

Abstract
Transferrin receptor-2 is a transmembrane protein whose expression is restricted to hepatocytes and erythroid cells. Transferrin receptor-2 has a regulatory function in iron homeostasis, since its inactivation causes systemic iron overload. Hepatic transferrin receptor-2 participates in iron sensing and is involved in hepcidin activation, although the mechanism remains unclear. Erythroid transferrin receptor-2 associates with and stabilizes erythropoietin receptors on the erythroblast surface and is essential to control erythrocyte production in iron deficiency. We identified a soluble form of transferrin receptor-2 in the media of transfected cells and showed that cultured human erythroid cells release an endogenous soluble form. Soluble transferrin receptor-2 originates from a cleavage of the cell surface protein, which is inhibited by diferric transferrin in a dose-dependent manner. Accordingly, the shedding of the transferrin receptor-2 variant G679A, mutated in the Arginine-Glycine-Aspartic acid motif and unable to bind diferric transferrin, is not modulated by the ligand. This observation links the process of transferrin receptor-2 removal from the plasma membrane to iron homeostasis. Soluble transferrin receptor-2 does not affect the binding of erythropoietin to erythropoietin receptor or the consequent signaling and partially inhibits hepcidin promoter activation only in vitro. Whether it is a component of the signals released by erythropoiesis in iron deficiency remains to be investigated. Our results indicate that membrane transferrin receptor-2, a sensor of circulating iron, is released from the cell membrane in iron deficiency.
Introduction
Transferrin receptor-2 (TFR2) is a type II transmembrane glycoprotein homologous to TFR1,1 able to bind diferric-transferrin (holo-TF), although with lower affinity than TFR1. In iron homeostasis TFR2 appears to have regulatory rather than transport functions, since TFR2 mutations in humans2 and Tfr2 inactivation in mice3–5 cause iron overload with low hepcidin.6 TFR2 is mainly expressed in the liver1,7 where it is essential for hepcidin control. Its expression is up-regulated during hepatic development but is not modulated by iron; indeed TFR2 mRNA has no detectable iron-responsive elements in its untranslated regions.8
TFR2 encodes an 801 amino acid protein with a short cytosolic tail that contains a potential internalization signal, a transmembrane domain and a large C-terminal ectodomain. This last has a protease-associated domain, a peptidase M28-like domain and a dimerization domain with two RGD motifs, important for protein-protein interactions and for transferrin binding. Mutations of TFR2 cause type 3 hemochromatosis: all reported mutations2 are rare, often private. Mutations cause loss of function and include frameshift, premature stop, small deletions and missense mutations prevalently affecting the protein C-terminus, especially the peptidase-like and the dimerization domains.9
Binding to holo-TF stabilizes TFR2 on the cell surface10 and this interaction redirects TFR2 towards the recycling instead of the lysosomal degradation pathway.4 Experiments in cultured hepatoma cell lines suggest that TFR2, bound to holo-TF, may simultaneously bind HFE. While the HFE- and the TF-binding sites on TFR1 overlap, in TFR2 they are different.10,11 The HFE-TFR2 complex has been proposed to act as a sensor of circulating iron and as an activator of hepcidin expression.12 However, recent data suggest that the two proteins may have separate functions and their interaction is also controversial.13 Iron overload is more severe in Tfr2−/− than in Hfe−/− mice and Tfr2-Hfe double knock-out mice show the most severe disease.14 Studies in patients indicate that TFR2 plays a prominent and HFE a minor role in hepcidin activation in response to an acute increase of transferrin saturation after a single dose of oral iron.15 As a further level of complexity, the TFR2 gene, which has two consensus sequences for the erythroid transcription factor GATA-1 in its promoter,12 is expressed in immature erythroid cells16 where it is a component of the erythropoietin receptor (EPOR) complex.17 The TFR2-EPOR association is required for the efficient transport of EPOR to the cell surface. Although neither TFR2-hemochromatosis patients nor Tfr2−/− mice have evident hematologic abnormalities, the erythroid progenitors from Tfr2−/− young mice appear less sensitive to erythropoietin (EPO) and have increased serum Epo levels. Moreover, TFR2 silencing in human erythroid progenitors delays their terminal differentiation.17
TFR1 sheds a soluble counterpart (sTFR1), in vitro and in vivo, which is measurable in serum from normal individuals.18 TFR1 is highly expressed on the surface of maturing erythroblasts and, when unbound to holo-TF, is released into plasma in high concentrations. This process occurs in iron deficiency anemia, in erythropoiesis expansion and in some hematologic malignancies.19 In contrast, sTFR1 levels are reduced in aplastic and hypoproliferative anemias. Although its function remains unexplained, sTFR1 has been proposed as a marker of iron deficiency and/or of total erythropoietic activity. The shedding of sTFR1 is regulated by its ligand holo-TF and, as recently shown, is mediated by the proprotein convertase subtilisin/kexin type 7 (PCSK7).20 Interestingly, PCSK7 genetic variants are associated with sTFR1 quantitative trait in genome-wide association studies.21
Here we characterize a previously unrecognized soluble form of TFR2 (sTFR2) that is shed from the plasma membrane both in transfected cell lines and in TFR2-expressing erythroid cells. We show that the process of TFR2 release is regulated by the ligand holo-TF, a regulation lost in a TFR2 mutant (TFR2G679A) unable to bind the ligand. Because of its low affinity for holo-TF, we suggest that the shedding of TFR2 signals iron deficiency simultaneously in hepatic and erythroid cells.
