

Follicular lymphoma in situ (FLIS) is composed of a clonal B-cell population harboring the typical t(14;18) hallmark of follicular lymphoma (FL), forming unconventional BCL2BrightCD10+ cell foci in an otherwise normal reactive lymph node (LN). The diagnosis of FLIS is made on the fortuitous discovery of unconventional BCL2BrightCD10+ cell foci.1 Several studies recently demonstrated that FLIS are already advanced precursors in follicular lymphomagenesis, but not necessarily committed to malignant transformation.2,3 However, the relationship between FLIS and FL still remains unclear, as only a minority (<5%) of FLIS patients eventually develop FL. This is in line with the usually indolent progression of the disease, and the genomic instability observed in FLIS cells, which can engage FL precursor cells either in an evolutionary malignant process, or to an evolutionary dead end.4
We report the case of a 35-year old male patient who presented with a cervical adenopathy. Histological examination of the excised LN displayed an altered architecture suggestive of FL, consisting of high number of monomorphic large follicles, uniformly spread in the cortical and medullary areas. Most follicles contained a predominant population of small cleaved cells with scant macrophages and mitoses. The mantle zone was reduced or absent. However, in a minor cortical area, a few follicles showed features mimicking residual classical germ cells (GC), including a smaller size, higher cell polymorphism, and a preserved mantle zone (Figure 1A).
Figure 1.
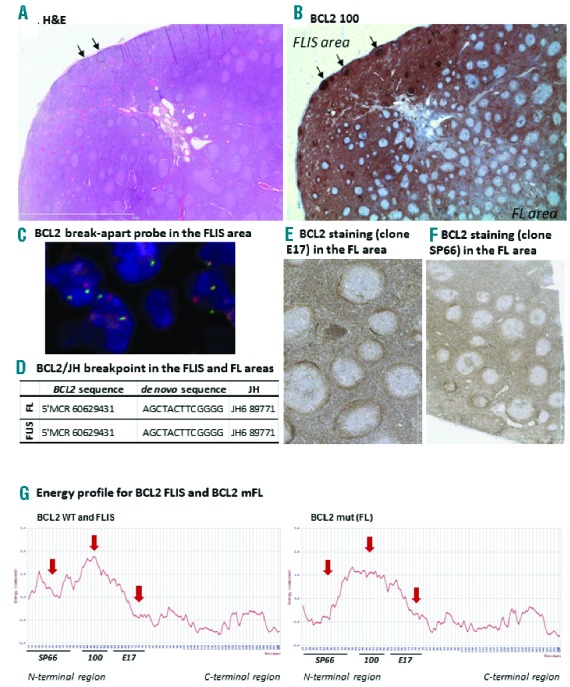
Description of BCL2 status in both the FLIS and the FL areas of a cervical lymph node. (A) Hematoxylin/eosin coloration showing the FLIS and FL zones. (B) Histochemical staining of BCL2 with the E100 clone. The staining was negative in the germinal center of the FL areas, whereas it was extremely intense within the GC of the FLIS containing area (stronger than BCL2+ cells of the extra follicular zones) (C) FISH staining with the BCL2 break apart probe (LSI BCL2 break-apart probe, Vysis®) in the FLIS area. Similar results were obtained in the FL area. (D) Sanger sequencing of the BCL2/JH breakpoint, and the de novo inserted sequence, in the FLIS and FL areas. (E) Histochemical staining of BCL2 with the E17 clone in the FL area (F) Histochemical staining of BCL2 with the SP66 clone in the FL area. (G) Energy profile obtained from the BCL2 sequence obtained from the FLIS and the FL. Fixation sites of the 3 tested antibodies are mentioned.
The BCL2 immunostaining (clone 100) was negative in follicles displaying a typical FL pattern. In contrast, follicles located in the pseudo-residual area were BCL2bright, i.e. more strongly stained than the surrounding mantle zone and reactive T cells (Figure 1B). Most follicles were only slightly positive for Ki67 (Online Supplementary Figure S1A). Both BCL2− and BCL2+ follicles were CD10 positive (Online Supplementary Figure S1B) and contained a BCL2/JH break-point evidenced by fluorescence in situ hybridization (FISH) (Figure 1C). Taken together these results suggested the diagnosis of simultaneous occurrence of BCL2− FL (grade I/II) and of BCL2+ FLIS in the same LN. We decided to further analyze those two lesions independently, and performed macrodissection in order to proceed with individual molecular analyses when required. Sanger sequencing revealed that both FLIS and FL shared the same BCL2/JH sequence at the t(14;18)+ breakpoint, and thus originated from the same clone (Figure 1D).
