

Abstract
Hypertension is a major risk factor for cardiovascular disease. While the cause of hypertension is multifactorial, renal dysregulation of salt and water excretion is a major factor. All components of the renin angiotensin system are produced locally in the kidney, suggesting that intrarenal generation of angiotensin II plays a key role in blood pressure regulation. Here, we show that two mouse models lacking renal angiotensin converting enzyme (ACE) are protected against angiotensin II and L-NAME induced hypertension. In response to hypertensive stimuli, mice lacking renal ACE do not produce renal angiotensin II. These studies indicate that the intrarenal renin angiotensin system works as an entity separate from systemic angiotensin II generation. Renal ACE appears necessary for experimental hypertension.
Keywords: ACE, hypertension, kidney, RAS
Introduction
Whether measured in terms of mortality or cost of treatment, hypertension is a huge problem in the United States. Approximately 31% of Americans are hypertensive [1]. This disease is a major risk factor for stroke, atherosclerosis and congestive heart failure. Thus, it is not surprising that the control of blood pressure markedly reduces the morbidity and mortality associated with these diseases, the major killers of Americans [2,3]. Hypertension is the second most prevalent cause of end stage renal disease [2]. While the cause of hypertension is multifactorial, an excess of dietary salt and a positive correlation between sodium intake and blood pressure, termed salt-sensitivity, is often a major factor. Among hypertensive patients, at least one in three are salt-sensitive [1]. Further, it is estimated that at least 30 million Americans with normal blood pressures are salt-sensitive. This is important because salt-sensitivity itself (i.e. independent of blood pressure) is a risk factor for cardiovascular morbidity and mortality [4,5].
In a salt sensitive individual, inappropriate renal sodium retention drives an increase in circulating volume and blood pressure; the pressure natriuresis response (increase sodium excretion in response to elevated blood pressure) restores the circulating volume to normal at the expense of an increase in blood pressure until the sodium retaining stimulus is removed [6,7]. As we will discuss below, there is emerging evidence that stimuli that activate intrarenal angiotensin II generation inappropriately stimulate renal sodium transport, and blunt the pressure natriuresis response, which drives an elevation in blood pressure.
Angiotensin II is the end product of the renin-angiotensin system (RAS). The systemic generation of angiotensin II results from liver production of angiotensinogen, which is cleaved into the 10 amino acid precursor angiotensin I by renin, a protease produced by the juxtaglomerular apparatus (JGA). In turn, plasma angiotensin I is rapidly converted to the 8 amino acid angiotensin II in the lung where it is cleaved by the peptidase angiotensin converting enzyme (ACE) located on the surface of endothelium [8]. Angiotensin II has effects throughout the body that coordinately act to raise blood pressure. While the major loci for the production of angiotensinogen, renin and ACE are the liver, kidney, and lung respectively, these proteins are made in a variety of other tissues including several locations within the kidney. This is reviewed in detail below but studies have suggested that angiotensin II is not only generated systemically in plasma but also locally within individual tissues. These findings have led to persistent questions about the relative physiologic importance of systemic generation of angiotensin II versus the local RAS generation of angiotensin II within kidney, the organ most critical for blood pressure control. This issue is particularly important given renal transplantation studies suggesting that susceptibility to experimental hypertension in rodents and clinical human hypertension follows the kidney and that this is due in part to intrarenal effects of angiotensin II [9–11]. Here, we review studies in genetically modified mice indicating that the intrarenal generation of angiotensin II is essential in the development of several models of experimental hypertension. These studies support the concept that the intrarenal RAS functions as a separate entity, and not as a simple extension of systemic angiotensin II generation. Further, they show that angiotensin II generated locally in the kidney regulates renal function, including the handling of salt at several different locations along the nephron. The observation that local angiotensin II production and local regulation of renal sodium transporters are linked and indeed are obligatory in the development of hypertension, represents a new understanding of the mechanism underlying inappropriate regulation of salt and water balance by the kidney. This, in turn, directly leads to hypertension and the progressive cardiovascular disease that is so debilitating and lethal in modern society.
Local renal production of RAS proteins
Angiotensinogen
The presence of angiotensinogen in proximal tubules has been widely documented [12–14]. Studies in mice demonstrated increased renal angiotensinogen mRNA during angiotensin II-induced hypertension suggesting de novo synthesis in the kidney [15,16]. However, recently published data demonstrated that, under both normal and pathologic conditions, most proximal tubular angiotensinogen originates in the liver [17,18]. According to these studies, plasma angiotensinogen can be filtered and reabsorbed through a megalin-dependent mechanism [17,19]. Indeed, angiotensinogen accumulation in segments 1 and 2 of the proximal tubule results from systemic uptake, while angiotensinogen in segment 3 seems to be locally produced [20]. Although the relative contribution of systemic vs. renal angiotensinogen to hypertension and kidney disease has not been clearly elucidated, a recent manuscript suggests that the systemic RAS can stimulate renal angiotensinogen production and increase urinary angiotensinogen excretion [21]. Regardless of its origin, the intrarenal accumulation of angiotensinogen seems to play a pivotal role in hypertension. Consistent with this, transgenic mice over-expressing angiotensinogen in proximal tubular epithelium develop hypertension and renal injury without changes in the systemic RAS [22].
