non-classic VR is not associated with improved survival.
Introduction
Pulmonary arterial hypertension (PAH) is a disease characterized by increased pulmonary vascular resistance (PVR) leading to right heart failure and death1-7. All patients with PAH are recommended to undergo right heart catheterization (RHC) for definitive diagnosis and determination of appropriate therapy through acute vasodilator testing1,8-10. “Classic” criteria for an acute vasodilator response (VR) are defined as a drop in mean pulmonary arterial pressure (mPAP) by >10mmHg to an mPAP <40mmHg with preserved cardiac output. Patients meeting these criteria often respond well to long-term CCB monotherapy and have a markedly improved prognosis1,8,9,11-13.
Acute VR is thought to be rare among non-idiopathic PAH subtypes and long term CCB response has not been well described in these patients8,14,15. Therefore, the clinical utility of acute VR testing is less well-established in non-idiopathic PAH, and in fact is not recommended in the most recent PH guidelines1,8,11,15-17. The degree of decrease in mPAP and PVR during acute VR testing, however, may have prognostic value5,18-22.
The purpose of this study was to describe long-term outcomes in acute VR responders with non-idiopathic PAH or chronic thromboembolic pulmonary hypertension (CTEPH) and to determine the prognostic value of an acute VR that does not meet classic criteria. We hypothesized that a significant but ‘non-classic’ response to acute vasodilator testing, defined a priori as a decrease in mPAP > 10mmHg to a mean mPAP > 40mmHg with preserved cardiac output, confers a better prognosis than no response to vasodilators.
Methods
This study was approved by the Vanderbilt University Medical Center Institutional Review Board, IRB#130268. A consent waiver was obtained for this retrospective analysis. Consent was waived for all patients.
Study population
Data for this study was obtained from a database of patients within Vanderbilt University Medical Center's institutional PH registry who underwent RHC for evaluation for PH between June 2004 and December 201123. RHC was performed either as an initial diagnostic study or repeat evaluation, including both treatment naïve patients and patients already on single or combination therapy with PDE5 inhibitors, CCBs, endothelin receptor antagonists (ERAs) and prostanoids. The results of fluid challenge in this cohort have previously been published, but this analysis has not previously been reported23. For the purposes of this analysis, only patients meeting criteria for WHO group I and IV PH by the Dana Point criteria were included24. CTEPH patients were included in the analysis due to similar histopathology with Group I PAH and proven response to PAH-approved therapies25-27. The diagnosis and etiology of PAH was made by experienced physicians according to consensus guidelines28 including mPAP >25mmHg, PVR >3 wood units, and pulmonary wedge pressure (PWP) ≤ 15mmHg in the absence of significant parenchymal lung disease. CTEPH was defined by the same hemodynamics but also with confirmatory imaging for chronic thromboembolic disease. RHC was performed by one of three cardiologists with experience with this procedure in patients with PH. Hemodynamic tracings were personally reviewed by physicians with extensive experience in the evaluation and management of PH (I.M.R. and A.R.H.). Heart rate (HR), right atrial pressure (RAP), pulmonary artery pressure (PAP) (mean, systolic, and diastolic), and PWP were recorded and cardiac index (CI), PVR, and stroke volume (SV) were calculated according to standard formulae. Cardiac output was measured using either thermodilution or Fick calculation, the latter using estimated oxygen uptake based on age and heart rate29. VR testing was performed using the Ikaria INOmax DSIR® system to deliver inhaled nitric oxide at 40ppm along with 10L continuous flow oxygen for 10 minutes followed by repeat hemodynamic measurements. Demographic data and clinical outcomes were extracted from the medical record.
Vasodilator-responder categories
Classic vasodilator responsiveness was defined as a decrease in mPAP of ≥ 10mmHg to an absolute mPAP of < 40mmHg, associated with no change or an increase in cardiac output1,13. A ‘non-classic’ VR was defined as a decrease in mPAP ≥10mmHg with preserved CO, but mPAP remaining >40mmHg (Table 1). These cutoffs were used to define a response pattern that, like the classic response definition, is easily applied to patients at the time of RHC. We also assessed the response of mPAP and PVR to acute vasodilator challenge as continuous variables to better understand the potential relationship between more subtle hemodynamic changes and clinical outcomes.