Methods
Wild-type and mutant constructs
A TFR2 wild-type cDNA was cloned in pCMV-TAG4 vector with a FLAG-tag at the C-terminus (TFR2WT-C-FLAG). TFR2 wild-type (TFR2WT-N-FLAG) and TFR2G679A constructs in pcDNA3.1(+) have a FLAG-tag at the N-terminus. A “soluble” artificial isoform of TFR2 (sTFR2*) was generated by amplification of the coding region from nucleotide 313 (Arg105) to nucleotide 2403 (Phe801) and in-frame-cloned in pSEC-TAG (B), with a MYC-HIS-tag at the C-terminus, using the following oligonucleotides:
sTFR2-KpnI-fw: 5′-GGGGTACCCGAGGGTCCTGCCAGGCG-3′
sTFR2-EcoRI-rev: 5′-GGAATTCCAGAAGTTGTTATCAAT-GTTCC-3′
Mammalian expressing vectors encoding for wild-type dynamin (DynWT) and the dominant negative variant DynK44A are described elsewhere.22 Hemojuvelin (HJV)-expressing vector and the hepcidin promoter luciferase reporter construct (Hep-Luc) were as described previously.23 FURIN and PCSK7 expressing vectors are described in the Online Supplementary Methods.
Cell culture and reagents
HeLa, HEK293, CHO-Trvb-0, the erythroleukemic cell line UT7 and the hepatoma cell lines HuH7 and Hep3B, culture conditions and reagents are described in the Online Supplementary Methods.
CD34+ cells were obtained from human donors who gave informed consent in accordance with the principles of the Declaration of Helsinki. Approval for their use was obtained from the La Pitié-Salpétrière Hospital Institutional Ethic Committee. CD34+cells were purified from the peripheral blood after cytapheresis. CD34+ and CD36+ cells were isolated by positive selection as described in the Online Supplementary Methods. CD36+ cells were cultured in the presence of 2 U/mL EPO, 100 ng/mL stem cell factor and 10 ng/mL interleukin-3 for up to 12 days for erythroid differentiation.
When indicated, cells were treated with different concentrations (0.5, 1.25, 5 and 25 μM) of human or bovine diferric-transferrin (H-holo-TF and B-holo-TF, respectively) and human apo-transferrin (apo-TF) (Sigma-Aldrich, St. Louis, MO, USA) in serum-free media; erythroid cells were treated with 25 μM of human apo-TF or holo-TF.
Brefeldin-A (Sigma-Aldrich), a fungal antibiotic that alters intracellular trafficking by blocking protein maturation across the post-endoplasmic reticulum compartment, was used at a concentration of 100–500 ng/mL.
Procedures for pull-down of membrane and soluble TFR2, western blot analysis of conditioned cell culture media and total cell lysates, and the luciferase reporter assay for the effect of sTFR2 on the hepcidin promoter are described in the Online Supplementary Methods.
Scatchard analysis
UT-7 cells were suspended in binding medium (alpha-MEM containing 20 mM Hepes, 10% fetal calf serum, 0.1% sodium azide, pH 7.2). Twenty-five microliters of the cell suspension, containing 4×106 cells, were incubated with 25 μL of conditioned medium from HEK293 cells transfected with the empty or sTFR2* expressing vectors. Samples were equilibrated at 37°C for 60 min. Twenty-five microliters of binding medium alone or 25 μL of a 100-fold excess of unlabeled EPO were added to the binding assay. Then the chosen amount of 125I-EPO was added in a volume of 25 μL and the cells were incubated for a further 30 min at 37°C. The content of each well was carefully layered over 900 μL of a cold solution of phosphate-buffered saline and centrifuged at 4°C for 10 min at 600 g. Unbound radioactivity was counted and after three washes in phosphate-buffered saline, cell-bound radioactivity was counted in the pellet. The non-specific binding of 125I-EPO, determined by adding a 100-fold excess of unlabeled EPO to the binding assay, was subtracted from the total binding to give the specific binding.
Two independent experiments were performed using two different batches of 125I-EPO and control conditioned medium or sTFR2*-containing conditioned medium from two independent experiments.
Results
Identification of a soluble form of transferrin receptor-2
To study the cell localization and processing of TFR2, HeLa cells, which do not express endogenous TFR2 (data not shown), were transiently transfected with an expressing vector that encodes for a N-FLAG tagged form of TFR2 (TFR2WT−N-FLAG). Since TFR1 and other iron-related proteins present on the cell membrane release soluble forms, we investigated whether this would also occur with TFR2. In the media of TFR2 transfected cells we identified a single band (sTFR2) of about 80 kDa, which migrated faster than the cell-associated isoforms, suggesting that it was the result of a cleavage mechanism. Since sTFR2 can be detected using an anti-TFR2 antibody, recognizing residues from amino acid 150 to 250, but not with the anti-FLAG antibody when using an N-terminal flagged construct, we concluded that the cleavage occurs at the N-terminal part of the protein, close to the transmembrane domain, as occurs for sTFR1.24 To confirm this finding, sTFR2 from cells transfected with TFR2WT-C-FLAG, which has a FLAG-tag at the C-terminus, was indeed detected with the anti-FLAG antibody (Figure 1A). The size of sTFR2 is compatible with that of a protein lacking intracellular and transmembrane regions. Indeed the artificial form of sTFR2 (sTFR2*), generated by cloning the TFR2 ectodomain (amino acids 105-801) downstream of the Ig k-chain leader sequence, migrates as the isoform shed from the full length TFR2 transfected cells (Figure 1A).