Besides its presence in the renal tubules, significant levels of angiotensinogen can also be found in the tubular fluid and the urine during hypertension and kidney disease [14,23]. It was previously demonstrated that plasma angiotensinogen can be filtered through the glomerulus [19]. The filtration fraction depends on the normal functioning of the glomerular filtration barrier. Indeed, impairment of the glomerular capillary functional integrity generated after podocyte injury significantly increased renal angiotensinogen [18]. Although not directly demonstrated, these studies collectively suggested that an increase in the leakage of plasma angiotensinogen into the tubular lumen could be the major mechanism of increased renal and urinary angiotensinogen in glomerular diseases [17,18]. However, virtually all filtered angiotensinogen is taken-up in the S1 and S2 segments of the proximal tubule [17], suggesting that urinary angiotensinogen is produced in the tubules through de novo pathways and secreted into the urine during renal injury. In line with these observations, hypertensive rats displayed an important elevation in endogenous, intrarenal-produced angiotensinogen in the urine while only a small increase in angiotensinogen of systemic (filtered) origin was observed [24].
Multiple studies show that patients with hypertension and renal disease display increased urinary angiotensinogen compared to normal subjects [25]. These findings support the concept that urinary angiotensinogen can be used as an index of intrarenal RAS activation, even before the development of proteinuria [26–28]. Clinical studies are exploring urinary angiotensinogen excretion in humans as a useful biomarker that closely correlates with systolic blood pressure and kidney angiotensin II content in the absence of elevated systemic angiotensin II. Because renin and ACE are also present in tubular fluid (see below), the formation of angiotensin I and angiotensin II in this part of the nephron is entirely possible, even when plasma renin is suppressed [29–31]. In line with this, studies in rats demonstrated a significant relationship between urinary angiotensinogen and renal angiotensin II content during angiotensin II-induced hypertension [32]. Although high tissue angiotensin II levels is thought to be primarily due to increased local formation [33,34], it recently became clear that AT1 receptor-mediated uptake of circulating angiotensin II may also contribute to the local angiotensin II accumulation in the kidney during hypertension [35,36].
Renin
Both plasma and intrarenal levels of renin are mainly produced by the granular cells of the JGA. However, the presence of renin in tubular fluid may be a consequence of renin production by cells other than juxtaglomerular cells. Indeed, certain kidney regions such as glomeruli, proximal tubules, connecting tubules and collecting ducts display positive renin immunoreactivity [14,37–39]. Since renin can be filtered, the presence of renin in areas different from the JGA was originally interpreted as simple uptake of the filtered protein. However, the observation of renin mRNA in mesangial and tubular cells confirms renin synthesis by cells other than those of the JGA [40]. Irrespective of the source, the presence of renin in tubular fluids significantly contributes to the local generation of angiotensin II along the nephron [19], and can be treated as a biomarker for renal RAS activation [41].
Remarkably, angiotensin II can differently regulate renin production in the JGA and distal segments of the nephron. While chronic infusion of angiotensin II inhibits renin secretion by the JGA, this octapeptide up-regulates renin production by principal cells of connecting tubules and cortical and medullary collecting ducts [42]. Thus, during juxtaglomerular renin suppression, increased expression of renin in the distal nephron provides constant intrarenal angiotensin II formation that maintains hypertension [31,42]. In line with these observations, in chronic angiotensin II-infused rats and mice, despite marked suppression of juxtaglomerular cell renin and plasma renin activity, the intrarenal and urinary levels of angiotensin II remained elevated [43,44]. Further studies indicated that angiotensin II can increase renin production in distal tubules by a mechanism independent of high blood pressure [45].
Angiotensin converting enzyme
Angiotensin I is effectively converted to angiotensin II by ACE, a membrane-bound metalloproteinase, predominantly expressed on the surface of endothelial cells [8]. In the kidney, ACE expression can be found in multiple cell types including endothelial cells, mesangial cells and podocytes [46,47]. However, by far, it is the brush border of proximal tubular epithelial cells that contains the majority of renal ACE [48]. ACE activity was also observed in the collecting duct fluid and urine [49]. Whether distal ACE activity is a consequence of actual local production [50] or just the uptake of ACE produced in the proximal tubule remains unclear. Interestingly, the pattern of renal ACE expression significantly differs between humans and commonly used experimental rodents [51]. In humans, ACE is predominantly expressed in the brush border of proximal tubular cells, while very little is observed in renal vascular endothelium. Also, no ACE is detected in the glomerular vasculature or the basolateral membrane of epithelial cells [51]. In contrast, relatively high expression of ACE was present in the renal vasculature of rats [51].