We tested two other anti-BCL2 antibodies (E17, SP66) directed against other epitopes, but the staining remained BCL2- in the FL area of the LN, similar to the anti-BCL2 antibody (clone 100) staining (Figure 1E and F). We thus sequenced exons 1 to 3 of the BCL2 gene (B-cell CLL/lymphoma 2, NG_009361.1). Punctual mutations, resulting in amino acid substitutions, were found in the FL component (Online Supplementary Table S1), and were indeed located in the targeted aa41 to aa54 epitope of clone 100 (mutations found in aa45–47), in the aa61 to aa76 epitope of clone E17 (mutation found at position aa64) and in the N-terminal region epitope of clone SP66 antibodies (mutation in aa32). None of the registered mutations involved a stop codon. The FLIS and FL sequences were submitted to 3D-molecule Viewer (Vector NTI advanced 5.11.1®), which revealed that the acquired mutations in the FL dissected area resulted in an altered energy profile of the BCL2 protein, probably preventing a proper fixation of most anti-BCL2 antibodies (Figure 1G and Online Supplementary Figure S2).
We thus determined whether the FL cells expressed at least the BCL2 transcript. mRNA was extracted from microdissected FL follicles and qRT-PCR was performed. An approximately 7-fold relative increase in BCL2 transcripts was found compared to microdissected benign reactive lympadenitis [Log2(BCL2/GAPDH) was 3.7 in FL follicles compared to 0.5 in benign reactive lympadenitis], indicating that the absence of BCL2 staining was not due to transcriptional downregulation.
Taking into account that the FL follicles were composed of more than 80% of FL B cells, it is unlikely that contaminating T cells could be entirely responsible for this high level of BCL2 transcription, which is thus likely related to FL B cells. Although we cannot exclude that a post-transcriptional mechanism could have induced downregulation of the BCL2 protein, these data are in accordance with the view of the FL component presenting with multiple mutations inducing a conformational change of the BCL2 protein, which may or may not have altered BCL2 function. Sustained activity of activation-induced cytidine deaminase (AID) has been shown to be partly responsible for somatic mutations in FL. Indeed, AID expression was present in both FLIS and FL areas of our sample (Online Supplementary Figure S3A).
Despite the concomitant FL/FLIS localization, these alterations were lacking in the FLIS dissection, suggesting that they were acquired in the FL component after divergence from a common founder clone. To our knowledge, only rare cases of FLIS with concomitant FL or DLBCL have been reported, and are usually observed in distinct LNs.5 These reports suggested that FLIS clones were probably remnants of an earlier colonization by t(14;18)+ B cells that have preceded FL.5,6 Our case is reminiscent of previously reported cases of FLIS associated with FL, in which the associated FL was often negative for BCL2 protein.5–7 This suggests either that FLIS is more easily detected in those cases because of the lack of BCL2 in the FL area, or that BCL2 mutations are frequently associated with progression from FLIS to FL, when compared to de novo or sporadic FL.
To further establish the clonal hierarchy between the FLIS and FL lesions, we investigated the immunoglobulin variable heavy chain (VH) gene region of FL cells, a region frequently mutated in FL.8 The VH region of the FL clone was identified as IGHV3-48*03/IGHD3-22*01/IGHJ4*02, with approximately 85.4% homology (+/−0.27) among the various FL subclones (n=16 analyzed sequences, corresponding to 7 different subclones) (Figure 2A and Online Supplementary Table S2). We backtracked this specific IGHV3-48*03/IGHD3-22*01/IGHJ4*02 sequence in the FLIS and found the same rearrangement within 3 subclones (Online Supplementary Table S2). Surprisingly, 2 of the FLIS subclones were more mutated than the corresponding FL subclones (82.3%+/−2.3 of homology), confirming a strong and/or repeated somatic hyper mutation (SHM) activity. In contrast, when looking at non-identical IGHV3 sequences, i.e. in distinct VDJ clones isolated from the FLIS area that did not match the sequence of the FL clone (and possibly represent infiltrating normal mantle zone B cells), the homology was of 98.2%+/−2.4 (n=14 sequences) (Figure 2A). In addition, the intra-clonal variability was higher in the FLIS than in the FL component, which could be due to a dynamic trafficking, such as multiple GC re-entries of the FLIS clones. This is in line with two recent reports showing a subclonal heterogeneity among genomic alterations observed in FLIS.2,3 Overall, our analysis reveals that the FLIS and the FL clones have evolved through a divergent evolution model, which postulates the existence of unique co-existing lesions and subclone selection, in a way similar to that reported in FL and relapsed FL9 (Figure 2B).
Figure 2.