Figure 1.
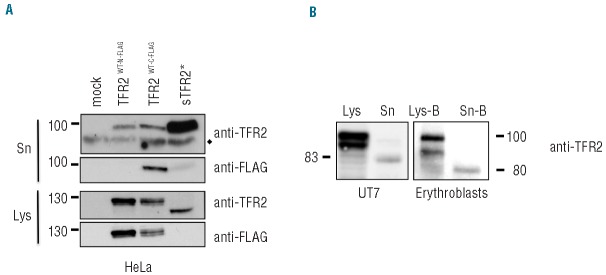
Identification of a soluble form of TFR2 (sTFR2). (A) HeLa cells were transiently transfected with empty vector (mock), wild-type TFR2 FLAG-tagged at the N-terminus (TFR2WT-N-FLAG), wild-type TFR2 FLAG-tagged at the C-terminus (TFR2WT-C-FLAG) and the artificial sTFR2 (sTFR2*) expressing vectors. Media were replaced 18 h after transfection with serum-free media. Twenty-four hours later media were collected and concentrated and cells were lysed. Fifty micrograms of proteins from supernatants and cellular lysates were analyzed by western blot, using anti-TFR2 (Abcam) and anti-FLAG antibodies. ♦ = non-specific bands. (B) TFR2 from cell lysates and sTFR2 from supernatant were detected by western blot with an antibody recognizing the C-terminal part of TFR2 (Santa Cruz). Endogenous expression of TFR2 and sTFR2 is shown in UT7 cells (left) and in primary erythroblasts (right). Because human primary erythroblasts cannot survive in albumin-free medium, a cell surface biotinylation technique was used to purify sTFR2 from the supernatant. Lys: total cell lysate, Lys-B: cell lysate pull-down with neutravidin agarose beads, Sn: concentrated media, Sn-B: concentrated media pull-down with neutravidin agarose beads. Scales refer to relative molecular mass in kilo Daltons.
sTFR2 is not an artefact due to cDNA overexpression since it is also released by TFR2-expressing cells, such as the erythroleukemic cell line UT7 and human primary erythroblasts. Both UT7 cells and primary erythroblasts release a band of sTFR2 of approximately 80 kDa in culture media (Figure 1B), compatible with the size of sTFR2 released by TFR2-transfected cells. In the total lysates of erythroid cells, TFR2 migrates as a doublet of about 102 and 92 kDa, as previously observed.17
Soluble transferrin receptor-2 is shed from the plasma membrane
It was previously reported that the receptor exposed on the cell surface is stabilized in the presence of holo-TF with a maximum effect at 25 μM holo-TF.10 We confirmed the same stabilization both in transfected cells and in TFR2-competent cells. HeLa cells transfected with TFR2WT-C-FLAG construct were treated with human holo-TF and apo-TF (25 μM) in serum-free media and were biotinylated to label membrane proteins. sTFR2 was pulled-down using anti-FLAG Sepharose-beads to avoid holo-TF interference with gel migration of concentrated culture media. In these conditions, as a consequence of holo-TF treatment, we observed a reduction of sTFR2, mirrored by an increase in membrane-exposed TFR2 (Figure 2A).
Figure 2.

sTFR2 is cleaved from the plasma membrane. (A) HeLa cells were transiently transfected with empty vector (mock) and TFR2WT-C-FLAG coding vector. Eighteen hours after transfection the media were replaced with serum-free media to which human holo-transferrin (H-holo-TF, 25 μM) or human apo-transferrin (H-apo-TF, 25 μM) was added or not. After 24 h media were collected and concentrated, cells were biotinylated to label membrane protein then lysed. sTFR2 was pulled down using anti-FLAG Sepharose-beads. Cellular lysates were precipitated with streptavidin to analyze proteins originating from the cell surface. Total lysates (Lys), biotinylated samples (Lys-B) and concentrated media (Sn) were analyzed by western blot using anti-TFR2 (Abcam), anti-FLAG antibody. Anti-pan-cadherin, which recognizes a plasma membrane protein, was used to normalize plasma membrane biotinylation and streptavidin pull-down. (B) An erythroid cell line UT7 (left panel) and erythro blasts (right panel) were incubated for 24 h in the presence of 25 μM human apo-TF or holo-TF. After biotinylation of cell surface proteins, cell lysates (Lys-B) and supernatant (Sn-B) were precipitated using neutravidin agarose beads. Western blots were performed using an anti-TFR2 antibody (Santa Cruz). Scales refer to relative molecular mass in kilo Daltons. (C) HeLa cells transiently transfected with empty vector (mock) and wild-type TFR2 coding vector (TFR2WT-N-FLAG) were treated with brefeldin-A (BFA, 100 ng/mL) to block intracellular trafficking and protein export to the plasma membrane. After 18 h cells were incubated with biotin to label membrane proteins and than re-incubated with BFA. Twenty-four hours later media were collected and concentrated, cells lysed (Lys) and both media (Sn-B) and lysates (Lys-B) were subjected to streptavidin pull-down and western blot analysis using an anti-TFR2 antibody (Abcam). (D) The cell culture media and the total lysates of HeLa cells, transiently transfected with wild type TFR2 (TFR2WT-N-FLAG) in combination with DynWT and DynK44A expressing vectors, were analyzed by western blot using anti-TFR2 (Abcam). (E) HeLa cells were transiently transfected with wild-type TFR2 (TFR2WT-N-FLAG) and DynK44A expressing vectors. After 18 h cells were incubated with biotin to label membrane proteins and were incubated in serum-free media, treated or not with H-holo-TF (0.5 μM). After 24 h media were collected and concentrated and cells were lysed. Both media (Sn-B) and lysates (Lys-B) were precipitated with streptavidin to analyze by western blot proteins originating from the cell surface using an anti-TFR2 antibody (Abcam). Loading was estimated with anti-β-actin antibody. The results were confirmed several times in different experiments. Western blots of representative experiments are shown.
