

Abstract
Glucose-stimulated insulin secretion (GSIS) in the pancreatic β-cells entails a variety of signaling mechanisms including activation of small GTP-binding proteins (G-proteins). Previous studies from our laboratory in human islets, rodent islets and clonal β-cells have demonstrated that G-proteins (e.g., Arf6, Cdc42 and Rac1) play novel roles in cytoskeletal remodeling, which is a critical step in the trafficking of insulin-laden secretory granules for fusion with plasma membrane and release of insulin. To further understand regulatory roles of Rac1 in GSIS, we utilized, herein, EHT 1864, a small molecule inhibitor, which attenuates Rac1 activation by retaining the G-protein in an inert/inactive state, thereby preventing activation of its downstream effector proteins. We demonstrate that EHT 1864 markedly attenuated GSIS in INS-1 832/13 cells. In addition, EHT 1864 significantly reduced glucose-induced activation and membrane targeting of Rac1 in INS-1 832/13 cells. This Rac1 inhibitor also suppressed glucose-induced activation of ERK1/2 and p53, but not Akt. Lastly, unlike the inhibitors of protein prenylation (simvastatin), EHT 1864 did not exert any significant effects on cell morphology (cell rounding) under the conditions it attenuated Rac1-sensitive signaling steps leading to GSIS. Based on these findings, we conclude that EHT 1864 specifically inhibits glucose-induced Rac1 activation and membrane association and associated downstream signaling events culminating in inhibition of GSIS.
Keywords: EHT 1864, small G-proteins, Rac1, insulin secretion, pancreatic islet
1. Introduction
It is well established that glucose-stimulated insulin secretion (GSIS) from the islet β-cell involves an interplay between a wide range of metabolic events and signal transduction pathways leading to the generation of second messenger molecules (cyclic nucleotides, adenine and guanine nucleotides and soluble second messengers) and mobilization of calcium ions [1–4]. Consequently, insulin-laden secretory granules are transported from distal sites to the plasma membrane for fusion and release of cargo into the circulation. Several lines of evidence suggest critical regulatory roles for small GTP-binding proteins (G-proteins; e.g., Cdc42 and Rac1) in the translocation of secretory vesicles to the plasma membrane [5, 6]. In this context, recent studies have demonstrated regulation of GSIS by small G-proteins including Arf6, Cdc42 and Rac1 [7–14]. Evidence from knockout animal models further affirms key functions of these signaling proteins (Rac1) in physiological insulin secretion [15].
Several mechanisms have been put forth for the regulation of small G-protein function, including post-translational modifications (e.g., prenylation, carboxylmethylation and acylation). These modifications are felt to increase the hydrophobicity of the target proteins thus rendering optimal membrane association [6, 12]. Previous studies from our laboratory have demonstrated requisite nature of these modification steps in GSIS. For example, using specific inhibitors of geranylgeranylation (e.g., GGTI-2147 and GGTI-2368), we reported significant inhibition of GSIS in clonal β-cells and normal rodent islets [6, 14]. Specific inhibitors of the carboxylmethylation of G-proteins (e.g., acetyl farnesyl cysteine) have also been shown to suppress GSIS in pancreatic β-cells [13, 16, 17]. Lastly, selective inhibitors of protein palmitoylation (e.g., cerulenin) also inhibit GSIS [13, 18]. These findings were further confirmed via the use of dominant negative mutants or siRNAs of the subunits of prenyltransferases [14] and carboxylmethyltransferases [17]. In addition, we have demonstrated that inhibition of Tiam1, a guanine nucleotide exchange factor (GEF) for Rac1, leads to inhibition of GSIS in insulin-secreting cells [9]. Together, the aforementioned observations implicate roles for small G-proteins (Rac1) in physiological insulin secretion.
Désiré and associates [19] have developed a small molecular weight (~ 582) inhibitor EHT 1864 [5-(5-(7-(Trifluoromethyl) quinolin-4-ylthio)pentyloxy)-2-(morpholinomethyl)-4H-pyran-4-one dihydrochloride; Figure 1], which inhibited Rac1 function in vivo. Further characterization of EHT 1864 by Shutes et al [20] suggested that EHT 1864 binds to Rac1 with high affinity, compared to GDP/GTP, and retains Rac1 in an inert and inactive state by displacement of pre-bound guanine nucleotide (GDP/GTP). Several recent studies have utilized EHT 1864 to deduce cellular roles of Rac1 function in health and disease [21–25]. Its unique inhibitory properties on Rac1 function have prompted us to undertake the current study to assess the roles of Rac1 in islet function and insulin secretion. Our findings suggest that EHT 1864 inhibits glucose-induced Rac1 activation and membrane targeting and associated signaling events (e.g., ERK1/2 and p53 activation) culminating in inhibition of GSIS.
Figure 1. Structure of EHT 1864.
Chemical structure of EHT 1864 is provided here.
2. Materials and Methods
2.1. Materials
EHT 1864 and PD0325901 were obtained from R&D systems (Minneapolis, MN). Simvastatin was from Sigma (St. Louis, MO). The rat insulin ELISA kit was purchased from American Laboratory Products Co (Windham, NH). Rac1 activation G-LISA kit was from Cytoskeleton Inc. (Denver, CO). Antibodies against phospho-p44/42 ERK1/2 (Thr202/Tyr204), total p44/42 ERK1/2, phospho-Akt (Ser473), total Akt, phospho-p53 (Ser15) and E-Cadherin were obtained from Cell Signaling Technology (Danvers, MA). Antisera directed against GAPDH and total p53 were from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibody directed against Rac1 was purchased from BD Bioscience (San Jose, CA).