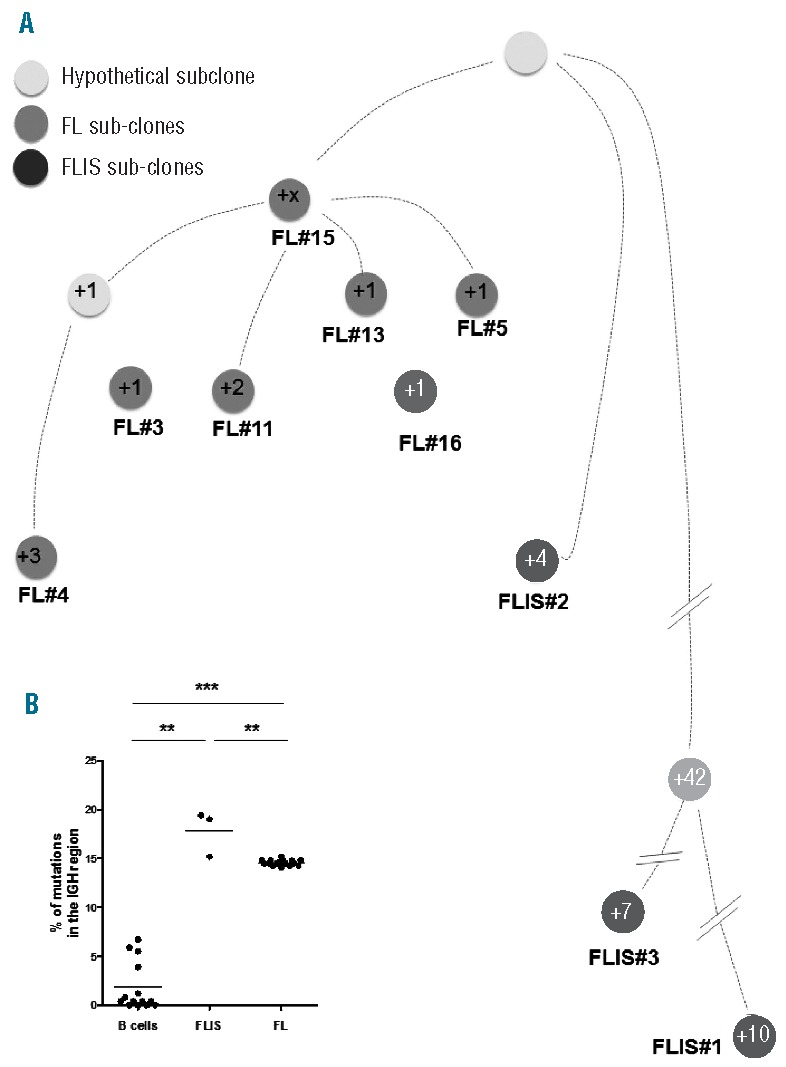
(A) The IGHV3-48*03/IGHD3-22*01/IGHJ4*02 sequences of the FL and FLIS were used to perform a hierarchical tree between clones. (B). Percentage of mutations found in all the IGHV3-48*03/IGHD3-22*01/IGHJ4*02 sequences of the FL and FLIS samples. Other germinal center B cells were used as control to have an idea of the percentage of mutations that can be observed on a similar population.
Finally, among the mutations at the VH IGHV3-48*03 sites, we observed that some of them were responsible for the introduction of a recurrent N-glycosylation motif (N-X-S/T) in both FL and FLIS lesions (Online Supplementary Table S3). These specific glycosylation sites introduce oligomannose glycans that are characteristic of FL, with an incidence of nearly 100%. They can occasionally be observed in some GC-derived tumors other than FL, but are very infrequent in normal B cells.10 Notably, a similar “N-I-S” motif was also found in the CDR2 region of a VH IGHV3-48*03 site sequenced from an FL sample.11
Functionally, added glycans terminate at high mannose, which might influence the behavior of FLIS or FL cells through opportunistic interactions between the B-cell receptor (BCR) with mannose-binding lectin bearing cells.12 The engagement of those lectins with the N-glycosylated sIg would substitute conventional antigen binding, and could represent a surrogate for antigen stimulation, providing necessary signaling for lymphoma cell survival.13 This chronic signaling through the BCR might not constitute an “oncogenic hit” per se, but might nonetheless favor the generation of long-lived FLIS clones, increasing the chance of accumulating the hits that will further drive clone fate.14 In addition to this role in B-cell survival, chronic engagement of N-glycosylated sIg may also contribute to a mucin-dependent evasion from immune surveillance and the induction of an immunosuppressive environment around the tumor.15 Since DC-SIGN and DC-SIGNR are two mannose-binding lectins able to bind mannosylated Igs in vitro and FL cells,13 and which were found, in public data sets, to be up-regulated in lymphoid organs (http://biogps.org/#goto=genereport&id=10332) (Online Supplementary Figure S4A and B), we have investigated their expression using IHC in our case. Positive immunostaining was only present in the lymphatic endothelium and sinus histiocytes, which were not directly in contact with FL cells (Online Supplementary Figure S3B). Although this result does not support a stimulation of FL cells by DC-SIGN and DC-SIGNR, one cannot rule out the possible role of other lectin-like receptors. In fact, lectins are rarely completely specific for particular sugars, and it is still unclear which oligosaccharides are responsible for lectin binding to lymphoma cells.12 To conclude, this case provided the opportunity to simultaneously analyze synchronous and contiguous FLIS and FL lesions sharing a common ancestor. This is the first report showing that N-glycosylation sites can already be present in an FLIS lesion, adding new information to help bridge the gap in our knowledge of the relationship between genomic instability and the dependency of FL cells on the micro-environment.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Cong P, Raffeld M, Teruya-Feldstein J, Sorbara L, Pittaluga S, Jaffe ES. In situ localization of follicular lymphoma: description and analysis by laser capture microdissection. Blood. 2002;99(9):3376–3382. [DOI] [PubMed] [Google Scholar]
