

Abstract
Objectives
In men, androgen deprivation contributes to the development of metabolic syndrome and type 2 diabetes (T2D). In women, androgen excess predisposes to insulin resistance and T2D. There is a bidirectional modulation of glucose homeostasis by androgen in males and females that we analyze in this review.
Methods
We review the literature in both rodents and humans on the role of androgens and the androgen receptor (AR) in the control of glucose and energy metabolism in health, obesity and T2D.
Results
Sex-specific activation of AR in the hypothalamus, skeletal muscle, liver, adipose tissue and pancreatic islet β cells accounts for maintenance or disruption in energy metabolism and glucose homeostasis.
Conclusion
We argue that AR is a target to prevent androgen-related metabolic disorders.
Keywords: Testosterone, androgen receptor, glucose homeostasis, diabetes
Introduction
With the increase in human longevity, men will spend a significant proportion of their live in a state of testosterone deficiency. This state increases type 2 diabetes (T2D) risk (1, 2). Men who are on androgen depletion therapy for prostate cancer are also at high risk to develop T2D, and add to the already large numbers of aging men with testosterone deficiency (3). The potential impact of decreased testosterone levels on T2D is not limited to men. Indeed, the association between testosterone excess and diabetes in women has been known for almost a century (4, 5). In women, hyperandrogenic conditions such as the polycystic ovarian syndrome (PCOS) are associated with insulin resistance, glucose intolerance and subsequent T2D. PCOS is the most common endocrine disorder in reproductive age women. Testosterone action is mediated by the androgen receptor (AR), a ligand-activated transcription factor. We need to gain a better understanding on the tissue selective actions of AR that influence glucose and fat metabolism in males and females. For example, selective androgen receptor modulators (SARMs) are a novel class of AR ligands intended to act like testosterone with tissue-selectivity in men (6). The ideal SARM is defined as an agent with anabolic activity on muscle and bone but without androgenic action in the prostate. SARMs allow extension of androgen therapy to geriatric functional decline in men that requires dual muscle and bone anabolism. Given that androgen treatment of T2D is not suitable because of prostate and cardiovascular risks, there is a need to design more selective SARMs for potential clinical application in the aging male population with obesity and T2D.
In this report, we review the literature in both rodents and humans on the role of testosterone and the androgen receptor (AR) in the control of glucose/energy homeostasis and the development of metabolic dysfunction in males and females with special emphasis on tissue selective actions in skeletal muscle, adipose, tissue, liver pancreatic β-cells and the central nervous system.
Role of AR in glucose homeostasis in the male (Figure 1)
Figure 1. Proposed mechanism of androgen action via AR in males.
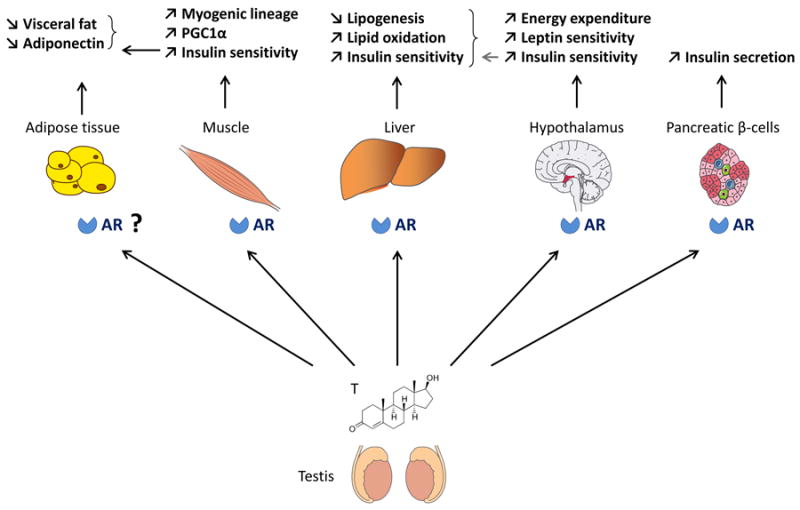
In males, androgens promote glucose and energy homeostasis via actions on AR in skeletal muscle, liver, pancreatic beta-cells and metabolic centers in the hypothalamus. Androgen actions on adipose tissue could be indirectly mediated via AR actions in muscle.
Testosterone deficiency predisposes to T2D in men
There is bidirectional interaction between androgen and diabetes in men in which low testosterone predisposes to diabetes and hyperglycemia leads to hypogonadism (7). There is substantial evidence that low testosterone is associated with and predicts diabetes in men. In a meta-analysis of 28 cross-sectional and prospective studies, Ding et al found that total testosterone levels were always lower in diabetic men than in non-diabetic controls, and that higher testosterone levels were associated with a lower risk of incident diabetes (8). A more recent meta-analysis of prospective and cross-sectional studies also found that T2D was independently associated with low total testosterone levels in men (7). In addition, the same meta-analysis of the metabolic effect of testosterone replacement therapy found that testosterone therapy was associated with a significant reduction in fasting plasma glucose, HBA1c, fat mass and triglyceride in men (7). Although, the association between low testosterone and obesity is clear, diabetic men still have lower testosterone levels even after adjustment for BMI (7, 9).
Very low testosterone levels during androgen deprivation therapy for prostate cancer have been associated with increased risk of diabetes in several large studies (10, 11). Recently, an observational study from the Veterans Healthcare Administration showed that among prostate cancer patients, androgen deprivation therapy with GnRH agonists was associated with an increased risk of T2D (12). Another study showed marked hyperglycemia and decreased pancreatic β-cell function among prostate cancer patients after androgen-deprivation therapy (13). Notably, testosterone deficiency has also been associated with impaired fasting glucose and glucose intolerance independently of obesity and metabolic syndrome in men (14). Altogether, these studies suggest that low testosterone predisposes to diabetes in men.