2.2. GSIS and KSIS from INS-1 832/13 cells
INS-1 832/13 cells (seeded at a density of 2.5 × 105 cells per well in a 24 well plate) were cultured in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation at varying concentrations of EHT 1864 (0–10 μM) for 1 h, cells were further incubated in the presence of low (2.5 mM) or varying concentrations of glucose (2.5–20 mM) or KCl (40 mM) for 30 min at 37°C in the continuous absence or presence of EHT 1864 at concentrations indicated in the text. The supernatant was then removed, centrifuged at 300g for 10 min, and the amount of insulin released was quantified by ELISA as we described previously [10,26].
2.3. Rac1 activation assay
INS-1 832/13 cells were cultured in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with EHT 1864 (10 μM) for 1 h, cells were incubated with low (2.5 mM) or high glucose (20 mM) for 20 min at 37°C in the continuous absence or presence of EHT 1864. Rac1 activation was quantified using a G-LISA kit as described in [27].
2.4. Subcellular fractionation and phase partitioning using Triton X-114
INS-1 832/13 cells were cultured in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with EHT 1864 (10 μM) for 1 h, cells were treated with low (2.5 mM) or high glucose (20 mM) in the continuous absence or presence of EHT 1864 for 20 min. Cell lysates were homogenized in a homogenization buffer (20 mM Tris-HCl, pH 7.5, 0.5 mM EGTA, 2 mM MgCl2 and protease inhibitor cocktail) and subjected to a single-step centrifugation at 100,000g for 60 min at 4°C. Total membrane (pellet) and soluble (supernatant) fractions were separated. Hydrophilic and hydrophobic phases of the total membrane fractions were isolated using Triton X-114 as we described earlier [28]. Relative abundance of Rac1 in cytosol and each of the individual hydrophilic and hydrophobic phases derived from the membranous fractions was determined by Western blotting. Additionally, the relative purity of these fractions was verified by probing for marker proteins such as GAPDH (a marker for the cytosolic fraction) and E-Cadherin (a marker for membrane fraction).
2.5. Western Blotting
INS-1 832/13 cells were cultured in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with EHT 1864 (10 μM) or PD0325901 (1 μM) for 1 h, cells were treated with low (2.5 mM) or high glucose (20 mM) in the continuous absence or presence of EHT 1864 or PD0325901 for 30 min. Cell lysates were separated by SDS-PAGE on 10 % (w/v) polyacrylamide gels and electrotransferred to nitrocellulose membrane. The membranes were then blocked with 0.1 % Casein in 0.2X PBS for 1 h at room temperature. Blots were then incubated overnight at 4 °C with appropriate primary antibody (Rac1, phospho-ERK1/2 (Thr202/Tyr204), total-ERK1/2, phospho-Akt (Ser473), total Akt, phospho-p53 (Ser15), E-Cadherin and GAPDH) in 0.2X PBS-T containing 0.1 % Casein. The membranes were washed 5X for 5 min each with PBS-T and probed with appropriate IRDye® 800CW secondary antibody in 0.1 % casein in PBS-T at room temperature for 1 h. After washing, the immune complexes comprised of the target proteins were detected under Odyssey® Imaging Systems.
2.6. Changes in β-cell morphology following inhibition of protein prenylation and Rac1 activation
To observe the effects of EHT 1864 on cell morphology, INS-1 832/13 cells were incubated overnight, in a 6-well plate, with RPMI (2.5 mM glucose and 2.5% fetal bovine serum) in the presence of diluent (de-ionized water) or simvastatin (30 μM). Cells were also incubated with EHT 1864 (10 μM) for 1 h following overnight incubation in RPMI with 2.5 mM glucose and 2.5% fetal bovine serum. Changes in cell morphology were then quantified visually under an Olympus IX71 microscope.
2.7. Statistical analysis of experimental data
The statistical significance of the differences between the experimental conditions was determined by ANOVA. p values < 0.05 were considered significant.
3. Results
3.1. EHT 1864 inhibits GSIS in INS-1 832/13 cells
At the outset, we quantified the effects of EHT 1864 on GSIS from INS-1 832/13 cells. Data depicted in Figure 2 demonstrate a modest (~ 13%; not significant) inhibition of GSIS following incubation with EHT 1864 at 5 μM concentration. Furthermore, we noticed a complete inhibition of GSIS by EHT 1864 at 10 μM concentration. It should also be noted that, while 5 μM EHT 1864 exerted no significant effects on basal secretion, it did modestly increase the basal secretion at 10 μM concentration (~ 50%). Therefore, in all the subsequent experiments, we studied the effects of EHT 1864 at 10 μM. Furthermore, EHT 1864 inhibited GSIS in INS-1 832/13 cells elicited at 10–20 mM glucose (Figure 3). Compatible with findings reported in Figure 2, a significant increase in insulin secretion was also noted in cells incubated with this inhibitor at 5 mM glucose concentration. Together, data depicted in Figures 2 and 3 demonstrate significant inhibitory effects of EHT 1864 on GSIS in INS-1 832/13 cells.
Figure 2. EHT 1864 inhibits GSIS in INS-1 832/13 cells.

INS-1 832/13 cells were cultured in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with EHT 1864 (0–10 μM) for 1 h, cells were incubated in the presence of low (2.5 mM) or high glucose (20 mM) in the continuous absence or presence of EHT 1864 for 30 min at 37°C. Insulin released into the medium was quantified by ELISA. The data was expressed as ng/ml of insulin released ± SEM. * p < 0.05 vs. 2.5 mM glucose and ** p < 0.05 vs. 20 mM glucose alone or in presence of 5 μM of EHT 1864.