A substantial increase in the expression of membrane TFR2 was also detected in biotinylated plasma membranes from erythroid UT7 cells and primary erythroblasts when cultured in the presence of holo-TF, while endogenous sTFR2 production was decreased (Figure 2B). This confirms that the inverse regulation of membrane and soluble TFR2 in the presence of the ligand is also maintained in cells expressing endogenous TFR2 (Figure 2A).
We asked whether sTFR2 originates from the plasma membrane or is released during receptor internalization. To this aim cell surface proteins were labeled using a cell-impermeable biotin and were treated with holo-TF. sTFR2 was detected in the culture media of transfected cells after streptavidin pull-down of biotin-labeled proteins, demonstrating that it originates from membrane-exposed TFR2 (Online Supplementary Figure S1B).
To further prove the membrane origin of sTFR2, we treated HeLa cells with brefeldin-A, a fungal antibiotic that alters intracellular trafficking by blocking protein maturation across the post-endoplasmic reticulum compartment.25 Brefeldin-A induced a strong reduction of both membrane-associated and soluble TFR2 (Figure 2C), consistent with the finding that TFR2 is shed from the cell surface.
To investigate the potential contribution of endocytosis to the release of sTFR2, we exploited the effect of dynamin dead-mutant K44A (DynK44A), which blocks the endocytosis process, thus “fixing” the proteins on the cell surface.26 The amount of sTFR2 in cells transfected with DynK44A was strongly increased compared to that of cells transfected with wild-type dynamin (DynWT), further strengthening the belief that the cleavage occurs at the cell surface (Figure 2D). Under these conditions, holo-TF was still able to reduce the shedding process (Figure 2E).
Transferrin receptor-2 shedding is inhibited by holo-transferrin in a dose-dependent manner
TFR2 can bind holo-TF and is highly homologous to TFR1 and the release of sTFR1 is reduced by its ligand.27 For these reasons we investigated whether sTFR2 release was affected by holo-TF in our in vitro system. HuH7 (Figure 3A) and HeLa (Online Supplementary Figure S1B,C) cells were transiently transfected with the TFR2-expressing vector and then treated with increasing concentrations of human holo- TF. To avoid holo-TF interference with gel migration of concentrated culture media, we biotinylated cell surface proteins and pulled-down sTFR2 in the supernatants using streptavidin beads. As shown in Figure 3A, human holo-TF inhibited the release of sTFR2 in a dose-dependent manner. This inhibition was not mediated by iron, since treatment of TFR2-transfected cells with iron donors (ferric ammonium citrate) or iron chelators (deferoxamine) did not modulate sTFR2 concentration in the culture media (Online Supplementary Figure S1A).
Figure 3.

Modulation of sTFR2 release by holo-transferrin. (A) HuH7 cells were transiently transfected with empty vector (mock) and wild-type TFR2WT-N-FLAG coding vector. Eighteen hours after transfection cells were biotinylated to label membrane protein, incubated in serum-free media and treated with increasing concentrations (0.5, 1.25, 5 and 25 μM) of human diferric-transferrin (H-holo-TF) or human apotransferrin 25 μM (H-apo-TF) for 24 h. Cells were then lysed and the media concentrated. The supernatant (Sn-B) was precipitated with streptavidin to analyze, by western blot, proteins originating from the cell surface. Western blots were performed using an anti-TFR2 antibody (Abcam). A densitometric analysis of sTFR2 vs. total TFR2 (expressed as % sTFR2) was performed in three independent experiments. The differences of the Holo-TF treatments vs. untreated conditions are statistically significant, **P<0.005. (B) HeLa cells were transiently transfected with empty vector (mock), TFR2WT-N-FLAG and TFR2G679A coding vectors. Eighteen hours after transfection cells were biotinylated to label membrane protein, were incubated in serum-free media and treated or not with H-holo-TF (5 μM). After 24 h cells were lysed, media were collected, concentrated and precipitated with streptavidin. Biotinylated media (Sn-B) were analyzed by western blot using an anti-TFR2 antibody (Abcam) to detect the proteins deriving from the cell surface. Lys: total lysates. Loading was estimated with anti-β-actin antibody. The results were confirmed several times in different experiments and representative western blots are shown.