Androgens prevents visceral fat accumulation and improves insulin sensitivity
The impact of testosterone deficiency on the development of visceral obesity, insulin resistance and metabolic syndrome in men is well established (1, 2, 15, 16, 17, 18). There is an inverse correlation between total serum testosterone and the amount of visceral adipose tissue (16). This is true in all situations of androgen deficiency, whether in the context of hypogonadism in older men (18), inherited testosterone deficiency as observed in Klinefelter's syndrome (19), or androgen deprivation during treatment for prostate carcinoma (15). Thus, in men, high serum testosterone is associated with insulin sensitivity (17). Of course, aromatization of testosterone into 17β-estradiol (E2) is critical to energy homeostasis in males, suggesting that testosterone functions as a prohormone in men to provide E2 for tissue metabolism. Indeed, orchidectomized male rodents treated with either testosterone or E2 remain lean, while those treated with the pure androgen DHT –that cannot be converted to E2– develop obesity demonstrating that following orchiectomy, the restoration of adiposity was due to testosterone conversion into E2 acting on ERs (20). This is also true in men for whom testosterone replacement suppresses adiposity, an effect that is blocked in the presence of an aromatase inhibitor (21). In addition, human and rodent studies have revealed that mutations in the aromatase or the ERα genes increase visceral obesity in males further demonstrating the importance of estrogen in male energy metabolism (22). Still, several lines of evidence demonstrate that in males, testosterone has anti-obesity properties mediated via AR actions. First, in men with genetic androgen resistance linked to CAG repeat polymorphisms in the AR gene –which decreases AR-mediated gene transcription– a low number of CAG repeats is independently associated with protective metabolic parameters such as low body fat mass and plasma insulin, suggesting that intact AR transcription favors metabolic homeostasis (23). Second, male mice with global deletion of the AR develop late onset visceral obesity with leptin resistance, insulin resistance, and increased lipogenesis in adipose tissue and liver (24, 25). Furthermore, AR regulates adiponectin production. Serum adiponectin levels are high in hypogonadal men and are reduced by testosterone therapy (26). Testosterone infusion also decreases adiponectin in mice (27). This effect is at least partially mediated via AR since serum adiponectin concentrations are elevated in AR-deficient mice (24). However, it is unclear whether AR suppression of adiponectin reflects increased adiponectin sensitivity, decreased adipocyte number, or improved adipose function.
Several lines of evidence suggest that the suppressing effect of testosterone on white adipose tissue (WAT) mass in males is indirectly mediated via AR signaling in skeletal muscle. First, in vitro, testosterone stimulates the commitment of pluripotent mesenchymal stem cells into myogenic lineage while at the same time suppressing the adipogenic lineage (28). This AR-dependent pathway involves non-canonical Wnt signaling (29). This androgenic anabolism induces the expression of IGF1, leading to nuclear accumulation of beta-catenin, a myogenic and anti-adipogenic stem cell factor (30). Second, overexpression of AR selectively in muscle cells of transgenic male rats increases their lean mass via hypertrophy of type IIb muscle fibers which is associated with increased oxidative metabolism and metabolic rate (31). This results in reduced adipocyte size and adipose tissue mass. Finally, and consistent with this model, male adipocyte-specific androgen receptor KO (ARKO) mice exhibit no increase in fat mass demonstrating that direct AR action in adipose tissue is not necessary for the control of fat mass (32). Surprisingly, these mice show an increased production of leptin by adipose tissue without leptin resistance. Thus, activation of AR in skeletal muscle may indirectly decrease adipose tissue mass by increasing muscle oxidative metabolism or through the release of a circulating factor. However, surprisingly, mice with myocyte-specific AR ablation have lower intra-abdominal fat. It should be noted that these mice exhibit a fast-to-slow fiber conversion, without major change in muscle mass and without affecting muscle strength, which could affect the adipose phenotype (33). In summary, testosterone action prevents fat accumulation in males via a combination of ER (after aromatization in E2) and AR mediated effects. The AR suppression of adiposity could be mediated via AR signaling in skeletal muscle
Testosterone action in skeletal muscle promotes insulin sensitivity in males
The mechanism of insulin resistance following androgen deficiency probably also involves an alteration in skeletal muscle insulin sensitivity. Castration of male rats is followed by a marked insulin resistance in skeletal muscle under euglycemic, hyperinsulinemic clamp conditions (34). Treatment with physiological doses of testosterone completely abolishes these perturbations in insulin sensitivity. Transcriptome analysis of skeletal muscle in mice demonstrates that testosterone regulates the expression of genes in glucose metabolism in a way that would promote insulin sensitivity (35). The mechanism of AR deficiency-induced insulin resistance in skeletal muscle probably involves a decrease in the transcription factor PGC1α (Peroxisome proliferator-activated receptor-gamma coactivator alpha). Indeed, PGC1α stimulates mitochondrial biogenesis and skeletal muscle oxidative fibers and is thus a molecular marker of muscle insulin sensitivity. A decrease in PGC1α expression in skeletal muscle of T2D subjects is associated with insulin resistance (36). Similarly, in men, low testosterone levels are associated with low PGC1α expression levels in muscle (17) and AR-deficient mice express low levels of PGC1α mRNA in tissues (24). Thus, testosterone deficiency promotes insulin resistance in skeletal muscle at least partially via an AR-dependent mechanism involving a decrease in PGC1α-mediated oxidative and insulin sensitive muscle fibers.
Androgen actions in liver favor lipid homeostasis and insulin sensitivity
Male hepatocyte-specific ARKO (HARKO) mice developed hepatic steatosis when fed a high-fat diet, but females did not (37). Increased hepatic steatosis in these obese male HARKO mice resulted from decreased hepatic peroxisome proliferator-activated receptor α (PPARα) expression leading to decreased fatty acid oxidation and increased hepatic sterol regulatory element binding protein 1c leading to increased de novo lipid synthesis. This ultimately led to hepatic insulin resistance. Mice deficient in 5α-reductase type 1 (5αR1-/-), the enzyme that converts testosterone to the active androgen DHT, develop hepatic steatosis and show decreased hepatic expression of genes involved in insulin signaling when fed a Western diet (38). Like the HARKO mice, male 5αR1-KO mice developed adiposity, hyperinsulinemia, hepatic steatosis, decreased mRNA transcript profiles for fatty acid β-oxidation, and increased genes for lipid storage. The non-selective 5α-reductase inhibitor finasteride induced hyperinsulinemia and hepatic steatosis in obese male Zucker rats, both intact and castrated (39). These rodent studies are supported by the observation that low testosterone levels are associated with hepatic steatosis in men (40). Together these studies show that AR actions in liver are important to prevent hepatic steatosis.
