

Abstract
The type II interleukin-4 receptor (IL4R) is expressed in human breast cancer, and in murine models thereof. It is activated by interleukin-4 (IL-4), a cytokine produced predominantly by immune cells. Previously, we showed that expression of IL4Rα, a signaling component of IL4R, mediates enhanced metastatic growth through promotion of tumor cell survival and proliferation. In lymphocytes, these processes are supported by increased glucose and glutamine metabolism, and B lymphocyte survival is dependent upon IL4/IL4R-induced glucose metabolism. However, it is unknown whether IL4R-mediated metabolic reprogramming could support tumor growth. Here, we show that IL4Rα expression increases proliferation thus enhancing primary mammary tumor growth. In vitro, IL4-enhanced glucose consumption and lactate production in 4T1 cells was mediated by IL4Rα. Expression of the glucose transporter GLUT1 increased in response to IL4 in vitro, and enhanced GLUT1 expression was associated with presence of IL4Rα in 4T1 mammary tumors in vivo. Although IL4 treatment did not induce changes in glucose metabolism in MDA-MB-231 human breast cancer cells, it increased expression of the main glutamine transporter, ASCT2, and enhanced glutamine consumption in both MDA-MB-231 and 4T1 cells. Pharmacologic inhibition of glutamine metabolism with compound 968 blocked IL4/IL4Rα-increased cell number in both cell lines. Our results demonstrate that IL4R mediates enhanced glucose and glutamine metabolism in 4T1 cancer cells, and that IL4-induced growth is supported by IL4/IL4R-enhanced glutamine metabolism in both human and murine mammary cancer cells. This highlights IL4Rα as a possible target for effective breast cancer therapy.
Keywords: cytokine, proliferation, survival, glucose, metabolism
1. Introduction
Second only to skin cancer, breast cancer remains the most commonly diagnosed cancer in women in the United States [1]. Cytokines and chemokines in the tumor microenvironment promote breast cancer progression and metastasis [2]. Interleukin-4 (IL4) is a Th2 immune cytokine that binds and activates the type 1 IL4R on lymphoid cells (composed of the IL4Rα and common gamma C chains) to promote proliferation and survival [3]. Normal epithelial tissues typically do not express IL4R, yet many epithelial cancers including breast cancer, upregulate a second type of IL4R, called the type II IL4R, which consists of the IL4Rα and IL13Ra1 chains [4]. Notably, interleukin-13 (IL13) can also activate the Type II IL4R. However, IL4 is the prototypical IL4R ligand, it binds with higher affinity [5], and is upregulated in the breast tumor microenvironment in patients [6].
Using two immune competent murine tumor models, we have defined IL4Rα expression in mammary cancer cells as a strong promoter of metastatic tumor growth mediating enhanced proliferation and survival [7]. Increased glucose consumption and utilization in activated lymphocytes supports these same pro-growth phenotypes [8,9]. Specifically, IL4 induces T cell proliferation [10], and IL4/IL4Rα-induced glucose metabolism is necessary to support the enhanced survival of B cells [8,11]. However, there is no data regarding whether IL4/IL4Rα-induced glucose metabolism serves as a novel mechanism to support tumor growth.
Normally, cells use glycolysis to metabolize glucose to pyruvate, which is fed into the tricarboxylic acid (TCA) cycle and used to generate ATP through oxidative phosphorylation. Highly proliferative cells, including activated lymphocytes and cancer cells, induce a comparatively high rate of aerobic glycolysis and metabolize the majority of glucose to lactate even when oxygen is present [12]. This phenomenon, termed the “Warburg effect” in cancer, is often accompanied by elevated glucose transporter (GLUT) expression to facilitate increased glucose uptake, as generation of ATP per glucose molecule from aerobic glycolysis is relatively inefficient. Enhanced aerobic glycolysis in tumors is often indicated by increased extracellular lactate production as it correlates proportionally with intracellular glycolytic activity [13]. The reprogramming of metabolism is now considered an emerging hallmark of cancer because of its critical role in supporting rapid biosynthesis during periods of stress and proliferation [14]. How cancer cells achieve such metabolic reprogramming is now an area of intense investigation.
There are 14 GLUT family members expressed in humans, of which GLUT1 is the most extensively studied in cancer for mediating upregulated glucose uptake and metabolism. Elevated expression of GLUT1 has been shown in many epithelial cancer types including breast cancer [15–18]. Importantly, IL4 signaling through the type I IL4R in B lymphocytes leads to increased expression of GLUT1 and other genes encoding glycolytic enzymes [19], and GLUT1 expression has been associated with IL4-increased glucose uptake [8].
In the setting of aerobic glycolysis, both activated lymphocytes and cancer cells often also upregulate glutamine uptake and metabolism to maintain the TCA cycle [9,20], and to provide purines and pyrimidines for DNA and RNA synthesis [21]. While colon and lung cancers depend on enhanced glutamine metabolism for survival and proliferation, little is known about glutamine metabolism in breast cancer cells [22,23]. Enhanced glutamine uptake may occur through increased ASC amino-acid transporter 2 (ASCT2) expression, the major cancer-related glutamine transporter [24,25]. Indeed, ASCT2 is expressed by a variety of breast cancer subtypes including human luminal A, luminal B, HER2 positive, and triple negative [26].
We have previously demonstrated that a traditionally immune signaling axis, IL4/IL4Rα, is a direct promoter of two cancer-acquired phenotypes in mammary cancer cells, survival and proliferation. Here, we investigate whether IL4 can also enhance glucose and/or glutamine metabolism in breast cancer cells expressing IL4Ra to support enhanced tumor growth.