As observed for every component of the RAS, renal ACE expression can also be upregulated during hypertension and in response to renal damage [52]. Indeed, previous reports showed increased kidney ACE activity in the two-kidney-one-clip Goldblatt hypertensive model and in chronic angiotensin II-infused rats [43,53]. More recently, our laboratory demonstrated that both angiotensin II- and L-NAME-induced hypertension are associated with significantly increased ACE expression in the kidney [54,55].
Almost 15 years ago, a catalytically active homolog of ACE, ACE2, was discovered [56]. ACE2 is not inhibited by ACE inhibitors and is widely expressed in several areas of the kidney, including glomerular and epithelial cells, smooth muscle cells and endothelium [56,57]. In contrast to ACE, ACE2 cleaves angiotensin I to generate the inactive metabolite angiotensin-(1–9) and cleaves angiotensin II to yield the vasodilator peptide angiotensin-(1–7) [58]. From a pathophysiological perspective, ACE2 may counteract the role of ACE during the inappropriate RAS activation associated with hypertension and chronic kidney disease [58].
Angiotensin II receptors
Angiotensin II actions are mediated predominantly by two different receptors termed angiotensin II type 1 (AT1) and type 2 (AT2) receptors [59]. Most of the pro-hypertensive actions of angiotensin II in the kidney are mediated by the AT1 receptor [23]. In rodents, two AT1 receptors subtypes have been cloned and characterized. The AT1a receptor is expressed in the luminal and basolateral membranes of several segments of the nephron, including epithelial cells of the proximal tubule, thick ascending limb, macula densa, distal tubules and collecting duct, as well as in mesangial cells, renal microvasculature in both cortex and medulla, and smooth muscle cells of afferent and efferent arterioles. The AT1b receptor is less abundant and is restricted to the glomerulus and the afferent arteriole [23]. The regulation of the AT1 receptor during hypertension is complex and different between the renal vasculature and tubules [60]. Typically, high angiotensin II levels associated with low sodium diets decrease AT1 receptor expression in the glomerulus while increasing its tubular expression [60].
The AT2 receptor is widely expressed during fetal life and decreases significantly after birth, suggesting a role in the prenatal period [61]. In the adult kidney, AT2 is mainly restricted to vascular elements and tubular segments, especially the proximal tubule [62]. Although its role and significance remains unclear, increasing evidence suggests that AT2 receptor-mediated actions counteract AT1 receptor effects by increasing bradykinin and nitric oxide production [63]. Angiotensin II infusion into AT2 receptor knock-out mice exacerbated hypertension and renal damage, probably as a consequence of decreased bradykinin production [64].
Mouse models lacking renal ACE
The use of ‘knockout mice’ lacking individual components of the RAS has repeatedly demonstrated that interruption of this pathway is associated with a marked reduction of blood pressure and kidney pathology [65]. In fact, global interruption of the RAS is even more severe in humans and is most often associated with embryonic lethality [66,67]. Our group prepared mouse models in which genetic manipulation resulted in mice lacking renal ACE but having sufficient ACE production in other tissues that basal blood pressure is normal. Two such mouse models are termed ACE 3/3 and ACE 10/10 [68,69]. In ACE 3/3 mice, control of ACE expression by the albumin promoter results in ACE production by hepatocytes. In this model, renal ACE expression is only 14% of normal levels. In the ACE 10/10 model, ACE is under the control of the c-fms promoter, resulting in ACE expression by myelomonocytic cells such as monocytes and macrophages. In this model, renal tissues essentially make no ACE. Both ACE 3/3 and ACE 10/10 mice produce sufficient ACE that serum levels of angiotensin II are near normal, there is normal kidney structure, and blood pressure levels are normal under basal conditions. These and other mouse models suggest that, given the kidney’s ability to regulate renin production, the localization of ACE expression in aberrant tissues is well tolerated with normal renal structure and function under baseline conditions [68,69].
We have generated experimental hypertension in both ACE 10/10 and ACE 3/3 mice [54,55]. Each strain of mice was tested with two separate models: the subcutaneous infusion of angiotensin II for two weeks and the administration in drinking water of the nitric oxide synthase inhibitor L-NAME for four weeks. Wild-type (WT) mice were matched for each strain. The blood pressure in ACE 10/10 and WT mice treated with angiotensin II was measured by telemetry, while all other blood pressure measurements were obtained by tail cuff analysis. These studies showed a huge difference in the responses of WT vs. ACE 10/10 or ACE 3/3 mice (Figure 1). For example, after 12 days of angiotensin II administration (400 ng/kg/min by minipump), the mean arterial blood pressure was 22 mmHg lower in ACE 10/10 mice as compared to WT. In ACE 3/3 mice, systolic blood pressure was 31 mmHg lower than in WT. After 4 weeks of L-NAME, systolic blood pressure was 22 and 25 mmHg lower in ACE 10/10 and ACE 3/3 than in WT mice (Figure 1). These data are highly significant, but even more profound is the overall observation that using two different hypertension protocols in two different strains of mice, the lack of renal ACE was associated with consistent protection against experimental hypertension.
Figure 1.