Central androgen actions regulate energy homeostasis in males
AR is more abundantly expressed in the brain of male rodents than that of females (41). Male whole-body AR-deficient mice develop obesity without increase energy intake but with decreased locomotor activity. These mice also display reduced brown adipose tissue thermogenesis which decreases energy expenditure (24). AR also functions in the male hypothalamus to favor central leptin action. Indeed, in AR-deficient male mice, leptin fails to promote STAT3 nuclear localization in arcuate nucleus (ARC) neurons and does not suppress food intake or reduce body weight even before the onset of overt obesity (41). Further, neuronal specific ARKO (NARKO) mice develop obesity, insulin resistance and glucose intolerance. These mice show hypothalamic insulin resistance by way of activation of hypothalamic NFκB that increases inflammation (42). Together, these observations demonstrate that in male rodents, AR is involved in the control of adipose tissue mass via central and peripheral effects.
Androgen action in β-cells in males
Early studies reported that when β-cell destruction is induced by streptozotocin in male mouse models of insulin-deficient diabetes (43, 44), testosterone accelerates the hyperglycemic decompensation in an AR-dependent manner. However, it was also reported that testosterone protects early apoptotic damage induced by streptozotocin in male rat pancreas through AR (45, 46). A previous report also suggested that testosterone stimulates islet insulin mRNA and content in culture and in vivo (47). Therefore, the role of AR in male β-cells is unclear. We have generated a β-cell specific AR knockout mouse to examine the direct role of AR in male β-cell physiology (βARKO-/y) (48). Male βARKO-/y mice exhibit decreased glucose-stimulated insulin secretion (GSIS) leading to glucose intolerance, and develop β-cell failure to compensate for diet-induced insulin resistance. The decreased GSIS is reproduced in cultured male βARKO-/y islets as well as in cultured human islets treated with flutamide, an AR antagonist. This suggests that AR is a physiological regulator of male β-cell function, a finding that has important implications for prevention of T2D in hypoandrogenic men.
Role of AR in glucose homeostasis in the female (figure 2)
Figure 2. Proposed mechanism of excess AR activation in women.
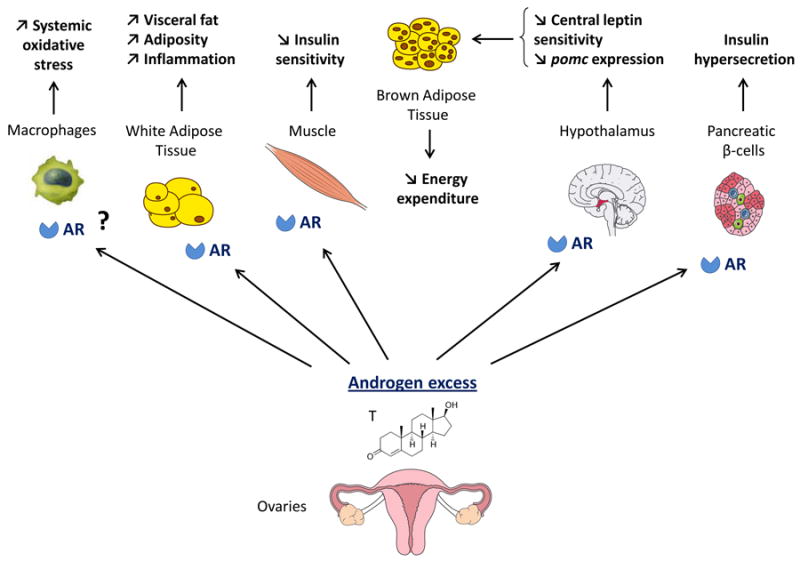
In females with hyperandrogenemia, excess AR activation in skeletal muscle, macrophages, pancreatic beta-cells and metabolic centers in the hypothalamus synergize to promote metabolic dysfunction, inflammation, visceral adiposity and T2D.
Hyperandrogenism predisposes to T2D in women
The role of AR deficiency in female metabolism is not well studied, though it is reported to have no effect on body weight in female mice (49). In contrast, the association between androgen excess and diabetes in women has been known since the early description of “diabetes in bearded women” by Achard and Thiers in 1921 (4).
In a prospective 12-year study of Swedish women, Bjorntorp and coworkers initially reported that low concentration of Sex-Hormone Binding Globulin (SHBG) –which increases free testosterone– was a strong independent risk factor for the development of T2D (50). Similarly, Larsson and Ahren showed that postmenopausal women with impaired glucose tolerance have higher androgen activity than women with normal glucose tolerance, and that the androgen activity correlates with the degree of glucose intolerance (51, 52). Higher levels of free testosterone and lower levels of SHBG have been repeatedly associated with glucose intolerance and insulin resistance in women (53, 54, 55, 56, 57). In a meta-analysis of available prospective and cross-sectional studies relating testosterone, SHBG, and estradiol levels with risk of T2D, Ding et al. reported that high free testosterone levels are associated with higher risk of T2D in women (8). However, estradiol levels were also elevated among postmenopausal women with diabetes suggesting that estrogen excess could also have played a role in T2D risk. In postmenopausal women, higher plasma levels of estradiol and testosterone were strongly and prospectively related to increased risk of developing T2D (58).
Hyperandrogenism promotes insulin resistance in women
To understand the role of androgen excess in insulin sensitivity, several groups have explored the metabolic response of androgen-exposed women during hyperglycemic and euglycemic-hyperinsulinemic clamps. Globally, these studies reported that high testosterone levels produce insulin resistance by decreasing insulin-stimulated whole body glucose uptake in healthy pre- and post-menopausal women (54, 59, 60, 61). Further, the reduced insulin-stimulated, whole body glucose uptake was not attributable to hepatic insulin resistance, which remained unchanged, supporting a role for skeletal muscle in insulin resistance (59). The role of excess testosterone in promoting skeletal muscle insulin resistance with fiber type switch has also been confirmed in studies of female rodents (62, 63). Still, the mechanisms by which androgen excess promotes muscle insulin resistance remains unknown.