2. Materials and Methods
2.1 Cell Lines and Culture
The MDA-MB-231 human breast cancer cell line, acquired from the American Type Culture Collection (ATCC), was transfected with SureSilencing™ small hairpin RNA (shRNA) plasmids directed toward three different regions of human IL4Rα (NM_000418) or with control non-target plasmids (SABiosciences) using Lipofectamine™ (Invitrogen). Individual sh-control and IL4Rα knockdown clones were selected by limiting dilution in 400 μg/mL hygromycin (Invitrogen). Immunoblotting followed by densitometry analysis in Adobe Photoshop® was used to calculate the percent of IL4Rα protein knockdown. The sources for PyVT-R221a (R221a) and 4T1 cells, and the validation of shRNA-mediated knockdown of IL4Rα in these murine mammary cancer cell lines was previously described [7]. Cells were maintained at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Corning) containing 10% fetal bovine serum (FBS, Atlanta Biologicals), and the appropriate selection agent. DMEM containing 2% FBS was used in all experimental assays shown.
2.2 Western Blot Analysis
Whole cell lysates collected with radioimmunoprecipitation assay (RIPA) lysis buffer containing protease inhibitors (Roche Applied Science), and sonicated as necessary, were used for western blots. Lysates used for GLUT1 blots were also deglycosylated by treatment with PNGase F (New England Biolabs). Primary antibodies were obtained from Santa Cruz Biotechnologies (human and murine IL4Rα, and murine ASCT2), Millipore (Human ASCT2), Abcam (GLUT1), or Cell signaling (β-actin) and immunoblotting performed as previously described [7].
2.3 Murine Tumor Models
All animal procedures were conducted in accordance with Guidelines for the Care and Use of Laboratory Animals following approval by the Institutional Animal Care and Use Committee. Age-matched female BALB/c and FVB/N mice (Jackson Laboratories) were used for orthotopic injections into the 4th mammary gland (2.5×105 4T1 cells per BALB/c mouse or 2.5×105 R221a cells per FVB mouse). Prior to injection, individual clones for each cell line (R221a or 4T1) were harvested and combined to generate combined sh-control or IL4Rα KD samples. The time to initial mammary tumor palpation was documented as tumor latency. Weekly caliper measurements were used to calculate tumor volume for growth rates. Mice were sacrificed when tumors reached 1 cm in any dimension. 4T1 injections were repeated with 2.5×105 cells for 6 days. At necropsy, these mammary tumors were formalin-fixed, paraffin-embedded, and sectioned for immunohistochemical analysis. In a separate study, R221a and 4T1 sh-control and IL4Rα knockdown cells were labeled with cell tracker red (Life Technologies) for one hour prior to injection. These mice were sacrificed 48 hours post-injection and mammary glands were homogenized and analyzed by flow cytometry to assess the initial seeding ability of tumor cells.
2.4 Immunohistochemical Analysis of Tissue Sections
Tissue sections of 4T1 orthotopic tumors or archival 4T1 lung metastases were stained by immunohistochemistry using citrate antigen retrieval, and primary antibodies, GLUT1 (Abcam) against Ki67 (Abcam), or cleaved Caspase-3 (Cell Signaling Technology) as previously described [7]. At least three separate orthotopic tumor areas and four separate metastatic lung tumors per mouse were imaged at 20X objective magnification where 372 pixels is equivalent to 100 microns. The positively stained area per tumor pixel area was measured by thresholding using Metamorph® software.
2.5 Analysis of Glucose and Glutamine Metabolism
Direct glucose uptake was measured using a glucose uptake cell-based kit (Cayman Chemical). 4T1 parental cells were seeded in 2% FBS DMEM (250,000 cells/well in 12-well plates). Post-attachment the media was replaced with 2% FBS DMEM with recombinant murine IL4 (BD Biosciences) or 0.1% BSA in PBS (BSA-PBS) as a vehicle control, and cells were allowed to grow for 22 hours. Media was removed and replaced with glucose-free and serum-free DMEM +/− 100 μg/mL 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose (2-NBDG) and +/− 10 ng/mL IL4 or vehicle control for 2 hours. 2-NBDG positive cells were detected by flow cytometry. In long-term metabolism assays, cells (4T1 200,000: cells/plate or MDA-MB-231: 300,000 cells/plate for 4 days, or 500,000 cells/plate for 3 days) were seeded in 10 cm2 plates in DMEM containing 2% FBS. Post attachment, human or murine IL4 diluted in BSA-PBS or the BSA-PBS vehicle control was added to each plate. IL4 or BSA-PBS was added to existing media every other day for the duration of each study. At each timepoint, media was collected from the plates, spun at 1,000 rpm for 5 min, aliquoted, and frozen at −80°C before being thawed, diluted, and analyzed for metabolites. Glucose consumption, glutamine consumption, and L-lactate production were measured using commercial kits per the manufacturer’s colorimetric protocol (glucose and glutamine kits from BioAssay Systems and L-lactate kit from Cayman Chemical). The metabolite consumption and production values from the media were normalized to the corresponding total amount of protein in each plate as determined by bicinchoninic acid (BCA, Thermo Scientific) assay of cell lysates. Lysates collected were also analyzed by western blot for GLUT1 and ASCT2 expression. RNA was extracted from a separate set of plates using QuickRNA Mini-prep (Zymo Research), and real time RT-PCR was performed using custom primers (Invitrogen) against human and murine GLUT1 and ASCT2 (Primer sequences and sources are provided in Supplemental Table 1). The ΔΔCT method was used to quantify changes in gene expression normalized to the expression of the human or murine ribosomal 18S subunit.
2.6 Assays with Pharmacologic Inhibitors
The glycolysis inhibitor, 2 Deoxy-D-glucose (2DG, Acros Organics), was dissolved in sterile water, and the glutaminase inhibitor, 968 (Calbiochem), was dissolved in DMSO. The compounds were then diluted in DMEM with 2% FBS for each experiment. Cell number in response to IL4 in the presence or absence of 2DG or 968 was quantified using a CyQUANT® proliferation assay (Life Technologies), per the manufacturer’s protocol. In all CyQUANT® assays, cells were seeded (4T1: 10,000 cells/well, MDA-MB-231: 8,000 cells/well, in 48-well plates) in 2% FBS DMEM, and allowed to attach before the media was removed and replaced with 2% FBS DMEM containing IL4 (BD Biosciences), the appropriate drug treatment, or vehicle control. Cells were allowed to grow for 48 hours in the presence of treatment prior to cell number analysis.