To understand whether the observed inhibition was a direct effect of the ligand binding, we used TFR2G679A, an artificial variant unable to bind holo-TF since the mutation inactivates the RGD binding domain.28 As shown in Figure 3B, TFR2G679A released sTFR2 in high concentrations and the process was not modulated by the ligand.
Shedding of transferrin receptor-2 occurs independently of transferrin receptor-1
To exclude that sTFR2 release might be influenced by binding of holo-TF to its high affinity receptor TFR1 we used two approaches. First, we used bovine holo-TF, which specifically binds TFR2 but is unable to bind human TFR1.28 As with human holo-TF, sTFR2 was strongly decreased in the presence of bovine holo-TF both in HeLa cells (Figure 4A) and in HuH7 cells (data not shown). Second, we analyzed the shedding process in TFR2-transfected CHO-Trvb-0 cells that lack TFR1.29 We confirmed that these cells did not express TFR1 (Online Supplementary Figure S2) and we showed that also in these cells holo-TF inhibited the release of sTFR2 and stabilized TFR2 on the cell surface (Figure 4B).
Figure 4.
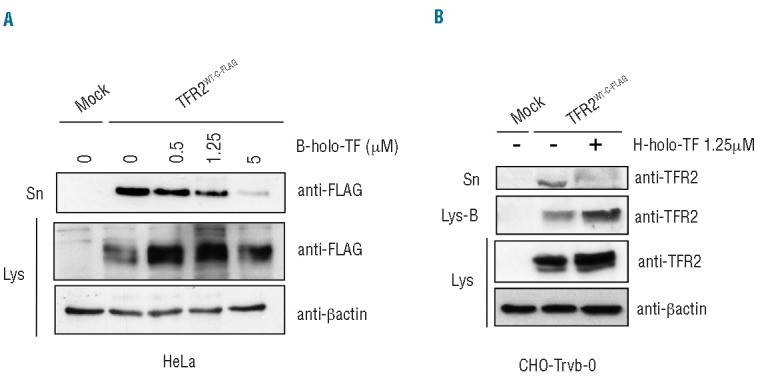
TFR2 shedding is independent of TFR1. (A) HeLa cells were transiently transfected with empty vector (mock) or TFR2WT-C-FLAG vector. Eighteen hours after transfection media were replaced with serum-free media and cells were treated or not for 24 h with increasing concentrations (0.5, 1.25, 5 μM) of bovine holo-transferrin (B-holo-TF), a ligand specific for TFR2 that does not bind TFR1. Media were collected, concentrated and sTFR2 was pulled down using Sepharose-beads anti-FLAG. Immunoprecipitated proteins were analyzed by western blot using the anti-FLAG antibody. The results were confirmed in three independent experiments and western blot of a representative one is shown. (B) CHO-Trvb-0 (TFR1 deficient) cells were transiently transfected with empty vector (mock) and TFR2WT-C-FLAG coding vector. Media were replaced 18 h after transfection with serum-free media containing or not 1.25 μM of human holo-TF (H-holo-TF), and surface proteins were biotinylated. Media were collected, concentrated and analyzed by western blot using anti-TFR2 antibody (Abcam). Sn= concentrated media; Lys= total cell lysate, Lys-B = cell lysate pull down with streptavidin-agarose beads. Loading was estimated with anti-β-actin antibody.
Overall our results demonstrate that the shedding of TFR2 does not require TFR1 and that the inhibition of sTFR2 release results directly from ligand-receptor binding.
Soluble transferrin receptor-2 does not affect erythropoietin receptor expression or erythropoietin binding in erythroid cells and mildly reduces hepcidin promoter activation in hepatoma cells
Because the TFR2-EPOR association is required for efficient transport of EPOR to the cell surface,17 we studied whether sTFR2 could modify the number of cell surface EPOR or the affinity of EPO for its receptor by Scatchard analysis in UT7 cells. To investigate this issue, we used sTFR2*, which is functional (see below) and more efficiently produced than the naturally cleaved TFR2 (Figure 1A). The conditioned medium of HEK293 cells transfected with sTFR2* (Sn sTFR2*) or with empty vector (Sn mock) did not significantly modify the number of EPOR expressed on the cell surface or the affinity for its ligand (Figure 5A). However, brefeldin-A treatment decreased the number of EPOR on the plasma membrane (Online Supplementary Figure S3). To investigate the activation of EPO-mediated intracellular signaling, the phosphorylation status of STAT5, AKT and ERK, major targets of this pathway, was assessed in UT7 cells treated or not with sTFR2*. As illustrated in Figure 5B and quantified in Figure 5C, EPO signaling was not significantly affected by sTFR2*. These data were supported by the observation that exogenous sTFR2* did not interact with EPOR when added to the culture medium (Online Supplementary Figure S4).
Figure 5.