Despite accumulated evidence that testosterone excess alters insulin sensitivity in females, it is not clear whether testosterone excess initiates insulin resistance or perpetuates it. Generally, insulin sensitivity improves when hyperandrogenism is reversed with anti-androgen therapy, in association with weight loss (40, 64, 65). These latter studies suggest androgen excess alone may be instrumental in insulin resistance. However, treatment with AR antagonists or suppression of ovarian androgen production with GnRH analogues in hyperandrogenic women does not always improve insulin resistance (66), suggesting that excess androgens in women may not be the cause of these metabolic abnormalities but rather an aggravating factor.
Hyperandrogenism increases visceral adiposity in females
Sexual dimorphisms in body fat distribution are well described. Men, on average, have less total body fat but more central/intra-abdominal adipose tissue, whereas women tend to have more total fat with gluteal/femoral and subcutaneous distribution (67, 68). In women with chronic androgen excess, plasma testosterone is positively correlated with waist circumference, an index of visceral obesity suggesting that testosterone is instrumental in this process (69). A recent proteomic study of the influence of androgen excess on human visceral and subcutaneous adipose tissue proteomes reported that the abundance in adipose tissue depots of several proteins involved in metabolism were similar in women with androgen excess and in men, suggesting that androgen excess has masculinized the function of female adipose tissue (70, 71). Nonetheless, little is known about the mechanism through which chronic androgen excess induces adiposity in females with a preferentially visceral distribution. We explored the mechanism by which chronic androgen excess in females induces visceral adiposity using female mice exposed to the non-aromatizable androgen receptor agonist DHT (72). We observed that androgen excess prevents leptin from activating brown adipose tissue (BAT) thermogenesis, which is associated with reduced energy expenditure and visceral obesity. We also observed decreased hypothalamic proopiomelanocortin (pomc) expression and POMC neuronal innervations into dorsomedial hypothalamus (DMH) (72). Therefore, the increase in visceral adiposity in hyperandrogenic females could have a central origin. In addition, these results suggest that androgen-induced visceral fat distribution and accumulation involves alteration of the melanocortin system between the ARC and DMH.
Hyperandrogenism predisposes to β-cell dysfunction in females
Women with hyperandrogenemia (testosterone excess) also show various degrees of pancreatic β-cell dysfunction. The mechanism(s) by which this develops is unknown. Women with functional hyperandrogenism have significantly higher basal insulin secretory rates and attenuated post-prandial insulin secretory responses (73). In addition, women with PCOS, have been reported to exhibit inadequate acute insulin release to the degree of insulin resistance (74) or an exaggerated early insulin response to glucose. These abnormalities are not accounted for by insulin resistance and are closely associated with hyperandrogenicity (75). In these women with PCOS, there is a robust relationship between β-cell function and free testosterone, raising the possibility that excess testosterone in women leads to insulin hypersecretion (76). Thus, women with hyperandrogenism display β-cell hyperfunction which may predispose to secondary β-cell failure. Consistent with this hypothesis, in female mice, testosterone accelerates hyperglycemic decompensation in experimental models of insulin-dependent diabetes in which β-cell destruction is induced by oxidative stress or inflammation (43, 77). In addition, hyperandrogenemia in women with PCOS is accompanied by systemic oxidative stress (78) and similarly, excess testosterone induces systemic oxidative stress in female mice (77). Further, in the presence of a prior β-cell injury induced by streptozotocin, female mice exposed to excess testosterone are predisposed to β-cell failure via an AR-dependent mechanism (77). Thus, excess AR activation in β-cells (and/or other tissues) may predispose to the β-cell dysfunction observed in women with androgen excess. This could be a direct islet effect since androgen exposure of isolated female pancreatic islets leads to an impaired response to glucose stimulation (77, 79). However, androgen excess without prior β-cell injury is not sufficient since testosterone infusion in healthy women does not produce β-cell dysfunction (59). Thus, in women with a prior β-cell defect, excess testosterone may predispose to β-cell failure through the cumulative action of various β-cell stresses, including insulin resistance and circulating oxidative stress.
To address the role of the β-cell AR in β-cell dysfunction in women with androgen excess, we generated mice with conditional knockout of AR in the β-cells. In female mice, chronic androgen excess induces β-cell hyperfunction, and islet failure to compensate for high fat feeding-induced insulin resistance that leads to T2D (80) similar to that observed in women with PCOS. However, androgen excess-induced T2D was not observed in female βARKO-/- mice. Thus, in women with androgen excess, predisposition to T2D is least partially mediated via excess AR activation in β-cells.
Conclusions
Clearly, there is a bidirectional modulation of glucose homeostasis by androgens in males and females. AR deficiency leads to dramatic metabolic dysfunction in aging males but in females it does not. Probably reflecting the lower concentration of testosterone and DHT in circulation and cells, AR activation is weaker in females and thus AR is less important to maintain energy homeostasis in female mammals. Still, when androgen concentrations increase to pathological levels in females, the resulting excess AR activation leads to metabolic dysfunction. Evidence presented in this review suggests that androgen deficiency in males and androgen excess in females produce metabolic dysfunction via deficient or excessive AR action, respectively, in multiple tissues including the central nervous system, liver, skeletal muscle, adipose and β-cells. In males the treatment could rely on selective androgen receptor modulators (SARMs). In the case of hyperandrogenic women, more studies are needed to define the exact role of androgen action in central nervous system, liver, skeletal muscle, adipose and β-cells to the development of T2D.
What is already known about this subject?
Androgen deficiency predisposes to metabolic syndrome and type2 diabetes in men.
Androgen excess predisposes to metabolic syndrome and type2 diabetes in women.
Androgen receptor deficient mice develop obesity and metabolic dysfunction.
What does this study add?
A dissection of the role of the androgen receptor in different tissues in males in the regulation of glucose homeostasis.
A dissection of the role of excess androgen receptor activation in different tissues in females in the pathophysiology of metabolic dysfunction.
A comparison of rodent and human studies.
Acknowledgments
We thank Loula Burton at Tulane University for editorial assistance. This work was supported by grants from the National Institutes of Health (DK074970, HD044405), the American Heart Association (11IRG5570010), the American Diabetes Association (7-13-BS-101), and the Price-Goldsmith Chair in Nutrition at Tulane University Health Sciences Center.
F.M-J received grant support and consulting fees from Pfizer.