2.7 EdU Proliferation Assays
4T1 cells or MDA-MB-231 cells were seeded in 8-well chamber slides (10,000 cells/well) in DMEM containing 2% FBS. After attachment, media was replaced with 2% FBS DMEM with IL4 or PBS vehicle control, and cells were allowed to proliferate for 46 hours. 10 μM EdU (Invitrogen) was added for the remaining two hours of the 48 hour studies. Cells were washed, fixed with 10% buffered formalin, and cells incorporating EdU (cells in S phase) were labeled by Alexa Fluor® azide dye (594 nm or 488 nm wavelength, Invitrogen) with 10 mM copper in a .5M ascorbic acid and 1.5M Tris-HCL (pH 8.5) aqueous solution. Hoechst 33258 was used to stain cell nuclei. Slides were imaged at 20X objective magnification, with three images taken per well, and four wells per condition. The number of azide dye and Hoescht positive cells were quantified after thresholding in ImageJ software, and used to calculate the percent of cells in S phase for each image.
2.8 Recombinant IL4
Each batch of recombinant human and murine IL4 obtained from BD Biosciences was tested for maximum effective dose using the CyQUANT® assay described above for each cell line prior to use in experiments since specific activity differed from lot to lot. In all experiments, the dose of IL4 ranged from 10–20 ng/mL depending on its measured mitogenic activity.
2.9 Statistical Analysis
GraphPad Prism® software was used to perform all statistical analyses. ANOVA analysis or Kruskal-Wallis with post-test was used when comparing more than two conditions. Otherwise, the parametric unpaired Student’s T test, or the nonparametric Mann-Whitney test were used. In graphs, error bars represent the standard deviation, and P values are represented by stars where: * ≤ .05, ** ≤ .01, and *** ≤ .001.
3. Results
3.1 IL4Rα promotes mammary tumor growth at the primary site
Having already established that IL4Rα is a strong promoter of metastatic mammary tumor growth in the lung and liver [7], we then asked whether IL4Rα expression can affect mammary tumor latency and growth at the primary tumor site. For these studies, we used orthotopic injection models with R221a and 4T1 sh-control or IL4Rα knockdown cells. In contrast to the large tumors formed by sh-control cells, FVB mice receiving R221a IL4Rα knockdown cells never formed palpable tumors (Figure 1A). Mice receiving sh-control 4T1 cells developed tumors on average 4 days earlier than mice receiving IL4Rα knockdown 4T1 cells, although this difference in latency did not reach significance (Figure 1A). For both the 4T1 and R221A cell lines, there was a significant reduction in IL4Rα knockdown tumor size at endpoint (Figure S1A). Analysis by flow cytometry showed no difference in the number of labeled sh-control or IL4Rα knockdown R221a or 4T1 cells in the mammary glands 48 hours post-injection (Figure S1B). This indicates that disparities in R221a tumor latency and R221a and 4T1 IL4Rα knockdown tumor size at endpoint were not due to fewer IL4Rα knockdown cells surviving the initial injection.
Figure 1. IL4Rα promotes the growth of mammary tumors at the primary site.

R221a or 4T1 sh-control (Ctl) or IL4Rα knockdown (KD) cells were orthotopically injected into the 4th mammary gland of mice. A) Representation of tumor latency for R221a (left, p < .0001) and 4T1 (right, p = NS) mammary tumors (R221a n = 16; 4T1 n = 14). B) Graphs of R221a (left) and 4T1 (right) mammary tumor volume over time calculated from tumor dimensions (R221a n = 16; 4T1 n = 14). C) Quantification of total Ki67 D) or cleaved caspase-3 positive area per 4T1 IL4Rα KD or sh-control mammary tumor area (n = 11). Murine tumor growth data represented in A&B was repeated with similar results obtained.
The growth rates of mammary tumors originating from IL4Rα knockdown clones were significantly reduced compared to sh-control tumors for both cell lines (Figure 1B). Immunohistochemistry for Ki67 and Caspase-3 was then performed on 4T1 sh-control and IL4Rα knockdown mammary tumors to assess whether IL4Rα-induced proliferation or survival could contribute to enhanced mammary tumor growth. There was a significant difference in Ki67 positivity between control and IL4Rα knockdown tumors (Figure 1C), but no difference in Caspase-3 positivity (Figure 1D). Collectively, these data demonstrate that IL4Rα expression promotes mammary tumor proliferation leading to enhanced growth at the primary tumor site.
3.2 IL4 induces glucose uptake in mammary cancer cells
IL4 stimulation results in the activation of Akt in both 4T1 and R221a cells [7], and PI3K/Akt signaling can elicit the translocation of glucose transporters to the cell surface, thus increasing glucose uptake and corresponding lactate production [27–29]. We therefore directly measured glucose uptake as 2-NBDG uptake by flow cytometry in 4T1 parental cells following 24 hours of IL4 exposure. Glucose uptake reported as mean 2-NBDG fluorescence was significantly increased in cells treated with IL4 (Figure 2A,B). Furthermore, using a sensitive multianalyte microphysiometer (MAMP) [30,31], we found that IL4 stimulated lactate production by ~8.5% from 4T1 cells within minutes of exposure (data not shown). These data confirmed that IL4 enhances short-term glucose uptake and possibly its metabolism in mammary cancer cells. However, tumor cells in the microenvironment would likely be exposed to stromally derived IL4 for longer periods of time. The question thus remained whether IL4 could induce long-term changes in glucose and glutamine metabolism to maintain enhanced tumor growth.
Figure 2. Glucose uptake in mammary cancer cells is increased following short-term IL4 exposure.