Figure 3. Effect of EHT 1864 on insulin secretion at different glucose concentrations in INS-1 832/13 cells.

INS-1 832/13 cells were cultured in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with EHT 1864 (10 μM) for 1 h, cells were incubated in the presence of different concentrations of glucose (0–20 mM) in the continuous absence or presence of EHT 1864 for 30 min at 37°C. The amount of insulin released was quantified by ELISA. The data was expressed as ng/ml ± SEM. * p < 0.05 vs. 10 mM or 20 mM glucose and ** p < 0.05 vs. 5 mM glucose.
3.2. EHT 1864 inhibits glucose-induced Rac1 activation in INS-1 832/13 cells
As stated above, several previous studies have demonstrated requisite roles for Rac1 activation in GSIS. Therefore, we asked if EHT 1864 inhibits glucose-induced Rac1 activation under conditions it inhibited insulin secretion. To test this, INS-1 832/13 cells were incubated in the absence or presence of EHT 1864 and glucose-induced activation of Rac1 was quantified. Data in Figure 4 suggested a significant increase in the activation of Rac1 (GTP-bound conformation) by stimulatory glucose (bar 1 vs. 3), which was inhibited by EHT 1864 (bar 3 vs. bar 4). Interestingly, EHT 1864 modestly increased Rac1 activation in the presence of basal glucose (2.5 mM; bar 1 vs. bar 2), but such an activation was not statistically significant. Taken together, these findings indicate that EHT 1864 inhibits glucose-induced Rac1 activation (Figure 4) resulting in inhibition of GSIS (Figures 2 and 3).
Figure 4. EHT 1864 significantly inhibits glucose-induced Rac1 activation in INS-1 832/13 cells.

INS-1 832/13 cells were incubated in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with EHT 1864 (10 μM) for 1 h, cells were incubated in the presence of low (2.5 mM) or high glucose (20 mM) in the continuous absence or presence of EHT 1864 for 20 min at 37°C. Rac1 activation was quantified by G-LISA method. Data are expressed as fold increase over 2.5 mM glucose, and are mean ± SEM. *p < 0.05 vs. 2.5 mM glucose and **p < 0.05 vs. 20 mM glucose.
3.3. EHT 1864 inhibits glucose-induced membrane association of Rac1 in INS-1 832/13 cells
Published evidence, including our own in pancreatic β-cells, supports the notion that activation and membrane association of small G-proteins (Cdc42 and Rac1) is necessary for optimal interaction of these proteins with their effector proteins [5,6]. Therefore, in the next series of experiments, we determined if glucose-induced membrane targeting is inhibited by EHT 1864 in INS-1 832/13 cells. To address this, the hydrophilic and hydrophobic compartments of the membrane fraction were isolated from cells exposed to basal or stimulatory glucose concentrations, incubated in the absence or presence of EHT 1864, by Triton X-114 phase extraction method (see section 2.4 for additional details). Relative abundance of Rac1 in the cytosolic and hydrophilic and hydrophobic compartments of the membrane fraction (above) was determined by Western blotting. As stated above, purity of the total soluble (cytosolic fraction) and the membrane fraction was assessed by marker proteins for these fractions. Data shown in Figure 5 (Panel A) indicate that Rac1 is predominantly localized in the cytosolic fraction under basal conditions. Treatment of these cells with EHT 1864 modestly increased the association of Rac1 with the hydrophobic fraction. As has been shown previously, a significant amount of Rac1 translocated to the hydrophobic and hydrophilic compartments from cells incubated with stimulatory glucose. Coprovision of EHT 1864 to these cells significantly inhibited glucose-induced translocation of Rac1 to the membrane fraction. Pooled data from multiple studies are provided in Panel B of Figure 5. These data indicate that glucose-induced activation and membrane association of Rac1 are prevented by EHT 1864 leading to inhibition of GSIS.
Figure 5. EHT 1864 significantly inhibits glucose-induced membrane targeting of Rac1.

INS-1 832/13 cells were incubated in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with EHT 1864 (10 μM) for 1 h, cells were stimulated with low (2.5 mM) or high glucose (20 mM) in the continuous absence or presence of EHT 1864 for 20 min at 37°C. Cells were then lysed and subjected to phase partitioning with Triton X-114, to obtain cytosolic and hydrophilic/hydrophobic fractions of the membrane. Rac1 localization in these fractions was determined by Western blotting, and a representative blot from three independent experiments is provided (A). The relative purity of cytosolic and hydrophilic/hydrophobic membrane fractions was verified by probing for GAPDH (cytosol) and E-Cadherin (Membrane). Rac1 band intensities were then quantified by densitometry (B). Data represent mean ± SEM from three independent experiments, and expressed as fold change over 2.5 mM glucose for each cell fraction. * p < 0.05 vs. 2.5 mM glucose-cytosol. ** p < 0.05 vs. 2.5 mM glucose-membrane hydrophilic. # p < 0.05 vs. 20 mM glucose-membrane hydrophilic. NS: not significant.