Effect of sTFR2 in erythroid cells. (A) Scatchard analysis of 125I erythropoietin (EPO) binding to UT7 cells in the presence of conditioned media from sTFR2* (Sn sTFR2*) or empty vectors (Sn mock) transfected HEK293 cells. (B) Growth factor-deprived UT-7 cells were pre-incubated for 1 h at 37°C with concentrated media from HEK293 cells transfected with empty vector (Sn mock) or sTFR2* (Sn sTFR2*). The cells were then stimulated with 10 U/mL EPO for 10 min. Media were concentrated and increasing concentrations of media were added to the UT7 cells. Cellular extracts were analyzed by western blot with anti-p-STAT5, anti-p-AKT and anti-p-ERK antibodies. Anti-STAT5, anti-AKT, anti-ERK were used as loading controls. One representative western blot is shown. ♦= non-specific band. (C) Western blot quantification with anti-β-actin antibody: values are the means of three independent experiments ±SD.
To evaluate the functional role of sTFR2 in the hepatic compartment, we studied the modulation of the hepcidin promoter in HuH7 cells treated with the conditioned medium of the same hepatoma-derived cells transfected with TFR2WT-N-FLAG. The supernatant of TFR2-transfected cells (Sn TFR2) mildly reduced hepcidin activation both in basal conditions and in the presence of the BMP co-receptor HJV (Online Supplementary Figure S5A). The artificial sTFR2* showed a similar activity when transfected in HuH7 and Hep3B cells, decreasing HJV-dependent hepcidin activation in a dose-dependent manner (Online Supplementary Figure S5B,C).
To analyze whether sTFR2 could interfere with BMP signaling we studied the response of the hepcidin promoter in Hep3B cells in the presence of both sTFR2* and BMP6 and we confirmed the same partial inhibitory effect observed both in basal and in HJV-mediated hepcidin activation (Online Supplementary Figure S5D). However, an effect of sTFR2 on the hepcidin promoter assay was observed only in vitro.
Discussion
From its discovery more than 10 years ago1 and its identification as the gene responsible for hemochromatosis type 3,2 the function of the second transferrin receptor (TFR2) has remained obscure. The role of TFR2 as a component of the liver iron-sensing machinery and hepcidin activator is well established, but the molecular mechanisms remain unclear. The role of TFR2 as a partner of EPOR in immature erythroid cells has been clearly defined, and our recent results have shown that it negatively controls erythropoiesis expansion in iron deficiency.30
In this study we have expanded our knowledge on the structure of TFR2, identifying its soluble form (sTFR2). sTFR2 was detected in the media of different cell lines transfected with TFR2 and most importantly was present in the culture media of both erythroid cell lines and primary human erythroid cells that endogenously express TFR2.16,17
We showed that sTFR2 is shed from the membrane-exposed receptor. This conclusion is supported by two lines of evidence: (i) the lack of sTFR2 shedding when the receptor does not reach the cell surface, as after brefeldin-A treatment; (ii) the increase of sTFR2 release in the opposite condition, when the receptor is stabilized on the membrane by a mutant dynamin.
TFR2 associates with different partners (EPOR in erythroid cells, HJV and HFE in the liver) and likely has different functions in the two tissues in which it is highly expressed. However, the shedding mechanism appears to be under a common iron-mediated regulation. Indeed binding of the holo-TF ligand to the receptor inhibits sTFR2 release, while in the absence of ligand binding the release is increased in both hepatoma and erythroid cells, providing a common iron sensing mechanism.
Holo-TF binds to the RGD motif in the TFR2 ectodomain. Importantly, the RGD mutant TFR2G679A, unable to bind the ligand,28 is insensitive to the shedding regulation. This phenomenon clearly indicates that the shedding process is directly controlled by the receptor-ligand molecular interaction and, as previously reported for TFR1, couples the TFR2 shedding process with iron deficiency. However, the release of TFR2 does not require TFR1. It is reasonable that the shedding of the two receptors occurs separately, since their expression is maximal at different stages of maturation of the erythroid cells, TFR2 being expressed in more immature erythroid precursors than TFR1.16,17 Also in the liver, Tfr2 expression increases during mouse development in parallel with the decrease of Tfr1 expression.8 Furthermore, the two receptors are likely cleaved by different proteases. The consensus cleavage site for PCSK7, the proconvertase that cleaves TFR1, is not conserved in TFR2 (Online Supplementary Figure S6). The observed inhibition of TFR2 shedding by the proconvertase inhibitor decanoyl-Arg-Val-Lys-Arg-chloromethylketone (CMK) indicates that a furin-like proconvertase operates the cleavage. However, both furin and PCSK7, the proteases involved in sHJV31 and sTFR120 cleavage are not involved in TFR2 shedding (Online Supplementary Figure S6). Overall, our results indicate that removal of TFR2 from the cell surface is not only due to lack of receptor stabilization, but is dependent on an active cleavage process. It is conceivable that membrane stabilization of TFR2 by holo-TF subtracts the receptor from the protease activity by changing its conformation or by hiding the cleavage site. The definition of these molecular mechanisms requires further studies. We cannot exclude that, as previously reported,4 decreased endocytosis and TFR2 lysosomal degradation might also occur in addition to the reduced shedding in the presence of the ligand.