Abbreviations
- 5αR1-KO
5α-reductase type 1 Knock-Out
- βARKO-/y
β-cell specific AR knockout
- AR
Androgen Receptor
- ARC
arcuate nucleus
- ARE
Androgen Response Elements
- ARKO
Androgen Receptor Knock-Out
- DHT
5α-dihydrotestosterone
- CREB
cAMP response element-binding protein
- DMH
Dorsomedial Hypothalamus
- E2
17β-estradiol
- ERK
Extracellular signal-regulated kinase
- GSIS
glucose-stimulated insulin secretion
- HARKO
Hepatocyte-specific ARKO
- Hsp
Heat Shock Protein
- MAPK
Mitogen-activated protein kinase
- NARKO
Neuronal specific ARKO
- PCOS
Polycystic Ovary Syndrome
- PGC-1α
Peroxisome proliferator-activated receptor-gamma coactivator 1α
- PKC
protein kinase C
- PI3K
Phosphatidylinositide 3-kinase
- POMC
Pro-opio-melano-cortin
- SHBG
Sex-Hormone Binding Globulin
- T2D
Type 2 Diabetes
- WAT
White Adipose Tissue
Footnotes
Author contributions: F.M-J conceived the manuscript. G. N., C. A., W. X. and FM-J wrote the manuscript. C. A. and FM-J revised the manuscript.
Conflicts of interest statement: Other authors declare no conflict of interest.
References
- 1.Zitzmann M. Testosterone deficiency, insulin resistance and the metabolic syndrome. Nature reviews Endocrinology. 2009;5:673–681. doi: 10.1038/nrendo.2009.212. [DOI] [PubMed] [Google Scholar]
- 2.Mauvais-Jarvis F. Estrogen and androgen receptors: regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol Metab. 2011;22:24–33. doi: 10.1016/j.tem.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keating NL, OM A, Freedland SJ, Smith MR. Diabetes and cardiovascular disease during androgen deprivation therapy: observational study of veterans with prostate cancer. J Natl Cancer Inst. 2012 doi: 10.1093/jnci/djs376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Achard C, Thiers J. Le virilisme pilaire et son association a l'insuffisance glycolytique (diabete des femmes a barbe) Bull Acad Natl Med Paris. 1921;86:51–55. [Google Scholar]
- 5.Stein J, Leventhal ML. Amenorreha associated with bilateral polycystic ovaries. Am J obstet Gynecol. 1935;20:181–187. [Google Scholar]
- 6.Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, He Y, et al. Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit. Journal of medicinal chemistry. 2009;52:3597–3617. doi: 10.1021/jm900280m. [DOI] [PubMed] [Google Scholar]
- 7.Corona G, Monami M, Rastrelli G, Aversa A, Sforza A, Lenzi A, et al. Type 2 diabetes mellitus and testosterone: a meta-analysis study. International journal of andrology. 2011;34:528–540. doi: 10.1111/j.1365-2605.2010.01117.x. [DOI] [PubMed] [Google Scholar]
- 8.Ding EL, Song Y, Malik VS, Liu S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: a systematic review and meta-analysis. Jama. 2006;295:1288–1299. doi: 10.1001/jama.295.11.1288. [DOI] [PubMed] [Google Scholar]
- 9.Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. The Journal of clinical endocrinology and metabolism. 2011;96:2341–2353. doi: 10.1210/jc.2011-0118. [DOI] [PubMed] [Google Scholar]
- 10.Grossmann M, Zajac JD. Androgen deprivation therapy in men with prostate cancer: how should the side effects be monitored and treated? Clinical endocrinology. 2011;74:289–293. doi: 10.1111/j.1365-2265.2010.03939.x. [DOI] [PubMed] [Google Scholar]
- 11.Keating NL, O'Malley AJ, Smith MR. Diabetes and cardiovascular disease during androgen deprivation therapy for prostate cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:4448–4456. doi: 10.1200/JCO.2006.06.2497. [DOI] [PubMed] [Google Scholar]
- 12.Keating NL, O'Malley A, Freedland SJ, Smith MR. Diabetes and cardiovascular disease during androgen deprivation therapy: observational study of veterans with prostate cancer. Journal of the National Cancer Institute. 2012;104:1518–1523. doi: 10.1093/jnci/djs376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inaba M, Otani Y, Nishimura K, Takaha N, Okuyama A, Koga M, et al. Marked hyperglycemia after androgen-deprivation therapy for prostate cancer and usefulness of pioglitazone for its treatment. Metabolism: clinical and experimental. 2005;54:55–59. doi: 10.1016/j.metabol.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 14.Ho CH, Yu HJ, Wang CY, Jaw FS, Hsieh JT, Liao WC, et al. Prediabetes is associated with an increased risk of testosterone deficiency, independent of obesity and metabolic syndrome. PloS one. 2013;8:e74173. doi: 10.1371/journal.pone.0074173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basaria S, Muller DC, Carducci MA, Egan J, Dobs AS. Hyperglycemia and insulin resistance in men with prostate carcinoma who receive androgen-deprivation therapy. Cancer. 2006;106:581–588. doi: 10.1002/cncr.21642. [DOI] [PubMed] [Google Scholar]
- 16.Khaw KT, Barrett-Connor E. Lower endogenous androgens predict central adiposity in men. Annals of epidemiology. 1992;2:675–682. doi: 10.1016/1047-2797(92)90012-f. [DOI] [PubMed] [Google Scholar]
- 17.Pitteloud N, Mootha VK, Dwyer AA, Hardin M, Lee H, Eriksson KF, et al. Relationship between testosterone levels, insulin sensitivity, and mitochondrial function in men. Diabetes care. 2005;28:1636–1642. doi: 10.2337/diacare.28.7.1636. [DOI] [PubMed] [Google Scholar]
- 18.Zitzmann M, Faber S, Nieschlag E. Association of specific symptoms and metabolic risks with serum testosterone in older men. The Journal of clinical endocrinology and metabolism. 2006;91:4335–4343. doi: 10.1210/jc.2006-0401. [DOI] [PubMed] [Google Scholar]