3.4. EHT 1864 inhibits glucose-induced ERK1/2 phosphorylation and activation in INS-1 832/13 cells
Recent evidence from our laboratory [26] and the Thurmond’s laboratory [29] implicated ERK1/2 activation as key to cytoskeletal remodeling, which is requisite for GSIS. Further, these studies suggested key regulatory roles for this signaling step in G-protein (Cdc42 and Rac1)-mediated insulin secretion in glucose-stimulated β-cell. Therefore, we quantified glucose-induced ERK1/2 phosphorylation in INS-1 832/13 cells following exposure to EHT 1864. At the outset, we assessed glucose-induced activation of ERK1/2 phosphorylation in the presence of PD0325901, a known inhibitor of MEK. Data in Figure 6 (Panel A) indicate complete inhibition by MEK inhibitor of glucose-induced phosphorylation of ERK1/2 in INS-1 832/13 cells. Data from multiple studies demonstrating inhibitory effects of PD 0325901 are provided in Figure 6 (Panel B). It is noteworthy that data depicted in Figure 6 (Panel C) demonstrate no significant effects of EHT 1864 on ERK1/2 phosphorylation under basal glucose conditions. However, a marked inhibition of high glucose-induced ERK1/2 phosphorylation was demonstrable in INS-1 832/13 cells exposed to EHT 1864. Pooled data from multiple studies are provided in Panel D of Figure 6. Based on the findings described above, we conclude that EHT 1864 efficiently inhibits glucose-induced ERK1/2 and Rac1 activation and its membrane association culminating in inhibition of GSIS.
Figure 6. Glucose-induced ERK1/2 activation is inhibited by PD 0325901 and EHT 1864.


INS-1 832/13 cells were starved in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with PD 0325901 (1 μM) or EHT 1864 (10 μM) for 1 h, cells were stimulated with low (2.5 mM) or high glucose (20 mM) in the continuous absence or presence of PD 0325901 or EHT 1864 for 30 min at 37°C. Cell lysate proteins were separated by SDS-PAGE, and then transferred onto a nitrocellulose membrane. The membranes were then blocked and probed with anti-phosphorylated ERK1/2 followed by incubation with anti-rabbit secondary antibody. The same blots were stripped and re-probed with anti-total ERK1/2 (A, C). Band intensities of phospho-ERK1/2 and total-ERK1/2 were quantified by densitometric analysis (B, D). Results are shown as mean ± SEM from three independent experiments and expressed as fold change in the ratios between phospho-ERK1/2 and total-ERK1/2. * p < 0.05 vs. 2.5 mM glucose alone or in presence of EHT 1864. # p < 0.05 vs. 20 mM glucose.
We next assessed if inhibition of glucose-induced Rac1 activation by EHT 1864 results in inhibition of other downstream signaling pathways involved in islet β-cell function. Data depicted in Figure 7 (Panel A) indicate a significant increase in the phosphorylation of Akt in INS 1-832/13 cells following exposure to stimulatory glucose. Interestingly, glucose-induced Akt phosphorylation was resistant to Rac1 inhibition by EHT 1864. Data from multiple experiments (Figure 7; Panel B) suggest that glucose-induced phosphorylation and activation of Akt does not require Rac1 activation signaling step. It is noteworthy, however, that p53 phosphorylation is significantly augmented by glucose in INS 1-832/13 cells (Figure 7; Panel C), which was abolished by EHT 1864-mediated inhibition of Rac1 (Figure 7; Panel C and D). Taken together, data in Figures 6 and 7 suggest specific roles of glucose-induced Rac1 activation in mediating downstream signaling steps (see section 4).
Figure 7. EHT 1864 significantly inhibited glucose-induced p53 phosphorylation but had no effect on phosphorylation of Akt.


INS-1 832/13 cells were starved overnight in RPMI media supplemented with 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with EHT 1864 (10 μM), cells were further stimulated with low (2.5 mM) or high glucose (20 mM) in the continuous absence or presence of EHT 1864 for 30 min at 37°C. Cells were then lysed and lysate proteins were separated by SDS-PAGE. Proteins were then transferred onto a nitrocellulose membrane, blocked and probed with antibody directed against phospho-Akt or phospho-p53. The membranes were then incubated with anti-rabbit secondary antibody. After detection, the same blots were stripped and re-probed with antibody directed against total-Akt or Actin (A, C). Band intensities were quantified by densitometric analysis (B, D). Results are shown as mean ± SEM from three independent experiments and expressed as fold change in the ratios between phospho-Akt and total-Akt or phospho-p53 and Actin. * p < 0.05 vs. 2.5 mM glucose alone or in presence of EHT 1864. # p < 0.05 vs. 20 mM glucose.
3.5. EHT 1864 elicits minimal effects on cell morphology under conditions it inhibits GSIS: Evidence for a lack of involvement of protein prenylation in the inhibitory effects of EHT 1864
Previous investigations from our laboratory have demonstrated that post-translational prenylation of small G-proteins is necessary for membrane targeting and effector activation in the pancreatic β-cell [6, 12]. It has also been suggested that inhibition of small G-protein function results in alterations in cell morphology (cell rounding) following inhibition of protein prenylation [6, 12]. Herein, we investigated if EHT 1864 elicits changes on cell morphology under conditions it inhibits ERK1/2-Rac1 activation and GSIS. To address this, we incubated INS-1 832/13 cells in the absence or presence of EHT 1864 and examined alterations in cell morphology by phase contrast microscopy. As a positive control, INS-1 832/13 cells were cultured in the presence of simvastatin, a known inhibitor of mevalonic acid, a precursor for farnesyl and geranylgeranyl pyrophosphates [6, 12]. Data shown in Figure 8 demonstrate significant cell rounding in INS-1 832/13 cells incubated in the presence of simvastatin (~57% of cells), but not in the presence of the diluent (~3% cells). However, incubation of these cells with EHT 1864, under conditions it inhibited cellular signaling events (ERK1/2 and Rac1 activation), led to no significant abnormalities in cell morphology (~3% of cells; Figure 8). These data indicate that EHT 1864 attenuates ERK1/2 and Rac1 activation and insulin secretion without exerting significant effects on cellular events leading to abnormal cell morphology.