- 2.Mamessier E, Song JY, Eberle FC, et al. Early lesions of follicular lymphoma: a genetic perspective. Haematologica. 2014;99(3):481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt J, Salaverria I, Haake A, et al. Increasing genomic and epigenomic complexity in the clonal evolution from in situ to manifest t(14;18)-positive follicular lymphoma. Leukemia. 2014;28(5):1103–1112. [DOI] [PubMed] [Google Scholar]
- 4.Mamessier E, Broussais-Guillaumot F, Chetaille B, et al. Nature and importance of follicular lymphoma precursors. Haematologica. 2014;99(5):802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jegalian AG, Eberle FC, Pack SD, et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood. 2011;118(11):2976–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonzheim I, Salaverria I, Haake A, et al. A unique case of follicular lymphoma provides insights to the clonal evolution from follicular lymphoma in situ to manifest follicular lymphoma. Blood. 2011;118(12):3442–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carbone A, Della Libera D, Zannier L, et al. In situ follicular lymphoma associated with overt B- or T-cell lymphomas in the same lymph node. Am J Hematol. 2011;86(12):E66–70. [DOI] [PubMed] [Google Scholar]
- 8.Kuppers R, Rajewsky K, Hansmann ML. Diffuse large cell lymphomas are derived from mature B cells carrying V region genes with a high load of somatic mutation and evidence of selection for antibody expression. Eur J Immunol. 1997;27(6):1398–1405. [DOI] [PubMed] [Google Scholar]
- 9.Pasqualucci L, Khiabanian H, Fangazio M, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6(1):130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu D, Ottensmeier CH, Du MQ, McCarthy H, Stevenson FK. Incidence of potential glycosylation sites in immunoglobulin variable regions distinguishes between subsets of Burkitt’s lymphoma and mucosa-associated lymphoid tissue lymphoma. Br J Haematol. 2003;120(2):217–222. [DOI] [PubMed] [Google Scholar]
- 11.McCann KJ, Ottensmeier CH, Callard A, et al. Remarkable selective glycosylation of the immunoglobulin variable region in follicular lymphoma. Mol Immunol. 2008;45(6):1567–1572. [DOI] [PubMed] [Google Scholar]
- 12.Radcliffe CM, Arnold JN, Suter DM, et al. Human follicular lymphoma cells contain oligomannose glycans in the antigen-binding site of the B-cell receptor. J Biol Chem. 2007;282(10):7405–7415. [DOI] [PubMed] [Google Scholar]
- 13.Coelho V, Krysov S, Ghaemmaghami AM, et al. Glycosylation of surface Ig creates a functional bridge between human follicular lymphoma and microenvironmental lectins. Proc Natl Acad Sci USA. 2010;107(43):18587–18592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCann KJ, Johnson PW, Stevenson FK, Ottensmeier CH. Universal N-glycosylation sites introduced into the B-cell receptor of follicular lymphoma by somatic mutation: a second tumorigenic event? Leukemia. 2006;20(3):530–534. [DOI] [PubMed] [Google Scholar]
- 15.Allavena P, Chieppa M, Bianchi G, et al. Engagement of the mannose receptor by tumoral mucins activates an immune suppressive phenotype in human tumor-associated macrophages. Clin Dev Immunol. 2010;2010:547179. [DOI] [PMC free article] [PubMed] [Google Scholar]