- 19.Bojesen A, Kristensen K, Birkebaek NH, Fedder J, Mosekilde L, Bennett P, et al. The metabolic syndrome is frequent in Klinefelter's syndrome and is associated with abdominal obesity and hypogonadism. Diabetes care. 2006;29:1591–1598. doi: 10.2337/dc06-0145. [DOI] [PubMed] [Google Scholar]
- 20.Moverare-Skrtic S, Venken K, Andersson N, Lindberg MK, Svensson J, Swanson C, et al. Dihydrotestosterone treatment results in obesity and altered lipid metabolism in orchidectomized mice. Obesity (Silver Spring) 2006;14:662–672. doi: 10.1038/oby.2006.75. [DOI] [PubMed] [Google Scholar]
- 21.Finkelstein JS, Lee H, Burnett-Bowie SA, Pallais JC, Yu EW, Borges LF, et al. Gonadal steroids and body composition, strength, and sexual function in men. The New England journal of medicine. 2013;369:1011–1022. doi: 10.1056/NEJMoa1206168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34:309–338. doi: 10.1210/er.2012-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zitzmann M, Gromoll J, von Eckardstein A, Nieschlag E. The CAG repeat polymorphism in the androgen receptor gene modulates body fat mass and serum concentrations of leptin and insulin in men. Diabetologia. 2003;46:31–39. doi: 10.1007/s00125-002-0980-9. [DOI] [PubMed] [Google Scholar]
- 24.Fan W, Yanase T, Nomura M, Okabe T, Goto K, Sato T, et al. Androgen receptor null male mice develop late-onset obesity caused by decreased energy expenditure and lipolytic activity but show normal insulin sensitivity with high adiponectin secretion. Diabetes. 2005;54:1000–1008. doi: 10.2337/diabetes.54.4.1000. [DOI] [PubMed] [Google Scholar]
- 25.Lin HY, Xu Q, Yeh S, Wang RS, Sparks JD, Chang C. Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes. 2005;54:1717–1725. doi: 10.2337/diabetes.54.6.1717. [DOI] [PubMed] [Google Scholar]
- 26.Lanfranco F, Zitzmann M, Simoni M, Nieschlag E. Serum adiponectin levels in hypogonadal males: influence of testosterone replacement therapy. Clinical endocrinology. 2004;60:500–507. doi: 10.1111/j.1365-2265.2004.02007.x. [DOI] [PubMed] [Google Scholar]
- 27.Nishizawa H, Shimomura I, Kishida K, Maeda N, Kuriyama H, Nagaretani H, et al. Androgens decrease plasma adiponectin, an insulin-sensitizing adipocyte-derived protein. Diabetes. 2002;51:2734–2741. doi: 10.2337/diabetes.51.9.2734. [DOI] [PubMed] [Google Scholar]
- 28.Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology. 2003;144:5081–5088. doi: 10.1210/en.2003-0741. [DOI] [PubMed] [Google Scholar]
- 29.Singh R, Artaza JN, Taylor WE, Braga M, Yuan X, Gonzalez-Cadavid NF, et al. Testosterone inhibits adipogenic differentiation in 3T3-L1 cells: nuclear translocation of androgen receptor complex with beta-catenin and T-cell factor 4 may bypass canonical Wnt signaling to down-regulate adipogenic transcription factors. Endocrinology. 2006;147:141–154. doi: 10.1210/en.2004-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gentile MA, Nantermet PV, Vogel RL, Phillips R, Holder D, Hodor P, et al. Androgen-mediated improvement of body composition and muscle function involves a novel early transcriptional program including IGF1, mechano growth factor, and induction of {beta}-catenin. J Mol Endocrinol. 2010;44:55–73. doi: 10.1677/JME-09-0048. [DOI] [PubMed] [Google Scholar]
- 31.Fernando SM, Rao P, Niel L, Chatterjee D, Stagljar M, Monks DA. Myocyte androgen receptors increase metabolic rate and improve body composition by reducing fat mass. Endocrinology. 2010;151:3125–3132. doi: 10.1210/en.2010-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu IC, Lin HY, Liu NC, Wang RS, Sparks JD, Yeh S, et al. Hyperleptinemia without obesity in male mice lacking androgen receptor in adipose tissue. Endocrinology. 2008;149:2361–2368. doi: 10.1210/en.2007-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ophoff J, Van Proeyen K, Callewaert F, De Gendt K, De Bock K, Vanden Bosch A, et al. Androgen signaling in myocytes contributes to the maintenance of muscle mass and fiber type regulation but not to muscle strength or fatigue. Endocrinology. 2009;150:3558–3566. doi: 10.1210/en.2008-1509. [DOI] [PubMed] [Google Scholar]
- 34.Holmang A, Bjorntorp P. The effects of testosterone on insulin sensitivity in male rats. Acta Physiol Scand. 1992;146:505–510. doi: 10.1111/j.1748-1716.1992.tb09452.x. [DOI] [PubMed] [Google Scholar]
- 35.Haren MT, Siddiqui AM, Armbrecht HJ, Kevorkian RT, Kim MJ, Haas MJ, et al. Testosterone modulates gene expression pathways regulating nutrient accumulation, glucose metabolism and protein turnover in mouse skeletal muscle. International journal of andrology. 2011;34:55–68. doi: 10.1111/j.1365-2605.2010.01061.x. [DOI] [PubMed] [Google Scholar]
- 36.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 37.Lin HY, Yu IC, Wang RS, Chen YT, Liu NC, Altuwaijri S, et al. Increased hepatic steatosis and insulin resistance in mice lacking hepatic androgen receptor. Hepatology. 2008;47:1924–1935. doi: 10.1002/hep.22252. [DOI] [PubMed] [Google Scholar]
- 38.Dowman JK, Hopkins LJ, Reynolds GM, Armstrong MJ, Nasiri M, Nikolaou N, et al. Loss of 5alpha-reductase type 1 accelerates the development of hepatic steatosis but protects against hepatocellular carcinoma in male mice. Endocrinology. 2013;154:4536–4547. doi: 10.1210/en.2013-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livingstone DE, Barat P, Di Rollo EM, Rees GA, Weldin BA, Rog-Zielinska EA, et al. 5alpha-Reductase type 1 deficiency or inhibition predisposes to insulin resistance, hepatic steatosis and liver fibrosis in rodents. Diabetes. 2014 doi: 10.2337/db14-0249. [DOI] [PubMed] [Google Scholar]