Figure 8. EHT 1864 had no effect on INS-1 832/13 cell morphology.
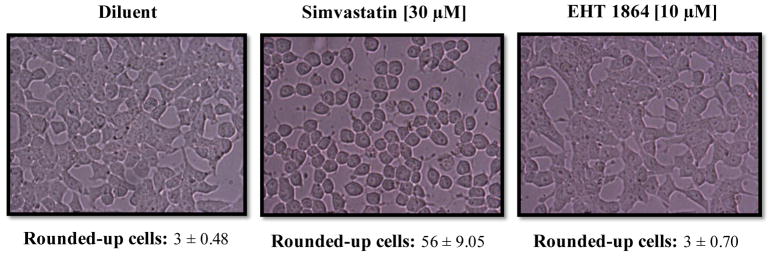
INS-1 832/13 cells were cultured overnight with RPMI (2.5 mM glucose and 2.5% fetal bovine serum) in the presence of diluent (de-ionized water) or Simvastatin (30 μM). The cells were also incubated with EHT 1864 (10 μM) for 1 h after overnight incubation in RPMI with 2.5 mM glucose and 2.5% fetal bovine serum. The cells were then visualized under the microscope to observe changes in morphology. Alterations in cell morphology were quantified by counting cells with morphological abnormalities (rounded-up cells), in the selected areas (0.5 cm2) and expressed as mean ± SEM (n=6).
3.6. In contrast to GSIS, KCl-stimulated insulin secretion (KSIS) is augmented by EHT 1864
Previous observations from the laboratories of Thurmond [5, 7, 8], Kowluru [6, 14, 28] and Asahara [15] have demonstrated that insulin secretion elicited by a membrane depolarizing concentration of KCl (KSIS) is resistant to Rac1 inhibition. In the last set of experiments, we quantified KSIS in INS-1 832/13 cells following exposure to EHT 1864 (5 or 10 μM). Interestingly, in contrast to GSIS, insulin secretion elicited by KCl was potentiated significantly by EHT 1864 (Figure 9). Such potentiating effects were more pronounced at 10 μM EHT 1864. These data raise an interesting possibility that Rac1 might mediate additional calcium-regulated signaling pathways, which are inhibitory to exocytotic secretion of insulin (see section 4).
Figure 9. EHT 1864 potentiates KSIS in INS-1 832/13 cells.

INS-1 832/13 cells were cultured in RPMI media overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation with EHT 1864 (0–10 μM) for 1h, cells were incubated in the presence of low glucose (2.5 mM) or KCl (40 mM) in the continuous absence or presence of EHT 1864 for 30 min at 37°C. The amount of insulin released was quantified by ELISA. The data was expressed as ng/ml of insulin released ± SEM and * p < 0.05 vs. 2.5 mM glucose, and # p < 0.05 vs. 40 mM KCl alone or in presence of 5 μM EHT 1864.
4. Discussion
The overall objective of this study was to further assess the role of Rac1 in the cascade of events leading to GSIS. To address this, we employed EHT 1864, a novel small molecule inhibitor, which attenuates Rac1 activation by retaining it in its inert and inactive conformation via inhibition of its association with guanine nucleotides. This class of inhibitors has not been tested before in the islet β-cell. Our findings indicate that EHT 1864 inhibits glucose-induced activation and membrane targeting of Rac1 and associated signaling steps, including ERK1/2, which has been shown to control cytoskeletal remodeling and insulin exocytosis [26, 29]. Compatible with these findings, we also observed significant inhibition of GSIS by EHT 1864 in INS-1 832/13 cells. Our findings also suggest no morphological changes in pancreatic β-cells incubated with EHT 1864 at least under conditions it suppressed glucose-sensitive signaling events. Thus, our findings provide the first evidence that inhibition of guanine nucleotide association on Rac1 GTPase impedes activation of key signaling steps necessary for the stimulus-secretion coupling of GSIS.
We demonstrated herein that glucose-induced ERK1/2 activation is inhibited by EHT 1864, thus establishing a link between Rac1 activation and ERK1/2 activation. These findings are supported by two recent studies that investigated regulatory roles of Raf-1 and ERK1/2 in glucose-induced cytoskeletal remodeling. First, using pharmacological approaches, Kalwat and associates demonstrated that glucose-induced activation of Cdc42 promotes activation of a signaling cascade involving activation of PAK1→Raf-1→MEK1/2→ERK1/2 to facilitate F-actin remodeling and insulin secretion [29]. Second, studies from our laboratory suggested that a protein farnesylation step is necessary for glucose-induced activation of Raf-1→ERK1/2 to promote Rac1 activation [26]. Together, data from the above and current studies place ERK1/2 as one of the key signaling steps in Cdc42/Rac1-mediated insulin secretion in the pancreatic β-cell. It should be noted that other downstream signaling steps involved in GSIS are also under the direct control of Rac1 since we observed in the current studies that glucose-induced phosphorylation of p53, but not Akt, was inhibited by EHT 1864. This will serve as one example, and it is likely that several downstream signaling pathways might be regulated by active Rac1. Future studies will address the identity of those signaling cascades.