Table 1. Definitions of Response Types.
| Response Type | ΔmPAP during VR | mPAP after VR testing |
|---|---|---|
| Classic | >10mmHg | <40mmHg |
| Non-Classic | >10mmgHg | ≥40mmHg |
| No Response | <10mmHg | >40mmHg |
Sustained Vasodilator-response
Patients were considered to have long-term CCB response if they met criteria for acute VR at initial RHC, demonstrated improvement in hemodynamic parameters on repeat evaluation via echocardiogram or RHC after initiation of CCB, and remained New York Heart Association (NHYA) functional class I-II after at least 1 year of CCB monotherapy.14
Patient Outcomes
Demographic and outcome data including six minute walk distance (6MWD) and NYHA functional class were gathered from the outpatient clinic visit preceding RHC. Hospitalizations at Vanderbilt University Hospital or outside institutions were reviewed and determined to be either PAH related or not. For purposes of this study, admission for catheter malfunction or infection were excluded, as well as elective admissions for research purposes. Hospitalizations due to one or more of the following were designated as PAH-related: heart failure (requiring adjustment of diuretic), syncope, or chest pain. In addition, because it is a marker of hemodynamic decompensation, initiation of epoprostenol was included as a PAH-related hospitalization. If throughout the course of admission and discharge no changes were made to PAH-directed therapy, the admission was deemed not PAH-related. Length of follow-up was determined by the date of last clinic visit or clinical communication, including patients still under care and those lost to follow-up. Date of death was determined from the medical record and query of the Social Security Death Index through May 16, 2013, at which time the databased was closed. The relationship between VR and death and a composite of death and PAH-related hospitalization was assessed.
Statistical analysis
Results are reported as mean ± standard deviation (SD) unless otherwise noted. The Mann Whitney U test was used to measure differences in continuous variables between groups. Categorical variables were compared between groups by means of the Chi-square test. Survival curves were constructed with the Kaplan-Meier method and survival differences compared with the log-rank test. Estimates of the variance in mean survival for each group were obtained using the Greenwood method30. Cox regression was used to determine the odds ratios (OR) and 95% confidence intervals for the change in hemodynamic variables before and after nitric oxide. Statistical analyses were performed with SPSS 20 software (SPSS, Chicago, IL) and Prism 5.0 (Graph Pad Software, La Jolla, CA).
Results
During the study period, 155 patients met diagnostic criteria for WHO group I or IV PH and comprised the study group. Among these, 20 patients (13%) displayed classic acute VR and 13 (8%) met our pre-specified criteria for non-classic VR response. Demographics, WHO functional class, baseline clinical data, and hemodynamics are presented in Table 2.
Table 2. Demographics and Hemodynamic Comparison of Classic, Non-classic, and Non-responders to Acute Vasodilator Challenge.
| Variable | Classic Response | Non-classic response | No Response (n = 122) |
|---|---|---|---|
| Age at RHC (years) | 49 (17) | 49 (12) | 52 (14) |
| Gender – female (%) | 16 (80) | 12 (86) | 99 (81) |
| Diagnosis (%) | |||
| IPAH | 12 (60) | 5 (38) | 38 (31) |
| FPAH | 0 (0) | 1 (8) | 9 (7) |
| CTD-PAH¶ | 8 (40) | 4 (31) | 39 (32) |
| CTEPH | 0 | 0 | 11 (9) |
| POPH | 0 | 1(8) | 17 (14) |
| CHD | 0 | 2 (15) | 8 (7) |
| NYHA Functional | |||
| Class (%) | |||
| I-II | 6 (30) | 5 (38) | 41 (34) |
| III-IV | 14 (70) | 8 (62) | 81 (66) |
| Six-minute walk distance (m) | 316 (132) | 324 (147) | 318 (132) |
| BNP (pg/mL) | 344 (315) | 526 (263) | 670 (729) |
| n = 10 | n = 8 | n = 71 | |
| Heart rate (beats/minute) | 74 (15) | 84 (15) | 78 (14) |
| RA pressure (mmHg) | 5 (3)* | 9 (5) | 8 (5) |
| Mean PAP (mmHg) | 46 (9)* | 64 (7)** | 49 (11) |
| Systolic PAP (mmHg) | 74 (15)* | 104 (12)** | 79 (17) |
| Diastolic PAP (mmHg) | 30 (8)* | 44 (8)** | 33 (9) |
| Cardiac index (L/min/m2) | 2.7 (0.7)* | 2.1 (0.5) | 2.4 (0.8) |
| PWP (mmHg) | 8 (3) | 10 (3)** | 9 (4) |
| PVR (WU) | 8 (4)* | 15 (5)** | 10 (5) |
| Delta mPAP (mmHg) | 17 (8) | 16 (6) | 2 (4)*** |
p<0.05 for Classic response vs. non-classic response,
p<0.05 for non-classic response vs. no response,
p < 0.001 for no response vs. either non-classic response or classic response.