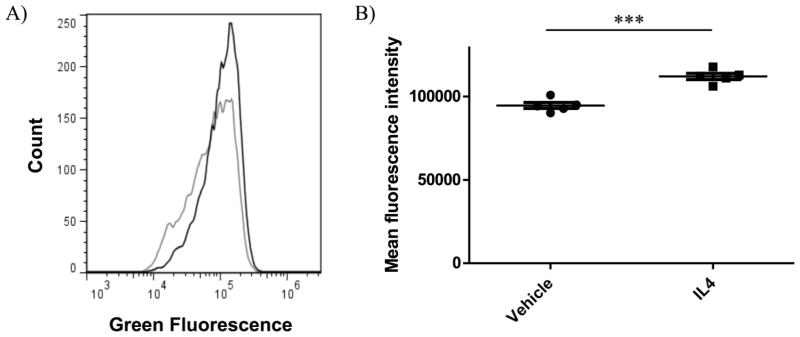
4T1 parental cells were treated with vehicle or 10 ng/mL murine IL4 for 22 hours prior to a 2 hour exposure to 2-NBDG +/− 10 ng/mL IL4, and immediate analysis by flow cytometry. A) Flow cytometry trace of 2-NBDG uptake in 4T1 cells treated with IL4 (black) or vehicle control (grey). B) Quantification of Mean fluorescence intensity by flow cytometry representative of 2-NBDG uptake in 4T1 cells with IL4 or vehicle control.
3.3 IL4Rα mediates enhanced GLUT1 expression in mammary cancer cells
IL4 treatment enhances the expression of GLUT1 and glucose uptake in B cells [8,19]. GLUT1 has also been identified as the most highly expressed GLUT isoform out of 12 GLUTs examined in several murine mammary cancer cell lines, including 4T1 cells [32]. As an initial screen for long-term changes in glucose metabolism in response to IL4, we examined changes in GLUT1 expression by western blot in 4T1 cells treated with IL4 every other day for a period of 8 days. Increased GLUT1 protein expression in response to IL4 was apparent by Western blot by day 8 (Figure 3A). We further confirmed IL4-induced expression of GLUT1 by RT-PCR in 4T1 cells compared to untreated controls (Figure 3B).
Figure 3. IL4Rα mediates enhanced expression of GLUT1 in mammary cancer cells.

4T1 sh-control clones were treated with vehicle or 20 ng/mL murine IL4 every other day for 8 days. A) Western blot analysis of deglycosylated glucose transporter 1 (GLUT1) upregulation in 4T1 sh-control clones over time. B) Quantification of GLUT1 mRNA expression normalized to murine ribosomal S18 in 4T1 sh-control clones 6 days after initial IL4 treatment. Quantification of IHC staining for GLUT1 in C) 4T1 sh-control (Ctl) and IL4Rα knockdown (KD) orthotopic mammary tumors (n = 8) and D) in metastatic lung tumors originating from sh-control (Ctl) or IL4Rα knockdown (IL4Ra KD) 4T1 cells in wild-type (WT) or IL4 knockout (IL4 KO) mice (n = 3–6). The upregulation of GLUT1 expression in response to IL4 was reproducible in three independent experiments, and IHC analysis of GLUT1 in 4T1 mammary tumors in two separate experiments.
To verify that IL4Rα mediates GLUT1 upregulation, and to demonstrate the in vivo relevance of our findings, we examined GLUT1 protein expression by immunohistochemistry (IHC) in orthotopic 4T1 mammary tumors. As expected, GLUT1 protein levels were reduced in IL4Rα knockdown tumors compared to sh-control (Figure 3C and S2A). We then examined whether GLUT1 expression was dependent on IL4 by performing IHC on archival metastatic 4T1 lung tumors established in wildtype (WT) or IL4-null (IL4 KO) mice. As expected, loss of IL4Rα in 4T1 tumor cells injected into WT mice resulted in the reduced expression of GLUT1 in lung metastases (Figure 3D and S2B). However, while GLUT1 levels were reduced, they were not significantly decreased in lung tumors of IL4 KO mice compared to WT mice receiving sh-control cells (Figure 3D and S2B). These results suggest that IL4Rα mediates enhanced GLUT1 expression at primary and metastatic tumor sites in vivo, but indicate that in addition to IL4, there may also be a role for the second IL4Rα-binding cytokine, IL13, in promoting GLUT1 expression through IL4Rα.
3.4 IL4 enhances glucose metabolism long-term in mammary cancer cells
We next assessed long-term functional changes in glucose metabolism in response to IL4/IL4Rα in 4T1 cells. IL4 was added to 4T1 cells every other day for 8 days, and samples of growth media were collected and analyzed for glucose and lactate levels. Both glucose consumption (Figure 4A) and lactate production (Figure 4B) significantly increased in response to IL4 over time. In addition, a comparison between glucose consumption (Figure 4C) and lactate production (Figure 4D) from 4T1 sh-control and IL4Rα knockdown clones treated with IL4 for 6 days confirmed that IL4Rα mediates IL4-induced glucose metabolism. These data confirm that IL4/IL4Rα increases glucose metabolism over extended periods of time in mammary cancer cells.
Figure 4. IL4Rα mediates enhanced glucose metabolism long-term in mammary cancer cells.

A) Glucose consumption (P < .0001) and B) lactate production (P < .0001) from 4T1 sh-control cells treated with vehicle or 20 ng/mL IL4 every other day for 8 days. C) Comparison of glucose consumption and D) lactate production from 4T1 sh-control (Ctl) or IL4Rα knockdown (KD) clones 6 days after initial IL4 treatment. Glucose consumption and lactate production were measured using enzyme based assays and normalized to total cellular protein content measured by BCA assay. Increased glucose uptake and lactate production in response to IL4 using the enzymatic assays was confirmed in three independent experiments.
3.5 IL4 treatment results in long-term increases in glutamine metabolism in breast cancer cells
Given our results with murine 4T1 mammary cancer cells, we next sought to determine whether IL4/IL4Rα could induce changes in glucose metabolism in human breast cancer cells using the MDA-MB-231 cell line. We first established that IL4 could induce growth in MDA-MB-231 cells, as it does in 4T1 cells (Figure S3). As before, we performed western blot analysis of GLUT1 protein expression in response to IL4 in MDA-MB-231 cells over a period of 8 days. Unlike 4T1 cells, MDA-MB-231 cells at similar seeding densities began to lose viability after day 4, which prompted us to look at earlier timepoints. At no point during a 4 day study did IL4 induce GLUT1 protein expression in MDA-MB-231 cells (Figure 5A). GLUT1 expression actually decreased over time in an IL4-independent manner, possibly in response to depleted glucose stores in the media over time. Furthermore, we saw no significant increase in glucose consumption when testing media from days 2, 3, or 4 (data not shown). We thus tested whether metabolism of the other major carbon and nitrogen source, glutamine, could be regulated by MDA-MB-231 cells.