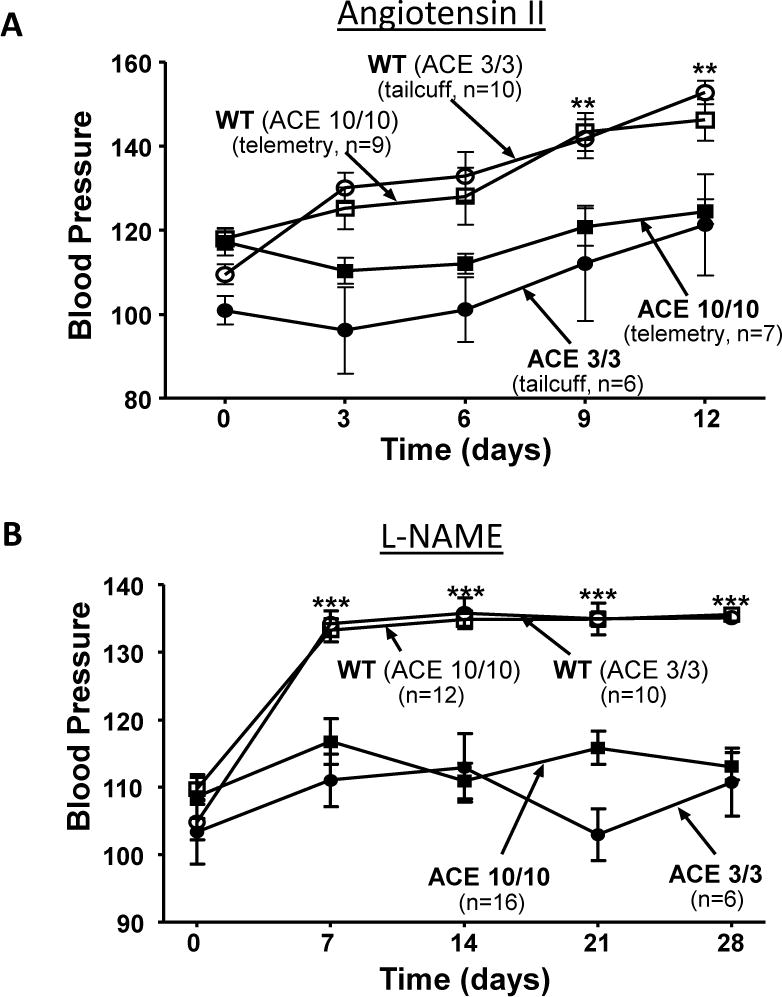
ACE 10/10 and ACE 3/3 mice were made hypertensive by either (A) subcutaneous angiotensin II (400 ng/kg/min by minipump) or by (B) oral administration of L-NAME (5 mg/ml in drinking water). Each line of genetically modified mice was matched to littermate wild type (WT) mice. The mean arterial blood pressure of ACE 10/10 and control WT mice treated with angiotensin II was determined by telemetry (A). The systolic blood pressure of all other groups of mice was determined by tail cuff (A and B). The two strains of mice lacking renal ACE were resistant to these two hypertensive stimuli. ** p<0.01, *** p<0.001
While both the administration of L-NAME and angiotensin II result in hypertension, there are some important differences between these two models. L-NAME inhibits nitric oxide generation, resulting in hypertensive changes that secondarily act to suppress plasma renin activity and systemic angiotensin II production [70,71]. Thus, this model is associated with low systemic angiotensin II levels. In contrast, administration of angiotensin II by osmotic minipump essentially clamps the animals at a high systemic concentration of angiotensin II. While the exogenous administration of angiotensin II suppresses the endogenous production of renin by the JGA, substantial data from Navar and colleagues show that angiotensin II has a paradoxical effect by stimulating renal tubular epithelium to produce angiotensinogen [23]. This and other components of the renal RAS increase local renal concentrations of angiotensin II. Perhaps, this was best demonstrated in a study by Shao et al. in which rats were infused with a form of angiotensin II having valine at position 5 (Val(5)-Ang II) in place of the endogenous isoleucine (Ile(5)-Ang II) [34]. While Val(5)-Ang II has all the vasoconstrictive and hypertensive effects of Ile(5)-Ang II, these two forms can be separated by HPLC. The systemic infusion of Val(5)-Ang II induced hypertension in rats and increased renal concentration and urinary excretion of endogenous Ile(5)-Ang II by 70% and 93%, respectively [34].
We have also measured mouse renal concentrations of angiotensin II and angiotensinogen by RIA and Western blot. As shown in Figure 2, WT mice increase renal concentrations of angiotensinogen and angiotensin II in response to exogenous angiotensin II administration. In contrast, ACE 10/10 mice treated with angiotensin II in an identical fashion showed a much smaller increase in renal angiotensinogen and renal angiotensin II peptide (Figure 2). Essentially, the identical pattern was found in mice made hypertensive by 4 weeks of L-NAME administration. Despite the fact that L-NAME is associated with low systemic angiotensin II levels (hypertension, in this instance, is induced by the inhibition of nitric formation), local kidney concentrations of angiotensinogen, ACE and angiotensin II are significantly higher in WT mice than in equivalently treated ACE 10/10 mice [55]. This is dramatically illustrated by staining kidney sections with a highly specific antibody that binds angiotensin II (Figure 3). In WT mice, hypertension induced by L-NAME resulted in a marked increase in renal angiotensin II reactivity. In contrast, ACE 10/10 mice, which lack renal ACE, have substantially lower levels of renal angiotensin II when treated in an identical fashion [55]. Thus, these data support the concept that renal concentrations of angiotensin II are critical in establishing hypertension. This appears to be true under conditions of both high and low systemic angiotensin II concentrations.