BNP = brain natriuretic peptide; CHD= Congenital heart disease; CTD= Connective tissue disease related PAH; CTEPH= Chronic thromboembolic PAH; FPAH= Familial PAH; IPAH= Idiopathic PAH; NYHA = New York Heart Association; POPH= Portopulmonary PAH; PVR = pulmonary vascular resistance; PWP = pulmonary wedge pressure; RA = right atrial; RHC = right heart catheterization;
CTD-PAH (n=51) included patients with scleroderma (n=30), SLE (n=9), rheumatoid arthritis (n=6), mixed connective tissue disease (n=4) and Sjögren's syndrome (n=2)
The mean age of the patients in the cohort was 51±14 years and 81% were female. IPAH was the most common diagnosis (35%) followed by connective tissue disease related PAH (33%). The distribution of ΔmPAP among the different subtypes of PH and the entire cohort is shown in Figure 1. None of the patients with chronic thromboembolic PH (Group IV) met criteria for a positive response to vasodilator challenge. Among the 11 CTEPH patients, 7 ultimately went on to have pulmonary thromboendarterectomy. The majority of patients were NYHA functional class III or IV at the time of initial evaluation and 83 patients (54%) were on one or more PAH-specific medications at the time of RHC. There were no significant differences in age, gender, or distribution of PAH sub-type among classic responders, non-classic responders, and non-responders (Table 2). Similarly, there was no significant difference in NYHA class or six-minute walk distance among classic, non-classic and non-responders.
Figure 1. Distribution of Delta Mean Pulmonary Artery Pressure Among PAH Subtypes.
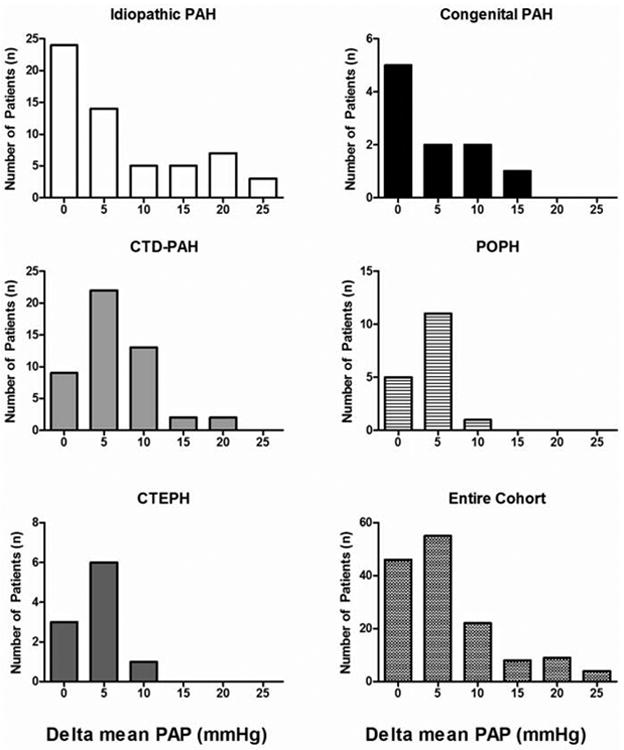
A decrease in mean pulmonary artery pressure > 10mmHg was rare in the entire cohort and was only observed in the idiopathic, connective tissue disease, and congenital subtypes. CTD-PAH = connective tissue disease associated pulmonary arterial hypertension; PAH = pulmonary arterial hypertension; PAP = pulmonary artery pressure; POPH = portopulmonary hypertension
Classic responders displayed several favorable hemodynamic parameters compared to non-classic responders prior to NO administration including lower mean RA pressure (5±3mmHg vs. 9±5mmHg, p<0.05), mPAP (46±9mmHg vs. 64±7mmHg, p<0.05), and PVR (8±4 WU vs. 15±5WU, p<0.05) and higher cardiac index (2.7±0.7L/min/m2 vs. 2.1±0.5L/min/m2, p<0.05). Non-responders also had lower mean PA pressure (49±11mmHg vs. 64±7mmHg p<0.05) and PVR (10±5 WU vs. 15±5 WU p<0.05) compared with non-classic responders. The drop in mPAP was similar between classic and non-classic responders during NO administration.