Figure 5. IL4 induces the expression of glutamine transporters and long-term glutamine uptake in breast cancer cells.

MDA-MB-231 parental cells were treated with vehicle or 10 ng/mL human IL4 every other day for 4 days. A) Western blot analysis of deglycosylated glucose transporter 1 (GLUT1) protein expression in MDA-MB-231 cells in response to IL4 over time. B) Western blot analysis of glutamine transporter ASC-amino acid transporter 2 (ASCT2) upregulation in MDA-MB-231 cells in response to IL4. C) Quantification of mRNA expression of ASCT2 normalized to ribosomal S18 in MDA-MB-231 cells treated with vehicle or 10 ng/mL IL4 for 24 hours. D) Quantification of glutamine consumption in MDA-MB-231 cells 3 days after initial IL4 exposure compared to a vehicle treated control. 4T1 sh-control clones were treated with vehicle or 20 ng/mL murine IL4 every other day for 6 days. E) Western blot analysis of ASCT2 upregulation in 4T1 cells. F) Quantification of mRNA expression of ASCT2 normalized to ribosomal S18 in 4T1 cells treated with vehicle or 20 ng/mL IL4 for 2 days. G) Quantification of glutamine consumption 6 days after initial IL4 exposure compared to a vehicle treated control. Glutamine consumption was measured using an enzyme-based assay and normalized to total cellular protein content by BCA assay. Upregulation of ASCT2 protein in response to IL4 was confirmed in two independent experiments, and the upregulation of glutamine metabolism in each cell line was reproducible in three independent experiments per cell line.
We performed western blot analysis for the expression of ASC-amino acid transporter 2 (ASCT2, also known as SLC1A5), a major cancer-related glutamine transporter [24,25], in response to IL4 in MDA-MB-231 cells. ASCT2 protein levels increased within 24 hours of IL4 exposure, and remained increased 2 days post exposure (Figure 5B). We then confirmed the transcriptional upregulation of ASCT2 in response to IL4 by RT-PCR (Figure 5C). We next examined the ability of IL4 to enhance glutamine uptake over an extended period of time by culturing MDA-MB-231 cells with IL4 and examining media glutamine levels using an enzyme-based assay. IL4 increased glutamine uptake in MDA-MB-231 cells approximately 2-fold (Figure 5D).
To examine whether changes in glutamine metabolism in response to IL4 are unique to the MDA-MB-231 cell line, we performed the same analyses for ASCT2 expression and glutamine uptake with 4T1 cells. We confirmed that IL4 was able to induce ASCT2 protein expression by western blot (Figure 5E), and mRNA expression by RT-PCR (Figure 5F). In addition, IL4 treatment nearly tripled glutamine uptake in 4T1 cells following a 6 day exposure (Figure 5G). Together, these results indicate that long-term enhancement of glutamine metabolism by IL4 is a reproducible phenotype between human and murine breast cancer cells.
3.6 IL4-induced glutamine metabolism supports breast cancer growth
Having established that IL4 is capable of enhancing glucose and glutamine metabolism in breast cancer cells, we then wanted to determine whether these processes are necessary to support IL4/IL4Rα-induced tumor growth. sh-RNA mediated knockdown of IL4Rα in MDA-MB-231 cells was performed to examine IL4/IL4R-mediated changes in human breast cancer. Western blot analysis confirmed a reduction (73–91%) in IL4Rα protein expression in MDA-MB-231 IL4Rα knockdown clones compared to sh-controls (Figure 6A).
Figure 6. Increased breast cancer growth in response to IL4/IL4Rα is dependent upon glutamine but not glucose metabolism in vitro.

A) Western blot analysis illustrating the percent knockdown of IL4Rα protein expression in MDA-MB-231 cells. MDA-MB-231 or 4T1 sh-control (Ctl) or IL4Rα knockdown (IL4Rα KD) clones were cultured +/− IL4 (MDA-MB-231 cells: 10 ng/mL human IL4; 4T1 cells: 20 ng/mL murine IL4) and +/− metabolic inhibitors for 48 hours before cell number was analyzed using a CyQUANT® proliferation kit and standard curves. Graphs in B–E represent data from individual experiments, and each of these experiments was repeated twice mores with similar results obtained. Treatment with the glycolysis inhibitor, 2DG, did not inhibit IL4-induced increases in cell number in B) 4T1 clones (+/− 1.5 mM 2DG) or C) MDA-MB-231 clones (+/− 12 mM 2DG). Treatment with the glutaminase 1 inhibitor, 968, blocked IL4-induced increases in cell number in both D) 4T1 clones (+/− 10 μM 968) and E) MDA-MB-231 clones (+/− 20 μM 968). F) The combined treatment of both 4T1 cells (1.5 mM 2DG and 10μM 968) and G) MDA-MB-231 cells (12 mM 2DG and 15 μM 968) with 2DG and 968 had an additive effect in reducing cell number compared to either drug or vehicle alone. Experiments represented in F–G were repeated with similar results obtained.
The glucose analog, 2 Deoxy-D-glucose (2DG), is commonly used to inhibit glycolysis by blocking the initial rate-limiting step mediated by the hexokinase 2 enzyme [33]. We determined the effective dose of drug that inhibited 4T1 (Figure S4A) and MDA-MB-231 (Figure S4B) growth in vitro by finding the dose that reduced cell number by ~50% (ED50) in CyQUANT® assays. We then examined the ability of the ED50 dose of 2DG to block IL4-induced growth in 4T1 and MDA-MB-231 sh-control and IL4Rα knockdown cells by again quantifying changes in cell number. As expected, IL4 treatment resulted in an increase in sh-control cell number for both cell lines, and this response was attenuated in IL4Rα knockdown cells (Figures 6B, C). This pattern was true for each of our cell number analyses (Figure 6B–E).