Figure 2.

Wild-type and ACE 10/10 mice were made hypertensive by either subcutaneous angiotensin II or by oral L-NAME administration. Angiotensin II quantification (A) was performed by radioimmunoassay and renal angiotensinogen abundance (B) was analyzed by Western blot in total kidney homogenates, *p<0.05; **p<0.01; ***p<0.001. WT mice displayed significant accumulation of renal angiotensin II and angiotensinogen. The absence of renal ACE blunted or significantly reduced the accumulation of these RAS components in the kidney.
Figure 3.
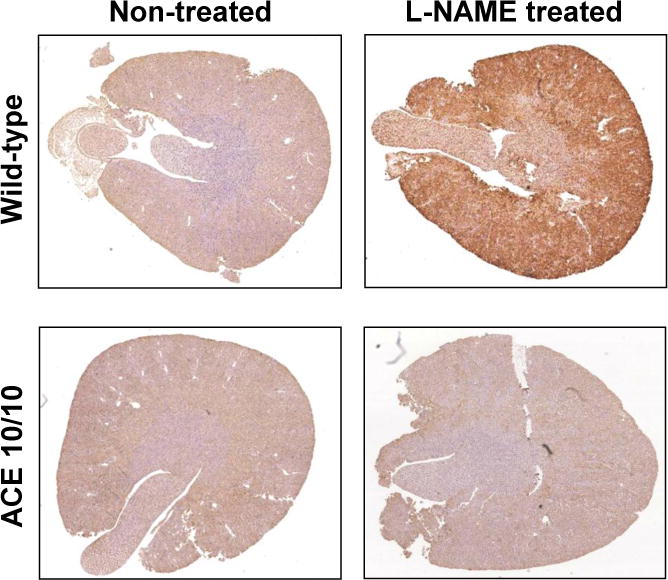
Kidney sections from wild type (WT) and ACE 10/10 mice were obtained before and after a 2 week treatment with L-NAME (5 mg/ml). The kidneys were immunostained with an antibody detecting angiotensin II (Phoenix Pharmaceuticals, Inc., Burlingame, CA). This antibody was previously shown to be very specific for angiotensin II [55]. Following L-NAME induced hypertension, ACE 10/10 mice have much less intrarenal angiotensin II than WT mice.
The concept that a local renal RAS has a major effect on blood pressure may strike some as unexpected. Additional support of this idea comes again from analysis of genetically modified mice called ACE 9/9 in which ACE production is only found in renal tubular epithelium [72]. In other words, the only ACE available to this animal is on the plasma membranes of tubular epithelial cells. Because these mice lack endothelial ACE and have very low concentrations of plasma ACE, they have very low sera concentrations of angiotensin II and are hypotensive. However, this model is perfectly suited to answer the question as to whether renal conversion of angiotensin I to angiotensin II can influence systemic blood pressure. Specifically, when this model was infused with systemic angiotensin I, these mice increased renal angiotensin II, increased urinary angiotensin II excretion and increased systemic blood pressure by 41 mmHg. When WT mice were treated in an identical fashion, systemic blood pressures increased by 36 mmHg. Thus, mice having only renal tubular ACE were able to convert angiotensin I to angiotensin II and raise systemic blood pressure as fast and as much as WT mice [72].
Given the evidence that the local generation of renal angiotensin II is important in blood pressure regulation, it is appropriate to ask what are the precise physiologic mechanisms regulating blood pressure. While systemic angiotensin II infusion is associated with vasoconstriction, many of the anti-natriuretic effects of angiotensin II are due to increases in renal tubular sodium transporter abundance and activation [41,73–75]. Detailed analysis of sodium transporter levels in WT and ACE 10/10 mice at baseline revealed no significant differences in abundance and/or activation (i.e. phosphorylation) of NHE, NKCC2, NCC or ENaC [54,55]. Treatment of WT mice with angiotensin II markedly increased expression and phosphorylation of transporters found in the thick ascending limb and in the distal nephron. WT mice had about a 3-fold increase in phosphorylated NKCC2 and a 1.6- and 5-fold increase in NCC abundance and phosphorylation. Remarkably, these changes were absent in ACE 10/10 mice [54]. In L-NAME induced hypertension, systemic angiotensin II levels are not elevated. However, in WT mice, kidney levels of angiotensin II are elevated and blunt the expected physiologic compensation of decreased sodium transporter abundance and activity. In contrast, ACE 10/10 mice maintain low intrarenal concentrations of angiotensin II and are able to further downregulate tubular sodium reabsorption, excrete more sodium, and maintain a normal blood pressure [55]. Thus, in both hypertension models, data support the hypothesis that the local concentration of angiotensin II in the kidney has a direct effect on renal sodium transporters that can both inappropriately raise sodium reabsorption as well as blunt pressure natriuresis response. In mice lacking renal ACE, reduced kidney angiotensin II reduces the anti-natriuresis, promotes salt loss, and thus protects against hypertension.