Treatment and Clinical Response
Eight out of 20 classic responders demonstrated sustained response to CCB monotherapy, representing 40% of the acute responders and 5.2% of the entire cohort. All of the long-term responders had IPAH except one who had systemic lupus erythematosus (SLE). Among the 12 classic responders without sustained clinical CCB response, 5 had IPAH and 7 had CTD-PAH. Seven of these 12 patients (3 IPAH and 4 CTD-PAH) were on CCBs as initial monotherapy but went on to require additional therapy. In the remaining 5 patients without sustained response, 2 were on other PAH therapy prior to initial RHC that was continued, 1 was started immediately on epoprostenol due to severe right heart failure, and 2 were lost to follow-up (1 each in the IPAH and CTD-PAH groups). Only one of the 8 CTD-PAH patients with classic acute VR displayed long-term response to CCB monotherapy; notably, this patient had SLE. Non-classic responders were treated with CCBs (5/13), ERAs (2/13), PDE5 inhibitors (6/13) and prostanoids (4/13). The proportion of patients meeting the pre-defined criteria for non-classic, classic, and no response was similar when comparing patients on no medications at the time of RHC versus those on therapy (6%, 15%, and 79% and 11%, 11%, and 78%, respectively, p = 0.39).
Demographics, WHO classification, baseline clinical data, and hemodynamics of the classic responders are displayed in Table 3. The long-term CCB responders were younger than those who displayed only acute VR without clinical response to CCBs (p <0.01). NYHA class, 6MWD, and all hemodynamic parameters were not significantly different between the two groups at initial evaluation.
Table 3. Comparison of Long-term Responders and Acute Only Responders Among Patients Meeting Classic Criteria for Acute Vasodilator Response.
| Variable | Classic Response (n = 20) | Long-term Responder (n = 8) | Acute Only Responder (n = 12) | P value (Long-term vs. Acute Only) |
|---|---|---|---|---|
| Age at RHC (years) | 49 (17) | 37 (9) | 56 (16) | 0.01 |
| Gender – female (%) | 16 (80) | 7 (100) | 9 (69) | 0.25 |
| Diagnosis (%) | ||||
| IPAH | 12 (60) | 7 (87) | 5 (42) | 0.02 |
| FPAH | 0 (0) | |||
| CTD-PAH | 1 (40) | 1 (13) | 7 (58) | |
| CTEPH | 0 | |||
| POPH | 0 | |||
| CHD | 0 | |||
| NYHA Functional | ||||
| Class (%) | 6 (30) | 3 (37) | 3 (25) | 0.39 |
| I-II | 14 (70) | 5 (63) | 9 (75) | |
| III-IV | ||||
| Six-minute walk distance (meters) | 316 (132) | 354 (113) | 289 (142) | 0.32 |
| Hemodynamics | ||||
| Heart rate (beats/minute) | 74 (15) | 78 (15) | 72 (16) | 0.30 |
| RA pressure (mmHg) | 5 (3) | 6 (3) | 4 (2) | 0.35 |
| Mean PAP (mmHg) | 46 (9) | 49 (12) | 43 (7) | 0.25 |
| Systolic PAP (mmHg) | 74 (15) | 77 (15) | 73 (15) | 0.64 |
| Diastolic PAP (mmHg) | 30 (8) | 34 (11) | 28 (5) | 0.32 |
| Cardiac index (L/min/m2) | 2.7 (0.7) | 2.6 (0.7) | 2.8 (0.7) | 0.59 |
| PWP (mmHg) PVR (WU) | 8 (3) 8 (4) | 8 (3) 9.8 (4.6) | 7 (2) 7.5 (2.5) | 0.85 0.40 |
6MWD = six minute walk distance; CHD= Congenital heart disease; CTD= Connective tissue disease related PAH, CTEPH= Chronic thromboembolic PAH, FPAH= Familial PAH; IPAH= Idiopathic PAH; PAP = pulmonary artery pressure; POPH= Portopulmonary PAH; PVR = pulmonary vascular resistance; PWP = pulmonary wedge pressure; RA = right atrial; RHC = right heart catheterization;
Outcomes
A non-classic response to acute VR testing was not associated with improved survival when compared to non-responders during median follow-up of 55 months (p=0.99) (Figure 2A). When all patients with classic acute VR, including 12 IPAH and 8 CTD-PAH patients, were compared to non-classic responders there was no improvement in survival (Figure 2B; p=0.33). However, after the exclusion of CTD-PAH patients from the classic responder group, the remaining classic responders demonstrated improved survival compared to non-classic responders (Figure 2C; p=0.014). The estimated mean survival of the non-responder group is 92 months (95% CI 79-105 months) versus 99 months (95% CI 59-138 months) among non-classic responders and 139 months (95% CI 99-179 months) among classic responders. Using Cox regression to take into account time-to-event, ΔmPAP (HR 0.98, 95% CI 0.96-1.01, p = 0.25) and ΔPVR (HR 0.99, 95% CI 0.92-1.07, p = 0.88) did not have an association with survival.