Different proteins and receptors acquire distinct functions when present either as membrane-bound or soluble forms, as shown for HJV.31–33 We cannot exclude a dual and opposite role also in the case of TFR2. Sensing the increased concentration of the ligand, membrane TFR2 is proposed to be involved in hepcidin activation to reduce the levels of circulating iron.12 Its soluble counterpart might act as an inhibitor to attenuate the signaling pathway, in conditions of reduced iron availability, to increase iron absorption. We consistently showed a modest inhibition of hepcidin promoter activation by sTFR2, but not of endogenous hepcidin in hepatic cell lines (data not shown). In addition, when iron deficiency is induced in double knock-out Tfr2−/−, Tmprss6−/− mice, hepcidin is inhibited, ruling out a significant contribution of sTfr2, which cannot be produced in these animals.34 Thus a decoy function on hepcidin activation is unlikely. However, irrespective of sTFR2 function, removal of membrane TFR2 in iron deficiency would negatively affect hepcidin activation.
An erythroid role for Tfr2 in conditions of low iron has recently been suggested by the observation that iron deficiency caused by ablation of the hepcidin inhibitor Tmprss6 induces erythrocytosis in mice knock-out for Tfr2, but not in mice with liver-specific Tfr2 deletion (which preserves the Tfr2 erythroid function.34 This event was directly confirmed by the increased erythropoiesis observed in mice that had Tfr2 exclusively deleted in the bone marrow.30
sTFR2 added to the culture media of transfected cells does not seem to modify the affinity of EPO for EPOR or the cell surface expression of EPOR. Consequently, EPO-mediated signaling appears not to be affected. Preliminary experiments show that the addition of sTFR2 to the culture medium of primary human erythroblasts does not affect their proliferation, survival or terminal differentiation (data not shown). However, irrespective of sTFR2 function, shedding membrane TFR2 in iron deficiency might serve to decrease the receptor’s interaction with EPOR and to increase EPO sensitivity of erythroid cells, as recently shown in mice with bone marrow-specific Tfr2 deletion.30 The lower affinity for holo-TF as compared with TFR1 makes TFR2 an ideal sensor of iron deficiency in the bone marrow which could anticipate the iron needs of maturing erythroblasts. It is possible that when holo-TF concentration starts decreasing, TFR2 would be shed from the membrane, whereas TFR1, because of its higher affinity, would still be able to bind holo-TF.
In conclusion we provide in vitro evidence of TFR2 shedding, a mechanism regulated by holo-TF, in analogy with the process described for TFR1. Although the in vivo role of sTFR2, as well as that of sTFR1, remains to be clarified, we suggest that while membrane TFR2 in the liver senses excess iron, TFR2 in the bone marrow has the opposite role of sensing iron-deficiency. This would explain why the effect of lack of TFR2 shedding is not evident in TFR2-hemochromatosis in which iron-deficiency is never achieved.6,15
Acknowledgments
We are indebted to Hiroshi Kawabata (Department of Hematology/Oncology, Kyoto University, Japan) and H. Phillip Koeffler (UCLA/Cedars-Sinai Medical Center, USA) for the kind gift of TFR2WT-N-FLAG and TFR2G679A constructs and to Caroline Enns (Oregon Health and Science University, Portland, Oregon, USA) for CHO-Trvb-0 cells. We thank Martina Muckenthaler (University of Heidelberg, Germany) for providing us with the HuH7 cell line and Jerry Kaplan (University of Utah, Salt Lake City, Utah, USA) for wild type and mutant Dynamin constructs. We are extremely grateful to Dr Nabih Azar (la Pitié Salpétrière Hospital, Paris, France) for providing the cytapheresis samples.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported in part by funds from “Telethon Onlus Foundation” (grant GGP12025), and Ricerca Finalizzata RF-2010-2312048, Ministero Sanità, MIUR-PRIN (Progetto di Rilevante Interesse Nazionale) 2010–2011 to CC and from the Agence National de la Recherche (ANR, ERYFER project) to PM. MV was funded by The Société Française d’Hématologie and the Fondation pour la Recherche Médicale.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Kawabata H, Yang R, Hirama T, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274(30):20826–20832. [DOI] [PubMed] [Google Scholar]
- 2.Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25(1):14–15. [DOI] [PubMed] [Google Scholar]
- 3.Wallace DF, Summerville L, Lusby PE, Subramaniam VN. First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut. 2005;54(7):980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson MB, Chen J, Murchison N, Green FA, Enns CA. Transferrin receptor 2: evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol Biol Cell. 2007;18(3):743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roetto A, Di Cunto F, Pellegrino RM, et al. Comparison of 3 Tfr2-deficient murine models suggests distinct functions for Tfr2-alpha and Tfr2-beta isoforms in different tissues. Blood. 2010;115(16):3382–3389. [DOI] [PubMed] [Google Scholar]
- 6.Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105(4):1803–1806. [DOI] [PubMed] [Google Scholar]