In neither cell line did the inhibition of glycolysis with 2DG attenuate the percent increase in IL4-induced cell number compared with vehicle control. This was true even though the increase in cell number between 2DG alone and IL4 + 2DG for MDA-MB-231 sh-control cells was not statistically significant. These experiments were done 3 times for each cell line and an average percent increase in cell number due to IL4 treatment in the presence or absence of 2DG was calculated. In 4T1 sh-control cells without 2DG, IL4 treatment increased cell number by 21.7% compared with vehicle. This was not statistically different from the 18.3 % percent increase in cell number induced by IL4 in the presence of 2DG. Similarly, in MDA-MB-231 sh-control cells without 2DG, IL4 treatment increased cell number by 10.2% compared with vehicle, which was not statistically different from the 10.7 % increase in cell number with IL4 in the presence of 2DG. This suggests that IL4 can induce breast cancer growth independently of enhanced glucose uptake.
In a separate dose response assay, we determined the ED50 of compound 968, an inhibitor of glutaminase 1 frequently used to block glutamine metabolism [34], to inhibit growth in both cell lines (Figure S4C, D), before combining this dose with IL4. In both 4T1 (Figure 6D) and MDA-MB-231 cells (Figure 6E), the ED50 of 968 significantly blocked IL4-increased cell number. Again, these experiments were done 3 times for each cell line. In 4T1 sh-control cells, IL4 treatment increased cell number by 16% compared with vehicle. However, the increase was significantly attenuated to only 1.2% (P = 0.02) when the assay was repeated in the presence of 968. Similarly, in MDA-MB-231 sh-control cells, IL4 treatment increased cell number by 11.5% compared with vehicle, which was significantly different (P = 0.04) to the 5.6% increase seen in the presence of 968. Collectively, our data demonstrates that glutamine metabolism, but not glucose metabolism, is necessary to support IL4-induced growth of breast cancer cells in vitro. Interestingly, the combined treatment of 4T1 cells (Figure 6F) and MDA-MB-231 cells (Figure 6G) with the ED50 of 2DG and 968 showed an additive effect in blocking growth compared to either drug alone.
Using EdU incorporation assays to measure proliferation as the percent of cells in S phase, we were able to demonstrate that IL4 treatment results in an increase in proliferation in both 4T1 (Figure S5A) and MDA-MB-231 (Figure S5B) cells in a dose dependent manner. Using sh-control and IL4Rα knockdown cell in similar assays, we confirmed that IL4Rα mediates this increase in proliferation in response to IL4 in both cell lines (Figure S5C, D). These data support the association between IL4Rα expression and enhanced proliferation in 4T1 mammary tumors in vivo, and indicate that IL4-induced proliferation could contribute to IL4-induced growth in vitro.
4. Discussion
The Th2 immune cytokine, interluekin-4 (IL4), is known to induce lymphocyte activation and differentiation, and regulate increased lymphocyte proliferation and survival through the type I IL4 receptor (IL4R) [3]. We previously defined a novel role for IL4 in directly promoting the proliferation and survival of mammary cancer cells expressing type II IL4Rs for enhanced growth at metastatic lung and liver sites [7]. Here, we used two orthotopic models of murine mammary cancer with R221a or 4T1 IL4Rα knockdown or sh-control clones, to demonstrate that expression of IL4Rα, a component of IL4Rs, also promotes mammary tumor growth at the primary site. Strikingly, shRNA-mediated knockdown of IL4Rα in the R221a cell line nearly abolished mammary tumor take and growth. This result prevented further analysis of knockdown tumors, as there were none at endpoint. While this stark result was not achieved with 4T1 IL4Rα knockdown clones, there was a reduction in average tumor latency, and a significant reduction in the rate of tumor growth compared to 4T1 sh-control clones. Disparities in latency between the 4T1 versus R221a cell lines may be attributable to the more aggressive nature of the 4T1 cell line, discussed previously [7].
Immunohistochemical analysis of 4T1 mammary tumors by Ki67 or Caspase-3 staining at endpoint demonstrated that increased proliferation but not survival contributed to IL4Rα-enhanced mammary tumor growth. This result is in accordance with our previous study showing that IL4Rα-enhanced proliferation rather than survival promotes the growth of allografted colon tumors in the cecum [35]. Given that IL4Rα expression consistently mediated enhanced tumor growth in all of our primary and metastatic mammary tumor models, we then sought to determine whether IL4/IL4Rα-induced metabolism could serve as a novel mechanism to support this IL4Rα-enhanced growth. This concept was based on previous reports of IL4-mediated metabolic changes in lymphocytes [8,11].
We show that elevated glucose transporter 1 (GLUT1) expression is associated with long-term increases in glucose metabolism (increased glucose uptake and lactate production) downstream of IL4/IL4Rα in murine mammary cancer cells. Elevated GLUT1 expression was also associated with IL4Rα expression in primary and metastatic 4T1 tumors in vivo. Interestingly, total protein levels of GLUT1 were not evidently increased until day 8 of IL4 treatment with 4T1 cells in vitro, while significant increases in glucose metabolism were evident by day 6 in the same study. This indicates that IL4/IL4Rα signaling may also influence the translocation of GLUT1 to the membrane for more immediate increases in glucose uptake and metabolism in addition to the later transcriptional upregulation of GLUT1 for maintaining enhanced glucose metabolism long-term. This concept is supported by additional experiments demonstrating increased glucose uptake by 2-NBDG uptake assay, and increased lactate production detected by multianalyte microphysiometry [30,31] (data not shown), after short-term IL4 exposure. However, more detailed assessment of GLUT1 translocation is required to substantiate this idea.