Figure 2. Survival Curves Comparing Vasodilator Response Groups.
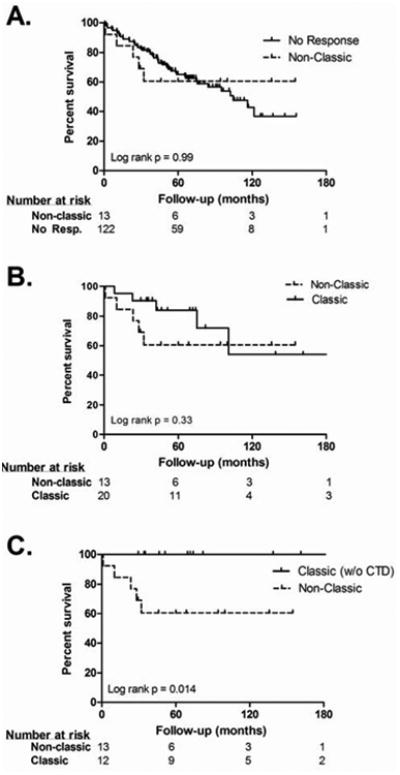
A. Analyzed from the time of first clinic visit, a non-classic response to VR testing was not associated with improved survival compared to non-responders (p= 0.99).B. ‘Classic’ responders did not have improved survival when all PAH subtypes were included (p= 0.33). C. After exclusion of CTD-PAH patients from the ‘Classic’ responder group, a ‘classic’ response was associated with improved survival (p=0.014) compared with non-classic responders.
Discussion
In a large cohort of well-phenotyped PAH and CTEPH patients, we demonstrate that classically defined acute vasoreactivity can be identified in both IPAH patients and CTD-PAH, but is uncommon in CTEPH and other types of group 1 PH. However, the identification of an acute response in non-IPAH is not associated with improved outcomes. Moreover, a non-classic response to acute vasodilators, defined by a decline in mPAP > 10mmHg to an absolute value above 40mmHg in response to inhaled NO is not associated with improved survival in a mixed cohort of PAH and CTEPH. The patients who displayed this ‘non-classic’ response had more severe disease at baseline hemodynamic assessment than both ‘classic’ responders and patients who had no significant VR (ΔmPAP <10mmHg).
Classically-defined vasoreactivity was observed in CTD-PAH, but did not predict sustained response to CCB monotherapy or survival in this sub-type of PAH in our cohort. In contrast, as demonstrated in figure 2C, classically-defined vasoreactivity in IPAH predicts excellent long-term outcomes in our cohort, consistent with prior reports13. Indeed, acute VR was relatively common in CTD-PAH (15.7%); however, only one, a patient with SLE, was a long-term CCB responder. These results are similar to a study performed by Montani et. al. who found that 10.1% of CTD-PAH patients displayed vasoreactivity, but only one patient (0.6%) had long-term CCB response14. They used different criteria for acute VR, defining it as decrease in mPAP and total pulmonary resistance of ≥20%, but reported similar findings when using the current acute VR criteria. These findings are clinically important because a classically-defined vasodilator responder with CTD-PAH would be unlikely to benefit from CCB therapy, confirming prior observations14. We found that there was no significant response (ΔmPAP ≥10mmHg) to nitric oxide administration in any patient with chronic thromboembolic pulmonary hypertension, a result consistent with prior evaluation of VR in CTEPH patients31, and only one patient in the subgroup with portopulmonary hypertension. A study of 103 CTEPH patients by Skoro-Sajer et. al. has previously shown that ΔmPAP during acute VR testing is predictive of outcomes and long-term survival after thromboendartectomy32. The mean ΔmPAP among patients with improved prognosis was 8mmHg (17%). Our study was underpowered with respect to CTEPH (n = 11) to detect a similar effect.