- 7.Fleming RE, Migas MC, Holden CC, et al. Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc Natl Acad Sci USA. 2000;97(5):2214–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawabata H, Germain RS, Ikezoe T, et al. Regulation of expression of murine transferrin receptor 2. Blood. 2001;98(6):1949–1954. [DOI] [PubMed] [Google Scholar]
- 9.Camaschella C, Roetto A. TFR2-related hereditary hemochromatosis. GeneReviews (Internet). 2011. update 2011. June 09. [Google Scholar]
- 10.Johnson MB, Enns CA. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood. 2004;104(13):4287–4293. [DOI] [PubMed] [Google Scholar]
- 11.D’Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J Hepatol. 2012;57(5):1052–1060. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Enns CA. Hereditary hemochromatosis and transferrin receptor 2. Biochim Biophys Acta. 2012;1820(3):256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rishi G, Crampton EM, Wallace DF, Subramaniam VN. In situ proximity ligation assays indicate that hemochromatosis proteins Hfe and transferrin receptor 2 (Tfr2) do not interact. PLoS One. 2013;8(10):e77267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace DF, Summerville L, Crampton EM, Frazer DM, Anderson GJ, Subramaniam VN. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology. 2009;50(6):1992–2000. [DOI] [PubMed] [Google Scholar]
- 15.Girelli D, Trombini P, Busti F, et al. A time course of hepcidin response to iron challenge in patients with HFE and TFR2 hemochromatosis. Haematologica. 2011;96(4):500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood. 2001;98(9):2714–2719. [DOI] [PubMed] [Google Scholar]
- 17.Forejtnikova H, Vieillevoye M, Zermati Y, et al. Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood. 2010;116(24):5357–5367. [DOI] [PubMed] [Google Scholar]
- 18.Beguin Y. Soluble transferrin receptor for the evaluation of erythropoiesis and iron status. Clin Chim Acta. 2003;329(1–2):9–22. [DOI] [PubMed] [Google Scholar]
- 19.Skikne BS. Serum transferrin receptor. Am J Hematol. 2008;83(11):872–875. [DOI] [PubMed] [Google Scholar]
- 20.Guillemot J, Canuel M, Essalmani R, Prat A, Seidah NG. Implication of the proprotein convertases in iron homeostasis: proprotein convertase 7 sheds human transferrin receptor 1 and furin activates hepcidin. Hepatology. 2013;57(6):2514–2524. [DOI] [PubMed] [Google Scholar]
- 21.Oexle K, Ried JS, Hicks AA, et al. Novel association to the proprotein convertase PCSK7 gene locus revealed by analysing soluble transferrin receptor (sTfR) levels. Hum Mol Genet. 2011;20(5):1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Domenico I, Ward DM, Langelier C, et al. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell. 2007;18(7):2569–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pagani A, Silvestri L, Nai A, Camaschella C. Hemojuvelin N-terminal mutants reach the plasma membrane but do not activate the hepcidin response. Haematologica. 2008;93(10):1466–1472. [DOI] [PubMed] [Google Scholar]
- 24.Shih YJ, Baynes RD, Hudson BG, Flowers CH, Skikne BS, Cook JD. Serum transferrin receptor is a truncated form of tissue receptor. J Biol Chem. 1990;265(31):19077–19081. [PubMed] [Google Scholar]
- 25.Traub LM, Kornfeld S. The trans-Golgi network: a late secretory sorting station. Curr Opin Cell Biol. 1997;9(4):527–33. [DOI] [PubMed] [Google Scholar]
- 26.Altschuler Y, Barbas SM, Terlecky LJ, et al. Redundant and distinct functions for dynamin-1 and dynamin-2 isoforms. J Cell Biol. 1998;143(7):1871–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dassler K, Zydek M, Wandzik K, Kaup M, Fuchs H. Release of the soluble transferrin receptor is directly regulated by binding of its ligand ferritransferrin. J Biol Chem. 2006;281(6):3297–3304. [DOI] [PubMed] [Google Scholar]
- 28.Kawabata H, Tong X, Kawanami T, et al. Analyses for binding of the transferrin family of proteins to the transferrin receptor 2. Br J Haematol. 2004;127(4):464–473. [DOI] [PubMed] [Google Scholar]
- 29.Kawabata H, Germain RS, Vuong PT, Nakamaki T, Said JW, Koeffler HP. Transferrin receptor 2-alpha supports cell growth both in iron-chelated cultured cells and in vivo. J Biol Chem. 2000;275(22):16618–16625. [DOI] [PubMed] [Google Scholar]
- 30.Nai A, Lidonnici MR, Rausa M, et al. The second transferrin receptor regulates red blood cell production in mice. Blood. 2015;125(7):1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silvestri L, Pagani A, Camaschella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood. 2008;111(2):924–931. [DOI] [PubMed] [Google Scholar]
- 32.Babitt JL, Huang FW, Wrighting DM, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38(5):531–539. [DOI] [PubMed] [Google Scholar]
- 33.Lin L, Nemeth E, Goodnough JB, Thapa DR, Gabayan V, Ganz T. Soluble hemojuvelin is released by proprotein convertase-mediated cleavage at a conserved polybasic RNRR site. Blood Cells Mol Dis. 2008;40(1):122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nai A, Pellegrino RM, Rausa M, et al. The erythroid function of transferrin receptor 2 revealed by Tmprss6 inactivation in different models of transferrin receptor 2 knockout mice. Haematologica. 2014;99(6):1016–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]