We also noted no clear changes in the morphology in INS-1 832/13 cells following exposure to EHT 1864 under the conditions it abrogated glucose-mediated signaling events in these cells. Our findings of significant alterations (rounding) in cells exposed to inhibitors of protein prenylation (simvastatin) suggest that they induce significant defects in the actin cytoskeleton leading to alterations in cell morphology (current studies) and inhibition of GSIS as shown earlier [13, 30]. It should be noted however that studies by Baier et al [31] and Shutes and coworkers [20] demonstrated significant inhibitory effects of EHT 1864 on membrane ruffling in mast cells and lamellipodia formation in U87 MG glioblastoma cells. The observed differences between these studies and our current investigations may, in part, be due to differences in cell types studied and the concentrations of EHT 1864 employed (20 μM in mast cells and 5 μM in glioblastoma cells) [20, 31].
It is noteworthy that while EHT 1864 inhibited GSIS at 5–10 μM concentrations (Figure 2), higher concentrations of this inhibitor potentiated insulin release from INS-1 832/13 cells in the absence (basal) or glucose-stimulated conditions. For example, at 20 μM concentration, insulin secretion under basal conditions was increased from 8 ± 0.8 ng/ml to 15 ± 1.2 ng/ml in the absence or presence of EHT 1864, respectively (n = 8). Likewise, GSIS values in the absence and presence of EHT 1864 represented 26 ± 1.5 vs. 31.5 ± 1.7 ng/ml (n = 8). While potential mechanisms underlying stimulatory effects of this inhibitor on insulin secretion under basal and glucose stimulated conditions remain to be determined, we also observed in the current studies the potentiating effects of this inhibitor on KCl-induced insulin secretion (Figure 9). These findings suggest additional Rac1-dependent regulatory mechanisms might underlie potentiation of calcium-induced insulin secretion. For example, previous studies from our laboratory have demonstrated significant potentiation of GSIS from INS-1 832/13 cells following siRNA-mediated knockdown of Tiam1, a GEF for Rac1 [9]. We also demonstrated that such a potentiation of GSIS was sensitive to extracellular calcium [9]. It is also likely that calcium-induced insulin secretion is negatively regulated by Rac1 or its downstream signaling events, inhibition of which by EHT 1864 relieves such an effect thereby further stimulating the secretion. Future studies will address these questions, including regulation, by Rac1, of putative inhibitory G-proteins (GEi) that regulate exocytosis that we proposed previously [32].
It is becoming increasingly clear that Rac1 contributes significantly towards the pathology of many disease states including diabetes. For example, several recent studies in pancreatic β-cells, retinal endothelial cells, and cardiomyocytes have demonstrated key roles for Rac1 in the induction of oxidative stress under conditions of glucotoxicity and diabetes [33–36]. These studies also demonstrated that such an increase in oxidative stress is due to activation of phagocyte-like NADPH oxidase (Nox2), and Rac1 represents one of the members of the Nox2 holoenzyme [27, 33–36]. Interestingly, data from in vitro and in vivo experiments suggested that inhibition of Tiam1, a GEF for Rac1 (using NSC23766), markedly prevented the damaging effects of glucotoxic and diabetic conditions [27, 33–37]. These data suggested that GTP/GDP exchange on Rac1 by Tiam1 is critical for Nox2 activation. Future studies will determine if EHT 1864 can prevent effects of high glucose and diabetes by inhibiting the activation of Nox2 and its downstream signaling steps. In this context, recent investigations by Yoshida et al [38] in cultured mesangial cells demonstrated significant inhibition of high glucose-induced Rac1 activation and mineralocorticoid receptor activation by over expression of dominant negative Rac1 or by coprovision of EHT 1864. Based on these observations, the authors concluded that Rac1 is a therapeutic target for the treatment of nephropathy in obesity-related diabetic population [38]. In addition, recent studies by Katz and associates, using EHT 1864, provided compelling evidence to implicate Rac1 as one of the key signaling proteins in the spread of human breast cancer [24]. Thus, it appears that Rac1 activation contributes to the pathogenesis of several disease states.
5. Conclusion
In conclusion, we present the first evidence to suggest that EHT 1864, which inhibits Rac1 function via inhibition of guanine nucleotide association, impedes glucose-induced Rac1-mediated signaling events leading to inhibition of GSIS. Based on our current findings we present a model detailing our current understanding of the signaling pathways involved in glucose-mediated activation of Rac1 (Figure 10). We propose that acute exposure of isolated β-cells to stimulatory glucose leads to the conversion of inactive (GDP-bound) Rac1 to its active configuration (GTP-bound) and targeting to the appropriate membranous compartment. Active Rac1 promotes phosphorylation of downstream signaling steps, including phosphorylation of ERK1/2 and p53. It appears that the stimulatory effects of GTP-bound Rac1 are specific, since phosphorylation of Akt was unaffected under conditions of Rac1 inhibition by EHT 1864. Studies are in progress to further determine the utility of this small molecule inhibitor in the prevention of metabolic dysfunction of the islet β-cell under conditions in in vitro and in vivo models of metabolic stress and diabetes.
Figure 10. Proposed model for glucose-stimulated insulin secretion via activation and membrane association of Rac1 and downstream activation of ERK1/2.

We propose that acute exposure of INS-1 832/13 cells to stimulatory glucose (20 mM) results in the activation and membrane association of Rac1. This results in the activation of downstream effectors including ERK1/2, culminating in insulin release from the pancreatic β-cell. Our findings suggest that EHT 1864, a selective inhibitor of Rac1 activation, disrupts this signaling mechanism, thereby attenuating GSIS (see section 4).
Highlights.
EHT 1864 suppressed glucose-stimulated insulin secretion in insulin-secreting INS-1 832/13 cells.
EHT 1864 inhibited glucose-induced activation and membrane translocation of Rac1.