A previous study by Malhotra et. al. has shown that in a treatment naïve population of 80 patients with PAH percent decrease in mPAP and PVR predicted long-term survival in univariate and age-adjusted multivariate analysis, including subgroup analysis of CTD-PAH over a mean follow up period of 2.4 years18. The criteria for vasoreactivity that predicted improved survival in this analysis was defined by 30% decrease in PVR or 12% decrease in mPAP, values which were determined by the mean VR in their study population. Our study did not replicate these findings in a larger cohort of 155 patients with mean follow up of 4.6 years either by univariate analysis or by dichotomizing the population by vasoreactivity defined as ΔmPAP of greater than or less than 10mmHg. Methodological differences are present between the two studies, for example the MGH cohort was entirely treatment naïve and VR testing was performed with 80ppm NO and 90% O2, however previous studies have suggested that VR to NO is not dose dependent33.
Our findings were consistent with prior studies showing that patients with ‘classic’ acute VR have less severe disease at baseline13. There are different hypotheses for why this is the case; one being that classic responders represent an earlier, more compensated form of PAH with some remaining vasoreactivity, prior to the loss of VR and right heart failure. This notion is supported by our findings that younger age and IPAH subtype were the only predictors of long-term CCB responsiveness. However, this does not explain our findings of a similar decrease in mPAP during VR testing among the ‘non-classic’ responders, who have worse hemodynamics at baseline and no survival advantage compared to patients with no VR. This leads to an alternative hypothesis that true vasodilator responsive PAH is a molecularly distinct disease that is physiologically identified through VR testing. The present study cannot differentiate between these two hypotheses.
Limitations
The retrospective nature and relatively small numbers of both classic and non-classic responders limit this study. This is evidenced by the wide and overlapping confidence intervals for mean survival in all groups. However, this is a comparatively large cohort of a very rare subset of a rare disease. Additionally, patient follow up was excellent and the longer follow up compared to prior studies strengthen our findings.
Conclusions
Classically defined acute VR occurs in both CTD-PAH and in IPAH, however only IPAH patients with classic VR have long-term response to CCB monotherapy and improved survival compared with non-responders. Our data suggest that in CTD-PAH, if the diagnosis is known prior to RHC, vasodilator testing might be deferred, thus saving time, cost and potentially risk. Additionally, identification of a significant but non-classic response does not confer improved prognosis or clinical response to CCB therapy. These patients are unlikely to improve on CCB monotherapy and should be instituted on other PAH directed therapy after diagnosis.
Acknowledgments
Grant support: American Heart Association Fellow-to-Faculty Award #13FTF16070002 (Brittain), P01 HL108800 (Robbins, Hemnes, Pugh).
Footnotes
Disclosures: Hemnes: Consulting for Pfizer, Bayer
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc.and the Pulmonary Hypertension Association. Circulation. 2009;119:2250–94. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 2.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–65. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 3.Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 4.Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Respir J. 2007;30:1103–10. doi: 10.1183/09031936.00042107. [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin VV, Presberg KW, Doyle RL, et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:78s–92s. doi: 10.1378/chest.126.1_suppl.78S. [DOI] [PubMed] [Google Scholar]
- 6.D'Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension.Results from a national prospective registry. Annals of internal medicine. 1991;115:343–9. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 7.Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 8.Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131:1917–28. doi: 10.1378/chest.06-2674. [DOI] [PubMed] [Google Scholar]
- 9.McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:14s–34s. doi: 10.1378/chest.126.1_suppl.14S. [DOI] [PubMed] [Google Scholar]