Conclusion
Given the kidney’s ability to lose large amounts of salt and water, a profound paradox is why so many people develop hypertension. While the solution to this paradox is not yet available, we now know that the RAS and the kidney are two of the major determinants of blood pressure. It was then not such a large step to wonder about the RAS in the kidney. There is a lively discussion of exactly how important is the systemic vs. the intrarenal RAS [76]. However, as discussed, several different mouse models suggest that, at least in rodents, the intrarenal RAS and the intrarenal generation of angiotensin II are critical in facilitating experimental hypertension. Perhaps a high salt diet, inflammation, or other pathologic processes initiate inappropriate activation of intrarenal RAS [77]. But ultimately, it seems that renal ACE and the intrarenal generation of angiotensin II inappropriately stimulate renal sodium handling and blunt pressure natriuresis, and that the resulting insufficient loss of salt and water mandates a rise in blood pressure to control effective circulating volume.
Highlights.
All components of the RAS are expressed in the kidney
Evidence suggests that the renal RAS functions as a separate entity from systemic RAS
Local generation of angiotensin II by renal ACE is essential for hypertension
Mice lacking renal ACE are protected against hypertension
Acknowledgments
The authors thank Tea Janjulia for technical support, and Brian Taylor for administrative support. This work was supported by NIH grants R00 DK083455, R03 DK101592, T32 DK007770, R01 HL110353, R01 DK083785, AHA Beginning Grant-in-Aid 13BGIA14680069.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Recently published papers of particular interest have been highlighted as:
* Of importance
** Of major importance
- 1.Felder RA, White MJ, Williams SM, Jose PA. Diagnostic tools for hypertension and salt sensitivity testing. Curr Opin Nephrol Hypertens. 2013;22:65–76. doi: 10.1097/MNH.0b013e32835b3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2*.Wadei HM, Textor SC. The role of the kidney in regulating arterial blood pressure. Nat Rev Nephrol. 2012;8:602–609. doi: 10.1038/nrneph.2012.191. This conprehensive review summerizes different disorders that impair kidney function and lead to hypertension. [DOI] [PubMed] [Google Scholar]
- 3.He FJ, Jenner KH, Macgregor GA. WASH-world action on salt and health. Kidney Int. 2010;78:745–753. doi: 10.1038/ki.2010.280. [DOI] [PubMed] [Google Scholar]
- 4.Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37:429–432. doi: 10.1161/01.hyp.37.2.429. [DOI] [PubMed] [Google Scholar]
- 5.Frisoli TM, Schmieder RE, Grodzicki T, Messerli FH. Salt and hypertension: is salt dietary reduction worth the effort? Am J Med. 2012;125:433–439. doi: 10.1016/j.amjmed.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 6.Guyton AC. Physiologic regulation of arterial pressure. Am J Cardiol. 1961;8:401–407. doi: 10.1016/0002-9149(61)90159-x. [DOI] [PubMed] [Google Scholar]
- 7.Guyton AC. The surprising kidney-fluid mechanism for pressure control–its infinite gain! Hypertension. 1990;16:725–730. doi: 10.1161/01.hyp.16.6.725. [DOI] [PubMed] [Google Scholar]
- 8**.Bernstein KE, Ong FS, Blackwell WL, Shah KH, Giani JF, Gonzalez-Villalobos RA, Shen XZ, Fuchs S, Touyz RM. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol Rev. 2013;65:1–46. doi: 10.1124/pr.112.006809. This review summerizes recent advances in understanding the pleotropic biological functions of angiotensin-converting enzyme. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianchi G, Fox U, Di Francesco GF, Giovanetti AM, Pagetti D. Blood pressure changes produced by kidney cross-transplantation between spontaneously hypertensive rats and normotensive rats. Clin Sci Mol Med. 1974;47:435–448. doi: 10.1042/cs0470435. [DOI] [PubMed] [Google Scholar]
- 10.Guidi E, Menghetti D, Milani S, Montagnino G, Palazzi P, Bianchi G. Hypertension may be transplanted with the kidney in humans: a long-term historical prospective follow-up of recipients grafted with kidneys coming from donors with or without hypertension in their families. J Am Soc Nephrol. 1996;7:1131–1138. doi: 10.1681/ASN.V781131. [DOI] [PubMed] [Google Scholar]