Acute exposure of INS-1 832/13 cells to stimulatory glucose concentrations resulted in phosphorylation of p44/42 ERK1/2 and p53 kinase, which was significantly inhibited by EHT 1864.
EHT 1864 exerted no effects on INS-1 832/13 cell morphology.
KCl-induced insulin secretion was potentiated by EHT 1864 in INS-1 832/13 cells.
Acknowledgments
This research was supported in part by a Merit Review award from the Department of Veterans Affairs, the National Institutes of Health, and the Juvenile Diabetes Research Foundation. AK is also the recipient of a Senior Research Career Scientist Award from the Department of VA. KS is the recipient of Rumble Pre-doctoral Fellowship from Wayne State University. We thank Prof. Chris Newgard for providing INS-1 832/13 cells.
Abbreviations used
- Arf6
ADP-ribosylation factor 6
- Cdc42
Cell division control protein 42
- ERK1/2
Extracellular signal-regulated kinases
- GEF
Guanine nucleotide exchange factor
- GDP
Guanosine diphosphate
- GTP
Guanosine triphosphate
- GGTI
Geranylgeranyl transferase inhibitor
- GSIS
Glucose stimulated insulin secretion
- HG
High glucose
- KSIS
KCl-stimulated insulin secretion
- LG
Low glucose
- Nox2
NADPH oxidase 2
- Rac1
Ras-related C3 botulinum toxin substrate 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prentki M, Matschinsky FM. Calcium, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev. 1987;67:1185–1248. doi: 10.1152/physrev.1987.67.4.1185. [DOI] [PubMed] [Google Scholar]
- 2.MacDonald MJ. Elusive proximal signals of β-cells for insulin secretion. Diabetes. 1990;39:1461–1466. doi: 10.2337/diab.39.12.1461. [DOI] [PubMed] [Google Scholar]
- 3.Komatsu M, Takei M, Ishii H, Sato Y. Glucose-stimulated insulin secretion: A newer perspective. J Diabetes Investig. 2013;27:511–516. doi: 10.1111/jdi.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang SN, Shi Y, Yang G, Li Y, Yu J, Berggren PO. Ionic mechanisms in pancreatic β cell signaling. Cell Mol Life Sci. 2014;71:4149–4177. doi: 10.1007/s00018-014-1680-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122:893–903. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kowluru A. Small G proteins in islet beta-cell function. Endocr Rev. 2010;31:52–78. doi: 10.1210/er.2009-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nevins AK, Thurmond DC. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am J Physiol Cell Physiol. 2003;285:C698–C710. doi: 10.1152/ajpcell.00093.2003. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Oh E, Thurmond DC. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J Biol Chem. 2007;282:9536–9546. doi: 10.1074/jbc.M610553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veluthakal R, Madathilparambil SV, McDonald P, Olson LK, Kowluru A. Regulatory roles of Tiam1, a guanine nucleotide exchange factor for Rac1, in glucose-stimulated insulin secretion in pancreatic β-cells. Biochem Pharmacol. 2009;77:101–113. doi: 10.1016/j.bcp.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayaram B, Syed I, Kyathanahalli CN, Rhodes CJ, Kowluru A. Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 β-cells and rat islets. Biochem Pharmacol. 2011;81:1016–1027. doi: 10.1016/j.bcp.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawrence JT, Birnbaum MJ. ADP-ribosylation factor 6 regulates insulin secretion through plasma membrane phosphatidylinositol 4, 5-bisphosphate. Proc Natl Acad Sci U S A. 2003;100:13320–13325. doi: 10.1073/pnas.2232129100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowluru A. Regulatory roles for small G proteins in the pancreatic beta-cell: lessons from models of impaired insulin secretion. Am J Physiol Endocrinol Metab. 2003;285:E669–E684. doi: 10.1152/ajpendo.00196.2003. [DOI] [PubMed] [Google Scholar]
- 13.Metz SA, Rabaglia ME, Stock JB, Kowluru A. Modulation of insulin secretion from normal rat islets by inhibitors of the posttranslational modifications of the GTP-binding proteins. Biochem J. 1993;295:31–40. doi: 10.1042/bj2950031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veluthakal R, Kaur H, Goalstone M, Kowluru A. Dominant negative α-subunit of farnesyl-and geranyl geranyltransferase inhibits glucose-stimulated, but not KCl-stimulated, insulin secretion in INS 832/13 cells. Diabetes. 2007;56:204–210. doi: 10.2337/db06-0668. [DOI] [PubMed] [Google Scholar]
- 15.Asahara S, Shibutani Y, Teruyama K, Inoue HY, Kawada Y, Etoh H, Matsuda T, Kimura-Koyanagi M, Hashimoto N, Sakahara M, Fujimoto W, Takahashi H, Ueda S, Hosooka T, Satoh T, Inoue H, Matsumoto M, Aiba A, Kasuga M, Kido Y. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia. 2013;56:1088–1097. doi: 10.1007/s00125-013-2849-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kowluru A, Seavey SE, Li G, Sorenson RL, Weinhaus AJ, Nesher R, Rabaglia ME, Vadakekalam J, Metz SA. Glucose- and GTP-dependent stimulation of the carboxylmethylation of Cdc42 in rodent and human pancreatic islets and pure β cells: evidence for an essential role for GTP-binding proteins in nutrient-induced insulin secretion. J Clin Invest. 1996;98:540–555. doi: 10.1172/JCI118822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jayaram B, Syed I, Singh A, Subasinghe W, Kyathanahalli CN, Kowluru A. Isoprenylcysteine carboxyl methyltransferase facilitates glucose-induced Rac1 activation, ROS generation and insulin secretion in INS 832/13 β-cells. Islets. 2011;3:48–57. doi: 10.4161/isl.3.2.15016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohammed AM, Chen F, Kowluru A. The two faces of protein palmitoylation in islet β-cell function: potential implications in the pathophysiology of islet metabolic dysregulation and diabetes. Recent Pat Endocr Metab Immune Drug Discov. 2013;7:203–212. doi: 10.2174/18722148113079990008. [DOI] [PubMed] [Google Scholar]