While other GLUT isoforms have been detected in breast cancer, only the overexpression of GLUT1 has been associated with poorer prognosis [15,36,37], and with increased [18F]-fluoro-deoxyglucose (FDG) uptake in human breast tumors [18]. However, we were unable to detect increases in GLUT1 expression or glycolysis (data not shown) in response to IL4 in the human MDA-MB-231 breast cancer cell line. One main difference between these cells and 4T1 mammary cancer cells is their expression of oncogenic K-Ras, which is known to drive enhanced glucose uptake and metabolism. In fact, metabolic flux analysis of MDA-MB-231 cells revealed a basal stoichiometric relationship between high glucose consumption and lactate production [38]. It is therefore feasible that IL4 treatment would have no added affect on glycolysis in this cell line as nearly all glucose consumed is already being shunted to lactate production. In addition, MDA-MB-231 cells required nearly 6 times the dose of glycolysis inhibitor, 2-Deoxy-D-glucose (2DG), to inhibit growth in vitro compared with 4T1 cells. MDA-MB-231 cells in general may be less reliant upon glucose metabolism for growth, and metabolism of glutamine as a carbon and nitrogen source during growth may be of greater importance. Glutamine metabolism has been shown to promote the viability and proliferation of MDA-MB-231 cells [38].
Although classified as a non-essential amino acid, glutamine is taken up disproportionally more by cancer cells than normal cells, and the therapeutic targeting of glutamine addiction is being considered for anti-cancer treatments [39]. ASC amino-acid transporter 2 (ASCT2), is a major transporter upregulated by breast cancer cells for enhanced glutamine uptake [24,25]. Here, we show that IL4 is a novel regulator of elevated ASCT2 mRNA and protein expression, and this was associated with enhanced long-term glutamine uptake in both 4T1 and MDA-MB-231 cells. In addition, inhibition of glutamine metabolism with compound 968 blocked IL4-induced growth in both cell lines. Although there appears to be a general need for glutamine metabolism independent of IL4 in breast cancer, our results provide evidence for a novel role of IL4-enhanced glutamine metabolism in supporting IL4-induced growth in both murine and human breast cancer cells.
Supporting the concept that enhanced glutamine metabolism may be the predominant metabolic phenotype backing breast cancer growth in response to IL4, IL4-induced growth was not attenuated when glycolysis was pharmacologically inhibited with 2DG treatment in either cell line. This result was not unexpected in the MDA-MB-231 cell line as IL4 did not induce glucose metabolism in these cells. While IL4 did enhance glucose metabolism in 4T1 cells, the inability of 2DG to block IL4-induced growth in these cells does not mean that IL4-enhanced glucose metabolism cannot support IL4-induced survival or proliferation specifically. We were able to show that IL4/IL4Rα-induced proliferation likely contributes to IL4Rα-induced growth in vitro, and similarly to B cells, there may still be a role for IL4-enhanced glucose metabolism in supporting survival for enhanced tumor growth, although this remains to be explored. In addition, the combined treatment of both 4T1 cells and MDA-MB-231 cells with 2DG and the glutamine metabolism inhibitor, compound 968, did have an additive effect in blocking growth compared to either drug alone. These data could indicate a possible compensatory mechanism where enhanced glutamine metabolism supports biosynthetic processes during growth when glycolysis is inhibited.
Here, we have shown for the first time that an immune cytokine, IL4, can reprogram glucose and glutamine metabolism in breast cancer cells expressing the type II IL4 receptor. Importantly, IL4-induced glutamine metabolism supports IL4/IL4Rα-enhanced growth in both human and mammary cancer cells. The benefits of therapeutically targeting the IL4/IL4Rα signaling axis may be three fold. We have already identified this axis as a strong promoter of mammary cancer survival and proliferation, and this work highlights a third targetable hallmark of cancer induced by IL4, IL4/IL4Rα-induced metabolism. Fortunately, therapies specifically targeting the type II IL4 receptor on epithelial cancer cells are currently being developed [40].
Supplementary Material
Highlights.
Expression of glucose and glutamine transporters increases in response to IL4.
IL4 induces both glucose and glutamine metabolism.
IL4-induced proliferation is dependent upon IL4-induced glutamine metabolism.
Acknowledgments
Financial support: The studies reported here were supported by R01 CA157781 (to BF). KTV was supported by the Cellular, Biochemical and Molecular Sciences training program (5T32GM008554) and an NRSA F31 Award (CA183539).
Footnotes
No conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cancer Facts and Figures 2013. American Cancer Society; Atlanta: 2013. [Google Scholar]
- 2.Ben-Baruch A. Host microenvironment in breast cancer development: inflammatory cells, cytokines and chemokines in breast cancer progression: reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2003;5:31–6. doi: 10.1186/bcr554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–38. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 4.Todaro M, Lombardo Y, Francipane MG, Alea MP, Cammareri P, Iovino F, et al. Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived interleukin-4. Cell Death Differ. 2008;15:762–72. doi: 10.1038/sj.cdd.4402305. [DOI] [PubMed] [Google Scholar]
- 5.LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X, Heller NM, et al. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. 2008;132:259–72. doi: 10.1016/j.cell.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camp BJ, Dyhrman ST, Memoli VA, Mott LA, Barth RJ. In situ cytokine production by breast cancer tumor-infiltrating lymphocytes. Ann Surg Oncol. 1996;3:176–84. doi: 10.1007/BF02305798. [DOI] [PubMed] [Google Scholar]
- 7.Venmar KT, Carter KJ, Hwang DG, Dozier EA, Fingleton B. IL-4 receptor ILR4α regulates metastatic colonization by mammary tumors through multiple signaling pathways. Cancer Res. 2014;74:4329–40. doi: 10.1158/0008-5472.CAN-14-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dufort FJ, Bleiman BF, Gumina MR, Blair D, Wagner DJ, Roberts MF, et al. Cutting Edge: IL-4-Mediated Protection of Primary B Lymphocytes from Apoptosis via Stat6-Dependent Regulation of Glycolytic Metabolism. J Immunol. 2007;179:4953–4957. doi: 10.4049/jimmunol.179.8.4953. [DOI] [PubMed] [Google Scholar]