- 11.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobori H, Ozawa Y, Suzaki Y, Prieto-Carrasquero MC, Nishiyama A, Shoji T, Cohen EP, Navar LG. Young Scholars Award Lecture: Intratubular angiotensinogen in hypertension and kidney diseases. Am J Hypertens. 2006;19:541–550. doi: 10.1016/j.amjhyper.2005.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobori H, Harrison-Bernard LM, Navar LG. Enhancement of angiotensinogen expression in angiotensin II-dependent hypertension. Hypertension. 2001;37:1329–1335. doi: 10.1161/01.hyp.37.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, Hillas E, Zhang S, Cheng T, Inagami T, Ward K, et al. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension. 1999;34:1265–1274. doi: 10.1161/01.hyp.34.6.1265. [DOI] [PubMed] [Google Scholar]
- 15.Kobori H, Harrison-Bernard LM, Navar LG. Expression of angiotensinogen mRNA and protein in angiotensin II-dependent hypertension. J Am Soc Nephrol. 2001;12:431–439. doi: 10.1681/asn.v123431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Villalobos RA, Seth DM, Satou R, Horton H, Ohashi N, Miyata K, Katsurada A, Tran DV, Kobori H, Navar LG. Intrarenal angiotensin II and angiotensinogen augmentation in chronic angiotensin II-infused mice. Am J Physiol Renal Physiol. 2008;295:F772–779. doi: 10.1152/ajprenal.00019.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, Nishiyama A, Ichikawa I. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol. 2012;23:1181–1189. doi: 10.1681/ASN.2011121159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsusaka T, Niimura F, Pastan I, Shintani A, Nishiyama A, Ichikawa I. Podocyte injury enhances filtration of liver-derived angiotensinogen and renal angiotensin II generation. Kidney Int. 2014;85:1068–1077. doi: 10.1038/ki.2013.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pohl M, Kaminski H, Castrop H, Bader M, Himmerkus N, Bleich M, Bachmann S, Theilig F. Intrarenal renin angiotensin system revisited: role of megalin-dependent endocytosis along the proximal nephron. J Biol Chem. 2010;285:41935–41946. doi: 10.1074/jbc.M110.150284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramkumar N, Kohan DE. Proximal tubule angiotensinogen modulation of arterial pressure. Curr Opin Nephrol Hypertens. 2013;22:32–36. doi: 10.1097/MNH.0b013e328359dbed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramkumar N, Stuart D, Ying J, Kohan DE. A possible interaction between systemic and renal angiotensinogen in the control of blood pressure. Am J Hypertens. 2013;26:473–480. doi: 10.1093/ajh/hps078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sachetelli S, Liu Q, Zhang SL, Liu F, Hsieh TJ, Brezniceanu ML, Guo DF, Filep JG, Ingelfinger JR, Sigmund CD, et al. RAS blockade decreases blood pressure and proteinuria in transgenic mice overexpressing rat angiotensinogen gene in the kidney. Kidney Int. 2006;69:1016–1023. doi: 10.1038/sj.ki.5000210. [DOI] [PubMed] [Google Scholar]
- 23**.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. This review summerizes the current understanding of the independent regulation of the intrarenal RAS and discusses how its inappropiate activation contributes to hypertension and renal injury. [DOI] [PubMed] [Google Scholar]
- 24.Nakano D, Kobori H, Burford JL, Gevorgyan H, Seidel S, Hitomi H, Nishiyama A, Peti-Peterdi J. Multiphoton imaging of the glomerular permeability of angiotensinogen. J Am Soc Nephrol. 2012;23:1847–1856. doi: 10.1681/ASN.2012010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Navar LG. Translational studies on augmentation of intratubular renin-angiotensin system in hypertension. Kidney Int Suppl (2011) 2013;3:321–325. doi: 10.1038/kisup.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saito T, Urushihara M, Kotani Y, Kagami S, Kobori H. Increased urinary angiotensinogen is precedent to increased urinary albumin in patients with type 1 diabetes. Am J Med Sci. 2009;338:478–480. doi: 10.1097/MAJ.0b013e3181b90c25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Kobori H, Alper AB, Jr, Shenava R, Katsurada A, Saito T, Ohashi N, Urushihara M, Miyata K, Satou R, Hamm LL, et al. Urinary angiotensinogen as a novel biomarker of the intrarenal renin-angiotensin system status in hypertensive patients. Hypertension. 2009;53:344–350. doi: 10.1161/HYPERTENSIONAHA.108.123802. This paper proposes urinary angiotensinogen is a potential novel biomarker of the intrarenal RAS status in hypertensive patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto T, Nakagawa T, Suzuki H, Ohashi N, Fukasawa H, Fujigaki Y, Kato A, Nakamura Y, Suzuki F, Hishida A. Urinary angiotensinogen as a marker of intrarenal angiotensin II activity associated with deterioration of renal function in patients with chronic kidney disease. J Am Soc Nephrol. 2007;18:1558–1565. doi: 10.1681/ASN.2006060554. [DOI] [PubMed] [Google Scholar]