- 10.Galie N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D60–72. doi: 10.1016/j.jacc.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 11.Tonelli AR, Alnuaimat H, Mubarak K. Pulmonary vasodilator testing and use of calcium channel blockers in pulmonary arterial hypertension. Respir Med. 2010;104:481–96. doi: 10.1016/j.rmed.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 12.Sitbon O, Humbert M, Jagot JL, et al. Inhaled nitric oxide as a screening agent for safely identifying responders to oral calcium-channel blockers in primary pulmonary hypertension. Eur Respir J. 1998;12:265–70. doi: 10.1183/09031936.98.12020265. [DOI] [PubMed] [Google Scholar]
- 13.Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105–11. doi: 10.1161/CIRCULATIONAHA.104.488486. [DOI] [PubMed] [Google Scholar]
- 14.Montani D, Savale L, Natali D, et al. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. European heart journal. 2010;31:1898–907. doi: 10.1093/eurheartj/ehq170. [DOI] [PubMed] [Google Scholar]
- 15.Elliott CG, Glissmeyer EW, Havlena GT, et al. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation. 2006;113:2509–15. doi: 10.1161/CIRCULATIONAHA.105.601930. [DOI] [PubMed] [Google Scholar]
- 16.Simonneau G, Fartoukh M, Sitbon O, Humbert M, Jagot JL, Herve P. Primary pulmonary hypertension associated with the use of fenfluramine derivatives. Chest. 1998;114:195s–9s. doi: 10.1378/chest.114.3_supplement.195s. [DOI] [PubMed] [Google Scholar]
- 17.Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D42–50. doi: 10.1016/j.jacc.2013.10.032. [DOI] [PubMed] [Google Scholar]
- 18.Malhotra R, Hess D, Lewis GD, Bloch KD, Waxman AB, Semigran MJ. Vasoreactivity to inhaled nitric oxide with oxygen predicts long-term survival in pulmonary arterial hypertension. Pulm Circ. 2011;1:250–8. doi: 10.4103/2045-8932.83449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klings ES, Hill NS, Ieong MH, Simms RW, Korn JH, Farber HW. Systemic sclerosis-associated pulmonary hypertension: short- and long-term effects of epoprostenol (prostacyclin) Arthritis Rheum. 1999;42:2638–45. doi: 10.1002/1529-0131(199912)42:12<2638::AID-ANR20>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 20.Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 21.Raffy O, Azarian R, Brenot F, et al. Clinical significance of the pulmonary vasodilator response during short-term infusion of prostacyclin in primary pulmonary hypertension. Circulation. 1996;93:484–8. doi: 10.1161/01.cir.93.3.484. [DOI] [PubMed] [Google Scholar]
- 22.McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106:1477–82. doi: 10.1161/01.cir.0000029100.82385.58. [DOI] [PubMed] [Google Scholar]
- 23.Robbins IM, Hemnes AR, Pugh ME, et al. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circulation Heart failure. 2014;7:116–22. doi: 10.1161/CIRCHEARTFAILURE.113.000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. Journal of the American College of Cardiology. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 25.Moser KM, Bloor CM. Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Chest. 1993;103:685–92. doi: 10.1378/chest.103.3.685. [DOI] [PubMed] [Google Scholar]
- 26.Jais X, D'Armini AM, Jansa P, et al. Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol. 2008;52:2127–34. doi: 10.1016/j.jacc.2008.08.059. [DOI] [PubMed] [Google Scholar]
- 27.Ghofrani HA, D'Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369:319–29. doi: 10.1056/NEJMoa1209657. [DOI] [PubMed] [Google Scholar]
- 28.Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 29.LaFarge CG, Miettinen OS. The estimation of oxygen consumption. Cardiovasc Res. 1970;4:23–30. doi: 10.1093/cvr/4.1.23. [DOI] [PubMed] [Google Scholar]
- 30.Greenwood MA. Report on the Natural Duration of Cancer. London: Her Majesty 's Stationery Office; 1926. [Google Scholar]
- 31.Suntharalingam J, Hughes RJ, Goldsmith K, et al. Acute haemodynamic responses to inhaled nitric oxide and intravenous sildenafil in distal chronic thromboembolic pulmonary hypertension (CTEPH) Vascular pharmacology. 2007;46:449–55. doi: 10.1016/j.vph.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 32.Skoro-Sajer N, Hack N, Sadushi-Kolici R, et al. Pulmonary vascular reactivity and prognosis in patients with chronic thromboembolic pulmonary hypertension: a pilot study. Circulation. 2009;119:298–305. doi: 10.1161/CIRCULATIONAHA.108.794610. [DOI] [PubMed] [Google Scholar]
- 33.Sitbon O, Brenot F, Denjean A, et al. Inhaled nitric oxide as a screening vasodilator agent in primary pulmonary hypertension.A dose-response study and comparison with prostacyclin. Am J Respir Crit Care Med. 1995;151:384–9. doi: 10.1164/ajrccm.151.2.7842196. [DOI] [PubMed] [Google Scholar]