- 9.Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol. 2012;13:907–15. doi: 10.1038/ni.2386. [DOI] [PubMed] [Google Scholar]
- 10.Or R, Renz H, Terada N, Gelfand EW. IL-4 and IL-2 promote human T-cell proliferation through symmetrical but independent pathways. Clin Immunol Immunopathol. 1992;64:210–7. doi: 10.1016/0090-1229(92)90202-y. [DOI] [PubMed] [Google Scholar]
- 11.Cho SH, Ahn AK, Bhargava P, Lee CH, Eischen CM, McGuinness O, et al. Glycolytic rate and lymphomagenesis depend on PARP14, an ADP ribosyltransferase of the B aggressive lymphoma (BAL) family. Proc Natl Acad Sci U S A. 2011;108:15972–7. doi: 10.1073/pnas.1017082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–82. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Griffin JL, Shockcor JP. Metabolic profiles of cancer cells. Nat Rev Cancer. 2004;4:551–61. doi: 10.1038/nrc1390. [DOI] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Brown RS, Wahl RL. Overexpression of Glut-1 glucose transporter in human breast cancer. An immunohistochemical study. Cancer. 1993;72:2979–85. doi: 10.1002/1097-0142(19931115)72:10<2979::aid-cncr2820721020>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 16.Binder C, Binder L, Marx D, Schauer A, Hiddemann W. Deregulated simultaneous expression of multiple glucose transporter isoforms in malignant cells and tissues. Anticancer Res. n.d;17:4299–4304. [PubMed] [Google Scholar]
- 17.Krzeslak A, Wojcik-Krowiranda K, Forma E, Jozwiak P, Romanowicz H, Bienkiewicz A, et al. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol Oncol Res. 2012;18:721–8. doi: 10.1007/s12253-012-9500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown RS, Goodman TM, Zasadny KR, Greenson JK, Wahl RL. Expression of hexokinase II and Glut-1 in untreated human breast cancer. Nucl Med Biol. 2002;29:443–453. doi: 10.1016/s0969-8051(02)00288-3. [DOI] [PubMed] [Google Scholar]
- 19.Zhu X, Hart R, Chang MS, Kim JW, Lee SY, Cao YA, et al. Analysis of the major patterns of B cell gene expression changes in response to short-term stimulation with 33 single ligands. J Immunol. 2004;173:7141–9. doi: 10.4049/jimmunol.173.12.7141. [DOI] [PubMed] [Google Scholar]
- 20.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678–84. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curi R, Newsholme P, Pithon-Curi TC, Pires-de-Melo M, Garcia C, Homem-de-Bittencourt PI, Júnior, et al. Metabolic fate of glutamine in lymphocytes, macrophages and neutrophils. Braz J Med Biol Res. 1999;32:15–21. doi: 10.1590/s0100-879x1999000100002. [DOI] [PubMed] [Google Scholar]
- 22.Mohamed A, Deng X, Khuri FR, Owonikoko TK. Altered glutamine metabolism and therapeutic opportunities for lung cancer. Clin Lung Cancer. 2014;15:7–15. doi: 10.1016/j.cllc.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang F, Zhao Y, Zhao J, Wu S, Jiang Y, Ma H, et al. Upregulated SLC1A5 promotes cell growth and survival in colorectal cancer. Int J Clin Exp Pathol. 2014;7:6006–14. [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol. 2005;15:254–66. doi: 10.1016/j.semcancer.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Collins CL, Wasa M, Souba WW, Abcouwer SF. Determinants of glutamine dependence and utilization by normal and tumor-derived breast cell lines. J Cell Physiol. 1998;176:166–78. doi: 10.1002/(SICI)1097-4652(199807)176:1<166::AID-JCP18>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 26.Kim S, Kim DH, Jung WH, Koo JS. Expression of glutamine metabolism-related proteins according to molecular subtype of breast cancer. Endocr Relat Cancer. 2013;20:339–48. doi: 10.1530/ERC-12-0398. [DOI] [PubMed] [Google Scholar]
- 27.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–9. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 28.Fan Y, Dickman KG, Zong WX. Akt and c-Myc differentially activate cellular metabolic programs and prime cells to bioenergetic inhibition. J Biol Chem. 2010;285:7324–33. doi: 10.1074/jbc.M109.035584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimmel DW, Dole WP, Cliffel DE. Application of multianalyte microphysiometry to characterize macrophage metabolic responses to oxidized LDL and effects of an apoA-1 mimetic. Biochem Biophys Res Commun. 2013;431:181–5. doi: 10.1016/j.bbrc.2012.12.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimmel DW, Meschievitz ME, Hiatt LA, Cliffel DE. Multianalyte microphysiometry of macrophage responses to phorbol myristate acetate, lipopolysaccharide, and lipoarabinomannan. Electroanalysis. 2013;25:1706–1712. doi: 10.1002/elan.201300121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young CD, Lewis AS, Rudolph MC, Ruehle MD, Jackman MR, Yun UJ, et al. Modulation of glucose transporter 1 (GLUT1) expression levels alters mouse mammary tumor cell growth in vitro and in vivo. PLoS One. 2011;6:e23205. doi: 10.1371/journal.pone.0023205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barban S, Schulze HO. The effects of 2-deoxyglucose on the growth and metabolism of cultured human cells. J Biol Chem. 1961;236:1887–90. [PubMed] [Google Scholar]
- 34.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–19. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koller FL, Hwang DG, Dozier EA, Fingleton B. Epithelial interleukin-4 receptor expression promotes colon tumor growth. Carcinogenesis. 2010;31:1010–7. doi: 10.1093/carcin/bgq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Younes M, Brown RW, Mody DR, Fernandez L, Laucirica R. GLUT1 expression in human breast carcinoma: correlation with known prognostic markers. Anticancer Res. 1995;15:2895–8. [PubMed] [Google Scholar]
- 37.Kang SS, Chun YK, Hur MH, Lee HK, Kim YJ, Hong SR, et al. Clinical Significance of Glucose Transporter 1 (GLUT1) Expression in Human Breast Carcinoma. Cancer Sci. 2002;93:1123–1128. doi: 10.1111/j.1349-7006.2002.tb01214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–33. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junttila IS, Creusot RJ, Moraga I, Bates DL, Wong MT, Alonso MN, et al. Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat Chem Biol. 2012;8:990–8. doi: 10.1038/nchembio.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.