- 40.Volzke H, Aumann N, Krebs A, Nauck M, Steveling A, Lerch MM, et al. Hepatic steatosis is associated with low serum testosterone and high serum DHEAS levels in men. International journal of andrology. 2010;33:45–53. doi: 10.1111/j.1365-2605.2009.00953.x. [DOI] [PubMed] [Google Scholar]
- 41.Fan W, Yanase T, Nishi Y, Chiba S, Okabe T, Nomura M, et al. Functional potentiation of leptin-signal transducer and activator of transcription 3 signaling by the androgen receptor. Endocrinology. 2008;149:6028–6036. doi: 10.1210/en.2008-0431. [DOI] [PubMed] [Google Scholar]
- 42.Yu IC, Lin HY, Liu NC, Sparks JD, Yeh S, Fang LY, et al. Neuronal androgen receptor regulates insulin sensitivity via suppression of hypothalamic NF-kappaB-mediated PTP1B expression. Diabetes. 2013;62:411–423. doi: 10.2337/db12-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maclaren NK, Neufeld M, McLaughlin JV, Taylor G. Androgen sensitization of streptozotocin-induced diabetes in mice. Diabetes. 1980;29:710–716. doi: 10.2337/diab.29.9.710. [DOI] [PubMed] [Google Scholar]
- 44.Paik SG, Michelis MA, Kim YT, Shin S. Induction of insulin-dependent diabetes by streptozotocin. Inhibition by estrogens and potentiation by androgens. Diabetes. 1982;31:724–729. doi: 10.2337/diab.31.8.724. [DOI] [PubMed] [Google Scholar]
- 45.Morimoto S, Mendoza-Rodriguez CA, Hiriart M, Larrieta ME, Vital P, Cerbon MA. Protective effect of testosterone on early apoptotic damage induced by streptozotocin in rat pancreas. The Journal of endocrinology. 2005;187:217–224. doi: 10.1677/joe.1.06357. [DOI] [PubMed] [Google Scholar]
- 46.Palomar-Morales M, Morimoto S, Mendoza-Rodriguez CA, Cerbon MA. The protective effect of testosterone on streptozotocin-induced apoptosis in beta cells is sex specific. Pancreas. 2010;39:193–200. doi: 10.1097/MPA.0b013e3181c156d9. [DOI] [PubMed] [Google Scholar]
- 47.Morimoto S, C M, Alvarez-Alvarez A, Romero-Navarro G, Díaz-Sánchez V. Insulin gene expression pattern in rat pancreas during the estrous cyle. Life Sci. 2001 doi: 10.1016/s0024-3205(01)01100-6. [DOI] [PubMed] [Google Scholar]
- 48.Navarro G, Mauvais-Jarvis F. The role of the Androgen Receptor in beta-cell function in male mice. Diabetes. 2013;62(Supl. 1):A571. [Google Scholar]
- 49.Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S. Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun. 2003;300:167–171. doi: 10.1016/s0006-291x(02)02774-2. [DOI] [PubMed] [Google Scholar]
- 50.Lindstedt G, Lundberg PA, Lapidus L, Lundgren H, Bengtsson C, Bjorntorp P. Low sex-hormone-binding globulin concentration as independent risk factor for development of NIDDM. 12-yr follow-up of population study of women in Gothenburg, Sweden. Diabetes. 1991;40:123–128. doi: 10.2337/diab.40.1.123. [DOI] [PubMed] [Google Scholar]
- 51.Larsson H, Ahren B. Androgen activity as a risk factor for impaired glucose tolerance in postmenopausal women. Diabetes care. 1996;19:1399–1403. doi: 10.2337/diacare.19.12.1399. [DOI] [PubMed] [Google Scholar]
- 52.Maggio M, Lauretani F, Ceda GP, Bandinelli S, Basaria S, Paolisso G, et al. Association of hormonal dysregulation with metabolic syndrome in older women: data from the InCHIANTI study. American journal of physiology Endocrinology and metabolism. 2007;292:E353–358. doi: 10.1152/ajpendo.00339.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andersson B, Marin P, Lissner L, Vermeulen A, Bjorntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes care. 1994;17:405–411. doi: 10.2337/diacare.17.5.405. [DOI] [PubMed] [Google Scholar]
- 54.Oh JY, Barrett-Connor E, Wedick NM, Wingard DL. Endogenous sex hormones and the development of type 2 diabetes in older men and women: the Rancho Bernardo study. Diabetes care. 2002;25:55–60. doi: 10.2337/diacare.25.1.55. [DOI] [PubMed] [Google Scholar]
- 55.Tok EC, Ertunc D, Evruke C, Dilek S. The androgenic profile of women with non-insulin-dependent diabetes mellitus. The Journal of reproductive medicine. 2004;49:746–752. [PubMed] [Google Scholar]
- 56.Page-Wilson G, Goulart AC, Rexrode KM. Interrelation between sex hormones and plasma sex hormone-binding globulin and hemoglobin A1c in healthy postmenopausal women. Metabolic syndrome and related disorders. 2009;7:249–254. doi: 10.1089/met.2008.0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dreaden EC, Gryder BE, Austin LA, Tene Defo BA, Hayden SC, Pi M, et al. Antiandrogen gold nanoparticles dual-target and overcome treatment resistance in hormone-insensitive prostate cancer cells. Bioconjugate chemistry. 2012;23:1507–1512. doi: 10.1021/bc300158k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding EL, Song Y, Manson JE, Rifai N, Buring JE, Liu S. Plasma sex steroid hormones and risk of developing type 2 diabetes in women: a prospective study. Diabetologia. 2007;50:2076–2084. doi: 10.1007/s00125-007-0785-y. [DOI] [PubMed] [Google Scholar]
- 59.Diamond MP, Grainger D, Diamond MC, Sherwin RS, Defronzo RA. Effects of methyltestosterone on insulin secretion and sensitivity in women. The Journal of clinical endocrinology and metabolism. 1998;83:4420–4425. doi: 10.1210/jcem.83.12.5333. [DOI] [PubMed] [Google Scholar]
- 60.Shamma FN, Rossi G, HajHassan L, Penzias AS, Connoly-Diamond M, Jones E, et al. The effect of Norplant on glucose metabolism under hyperglycemic hyperinsulinemic conditions. Fertility and sterility. 1995;63:767–772. [PubMed] [Google Scholar]