- 19.Désiré L, Bourdin J, Loiseau N, Peillon H, Picard V, De Oliveira C, Bachelot F, Leblond B, Taverne T, Beausoleil E, Lacombe S, Drouin D, Schweighoffer F. RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo. J Biol Chem. 2005;280:37516–37525. doi: 10.1074/jbc.M507913200. [DOI] [PubMed] [Google Scholar]
- 20.Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- 21.Dipaolo BC, Davidovich N, Kazanietz MG, Margulies SS. Rac1 pathway mediates stretch response in pulmonary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;305:141–153. doi: 10.1152/ajplung.00298.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stefanini L, Boulaftali Y, Ouellette TD, Holinstat M, Désiré L, Leblond B, Andre P, Conley PB, Bergmeier W. Rap1-Rac1 circuits potentiate platelet activation. Arterioscler Thromb Vasc Biol. 2012;32:434–441. doi: 10.1161/ATVBAHA.111.239194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raynaud F, Moutin E, Schmidt S, Dahl J, Bertaso F, Boeckers TM, Homburger V, Fagni L. Rho-GTPase-activating protein interacting with Cdc-42-interacting protein 4 homolog 2 (Rich2): a new Ras-related C3 botulinum toxin substrate 1 (Rac1) GTPase-activating protein that controls dendritic spine morphogenesis. J Biol Chem. 2014;289:2600–2609. doi: 10.1074/jbc.M113.534636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katz E, Sims AH, Sproul D, Caldwell H, Dixon MJ, Meehan RR, Harrison DJ. Targeting of Rac GTPases blocks the spread of intact human breast cancer. Oncotarget. 2012;3:608–619. doi: 10.18632/oncotarget.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Onesto C, Shutes A, Picard V, Schweighoffer F, Der CJ. Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods Enzymol. 2008;439:111–129. doi: 10.1016/S0076-6879(07)00409-0. [DOI] [PubMed] [Google Scholar]
- 26.Kowluru A, Veluthakal R, Rhodes CJ, Kamath V, Syed I, Koch BJ. Protein Farnesylation–Dependent Raf/Extracellular Signal–Related Kinase Signaling Links to Cytoskeletal Remodeling to Facilitate Glucose-Induced Insulin Secretion in Pancreatic β-Cells. Diabetes. 2010;59:967–977. doi: 10.2337/db09-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, Kowluru A. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes. 2011;60:2843–2852. doi: 10.2337/db11-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kowluru A, Veluthakal R. Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes. 2005;54:3523–3529. doi: 10.2337/diabetes.54.12.3523. [DOI] [PubMed] [Google Scholar]
- 29.Kalwat MA, Yoder SM, Wang Z, Thurmond DC. A p21-activated kinase (PAK1) signaling cascade coordinately regulates F-actin remodeling and insulin granule exocytosis in pancreatic β cells. Biochem Pharmacol. 2013;85:808–816. doi: 10.1016/j.bcp.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li G, Regazzi R, Roche E, Wollheim CB. Blockade of mevalonate production by lovastatin attenuates bombesin and vasopressin potentiation of nutrient-induced insulin secretion from HIT-T15 cells. Biochem J. 1993;289:379–385. doi: 10.1042/bj2890379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baier A, Ndoh VN, Lacy P, Eitzen G. Rac1 and Rac2 control distinct events during antigen-stimulated mast cell exocytosis. J Leukoc Biol. 2014 doi: 10.1189/jlb.0513281. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 32.Kowluru A, Metz SA. Stimulation by prostaglandin E2 of a high-affinity GTPase in the secretory granules of normal rat and human pancreatic islets. Biochem J. 1994;297:399–406. doi: 10.1042/bj2970399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kowluru A, Kowluru RA. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem Pharmacol. 2014;88:275–283. doi: 10.1016/j.bcp.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen E, Li Y, Li Y, Shan L, Zhu H, Feng Q, Arnold JM, Peng T. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes. 2009;58:2386–2395. doi: 10.2337/db08-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowluru RA, Kowluru A, Veluthakal R, Mohammad G, Syed I, Santos JM, Mishra M. TIAM1-RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia. 2014;57:1047–1056. doi: 10.1007/s00125-014-3194-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elnakish MT, Hassanain HH, Janssen PM, Angelos MG, Khan M. Emerging role of oxidative stress in metabolic syndrome and cardiovascular diseases: important role of Rac/NADPH oxidase. J Pathol. 2013;231:290–300. doi: 10.1002/path.4255. [DOI] [PubMed] [Google Scholar]
- 37.Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol. 2011;81:965–975. doi: 10.1016/j.bcp.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida S, Ishizawa K, Ayuzawa N, Ueda K, Takeuchi M, Kawarazaki W, Fujita T, Nagase M. Local mineralocorticoid receptor activation and the role of Rac1 in obesity-related diabetic kidney disease. Nephron Exp Nephrol. 2014;126:16–24. doi: 10.1159/000358758. [DOI] [PubMed] [Google Scholar]