- 61.Polderman KH, Gooren LJ, Asscheman H, Bakker A, Heine RJ. Induction of insulin resistance by androgens and estrogens. The Journal of clinical endocrinology and metabolism. 1994;79:265–271. doi: 10.1210/jcem.79.1.8027240. [DOI] [PubMed] [Google Scholar]
- 62.Holmang A, Larsson BM, Brzezinska Z, Bjorntorp P. Effects of short-term testosterone exposure on insulin sensitivity of muscles in female rats. The American journal of physiology. 1992;262:E851–855. doi: 10.1152/ajpendo.1992.262.6.E851. [DOI] [PubMed] [Google Scholar]
- 63.Holmang A, Svedberg J, Jennische E, Bjorntorp P. Effects of testosterone on muscle insulin sensitivity and morphology in female rats. The American journal of physiology. 1990;259:E555–560. doi: 10.1152/ajpendo.1990.259.4.E555. [DOI] [PubMed] [Google Scholar]
- 64.Zulian E, Sartorato P, Benedini S, Baro G, Armanini D, Mantero F, et al. Spironolactone in the treatment of polycystic ovary syndrome: effects on clinical features, insulin sensitivity and lipid profile. Journal of endocrinological investigation. 2005;28:49–53. doi: 10.1007/BF03345529. [DOI] [PubMed] [Google Scholar]
- 65.Gambineri A, Patton L, Vaccina A, Cacciari M, Morselli-Labate AM, Cavazza C, et al. Treatment with flutamide, metformin, and their combination added to a hypocaloric diet in overweight-obese women with polycystic ovary syndrome: a randomized, 12-month, placebo-controlled study. The Journal of clinical endocrinology and metabolism. 2006;91:3970–3980. doi: 10.1210/jc.2005-2250. [DOI] [PubMed] [Google Scholar]
- 66.Corbould A. Effects of androgens on insulin action in women: is androgen excess a component of female metabolic syndrome? Diabetes Metab Res Rev. 2008;24:520–532. doi: 10.1002/dmrr.872. [DOI] [PubMed] [Google Scholar]
- 67.Bouchard C, Despres JP, Mauriege P. Genetic and nongenetic determinants of regional fat distribution. Endocr Rev. 1993;14:72–93. doi: 10.1210/edrv-14-1-72. [DOI] [PubMed] [Google Scholar]
- 68.Bjorntorp P. Abdominal fat distribution and disease: an overview of epidemiological data. Ann Med. 1992;24:15–18. doi: 10.3109/07853899209164140. [DOI] [PubMed] [Google Scholar]
- 69.Evans DJ, Barth JH, Burke CW. Body fat topography in women with androgen excess. Int J Obes. 1988;12:157–162. [PubMed] [Google Scholar]
- 70.Montes-Nieto R, Insenser M, Martinez-Garcia MA, Escobar-Morreale HF. A nontargeted proteomic study of the influence of androgen excess on human visceral and subcutaneous adipose tissue proteomes. The Journal of clinical endocrinology and metabolism. 2013;98:E576–585. doi: 10.1210/jc.2012-3438. [DOI] [PubMed] [Google Scholar]
- 71.Gambineri A, Fanelli F, Tomassoni F, Munarini A, Pagotto U, Andrew R, et al. Tissue-specific dysregulation of 11beta-hydroxysteroid dehydrogenase type 1 in overweight/obese women with polycystic ovary syndrome compared with weight-matched controls. European journal of endocrinology / European Federation of Endocrine Societies. 2014;171:47–57. doi: 10.1530/EJE-13-1030. [DOI] [PubMed] [Google Scholar]
- 72.Nohara K, Laque A, Allard C, Munzberg H, Mauvais-Jarvis F. Central mechanisms of adiposity in adult female mice with androgen excess. Obesity (Silver Spring) 2014;22:1477–1484. doi: 10.1002/oby.20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O'Meara NM, Blackman JD, Ehrmann DA, Barnes RB, Jaspan JB, Rosenfield RL, et al. Defects in beta-cell function in functional ovarian hyperandrogenism. The Journal of clinical endocrinology and metabolism. 1993;76:1241–1247. doi: 10.1210/jcem.76.5.8496316. [DOI] [PubMed] [Google Scholar]
- 74.Dunaif A, Finegood DT. Beta-cell dysfunction independent of obesity and glucose intolerance in the polycystic ovary syndrome. The Journal of clinical endocrinology and metabolism. 1996;81:942–947. doi: 10.1210/jcem.81.3.8772555. [DOI] [PubMed] [Google Scholar]
- 75.Holte J, Bergh T, Berne C, Berglund L, Lithell H. Enhanced early insulin response to glucose in relation to insulin resistance in women with polycystic ovary syndrome and normal glucose tolerance. The Journal of clinical endocrinology and metabolism. 1994;78:1052–1058. doi: 10.1210/jcem.78.5.8175959. [DOI] [PubMed] [Google Scholar]
- 76.Goodarzi MO, Erickson S, Port SC, Jennrich RI, Korenman SG. beta-Cell function: a key pathological determinant in polycystic ovary syndrome. The Journal of clinical endocrinology and metabolism. 2005;90:310–315. doi: 10.1210/jc.2004-1006. [DOI] [PubMed] [Google Scholar]
- 77.Liu S, Navarro G, Mauvais-Jarvis F. Androgen excess produces systemic oxidative stress and predisposes to beta-cell failure in female mice. PloS one. 2010;5:e11302. doi: 10.1371/journal.pone.0011302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gonzalez F, Rote NS, Minium J, Kirwan JP. Increased activation of nuclear factor kappaB triggers inflammation and insulin resistance in polycystic ovary syndrome. The Journal of clinical endocrinology and metabolism. 2006;91:1508–1512. doi: 10.1210/jc.2005-2327. [DOI] [PubMed] [Google Scholar]
- 79.Roland AV, Nunemaker CS, Keller SR, Moenter SM. Prenatal androgen exposure programs metabolic dysfunction in female mice. The Journal of endocrinology. 2010;207:213–223. doi: 10.1677/JOE-10-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Navarro G, Suhuan Liu P, De Gendt K, Verhoeven G, Mauvais-Jarvis F. Importance of the beta - cell Androgen Receptor in Type 2 Diabetes. Endocr Rev. 2011;32:OR23–22. [Google Scholar]