- 29.Navar LG, Lewis L, Hymel A, Braam B, Mitchell KD. Tubular fluid concentrations and kidney contents of angiotensins I and II in anesthetized rats. J Am Soc Nephrol. 1994;5:1153–1158. doi: 10.1681/ASN.V541153. [DOI] [PubMed] [Google Scholar]
- 30.Kimbrough HM, Jr, Vaughan ED, Jr, Carey RM, Ayers CR. Effect of intrarenal angiotensin II blockade on renal function in conscious dogs. Circ Res. 1977;40:174–178. doi: 10.1161/01.res.40.2.174. [DOI] [PubMed] [Google Scholar]
- 31.Siragy HM, Carey RM. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. Am J Nephrol. 2010;31:541–550. doi: 10.1159/000313363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobori H, Harrison-Bernard LM, Navar LG. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int. 2002;61:579–585. doi: 10.1046/j.1523-1755.2002.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Kats JP, Schalekamp MA, Verdouw PD, Duncker DJ, Danser AH. Intrarenal angiotensin II: interstitial and cellular levels and site of production. Kidney Int. 2001;60:2311–2317. doi: 10.1046/j.1523-1755.2001.00049.x. [DOI] [PubMed] [Google Scholar]
- 34*.Shao W, Seth DM, Navar LG. Augmentation of endogenous intrarenal angiotensin II levels in Val5-ANG II-infused rats. Am J Physiol Renal Physiol. 2009;296:F1067–1071. doi: 10.1152/ajprenal.90596.2008. This manuscript shows that increases in intrarenal Ang II levels during chronic Ang II infusions involve substantial stimulation of endogenous Ang II formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li XC, Zhuo JL. In vivo regulation of AT1a receptor-mediated intracellular uptake of [125I]Val5-ANG II in the kidneys and adrenals of AT1a receptor-deficient mice. Am J Physiol Renal Physiol. 2008;294:F293–302. doi: 10.1152/ajprenal.00398.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zou LX, Imig JD, von Thun AM, Hymel A, Ono H, Navar LG. Receptor-mediated intrarenal angiotensin II augmentation in angiotensin II-infused rats. Hypertension. 1996;28:669–677. doi: 10.1161/01.hyp.28.4.669. [DOI] [PubMed] [Google Scholar]
- 37.Moe OW, Ujiie K, Star RA, Miller RT, Widell J, Alpern RJ, Henrich WL. Renin expression in renal proximal tubule. J Clin Invest. 1993;91:774–779. doi: 10.1172/JCI116296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang JJ, Toma I, Sipos A, Meer EJ, Vargas SL, Peti-Peterdi J. The collecting duct is the major source of prorenin in diabetes. Hypertension. 2008;51:1597–1604. doi: 10.1161/HYPERTENSIONAHA.107.107268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, Ozawa Y, Hering-Smith KS, Hamm LL, Navar LG. Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension. 2004;44:223–229. doi: 10.1161/01.HYP.0000135678.20725.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prieto MC, Gonzalez AA, Navar LG. Evolving concepts on regulation and function of renin in distal nephron. Pflugers Arch. 2013;465:121–132. doi: 10.1007/s00424-012-1151-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Lubbe N, Lim CH, Fenton RA, Meima ME, Jan Danser AH, Zietse R, Hoorn EJ. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int. 2011;79:66–76. doi: 10.1038/ki.2010.290. [DOI] [PubMed] [Google Scholar]
- 42.Prieto-Carrasquero MC, Kobori H, Ozawa Y, Gutierrez A, Seth D, Navar LG. AT1 receptor-mediated enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Am J Physiol Renal Physiol. 2005;289:F632–637. doi: 10.1152/ajprenal.00462.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonzalez-Villalobos RA, Satou R, Ohashi N, Semprun-Prieto LC, Katsurada A, Kim C, Upchurch GM, Prieto MC, Kobori H, Navar LG. Intrarenal mouse renin-angiotensin system during ANG II-induced hypertension and ACE inhibition. Am J Physiol Renal Physiol. 2010;298:F150–157. doi: 10.1152/ajprenal.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L, Gonzalez AA, McCormack M, Seth DM, Kobori H, Navar LG, Prieto MC. Increased renin excretion is associated with augmented urinary angiotensin II levels in chronic angiotensin II-infused hypertensive rats. Am J Physiol Renal Physiol. 2011;301:F1195–1201. doi: 10.1152/ajprenal.00339.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prieto-Carrasquero MC, Botros FT, Pagan J, Kobori H, Seth DM, Casarini DE, Navar LG. Collecting duct renin is upregulated in both kidneys of 2-kidney, 1-clip goldblatt hypertensive rats. Hypertension. 2008;51:1590–1596. doi: 10.1161/HYPERTENSIONAHA.108.110916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alhenc-Gelas F, Baussant T, Hubert C, Soubrier F, Corvol P. The angiotensin converting enzyme in the kidney. J Hypertens Suppl. 1989;7:S9–13. doi: 10.1097/00004872-198909007-00003. discussion S14. [DOI] [PubMed] [Google Scholar]
- 47.Liebau MC, Lang D, Bohm J, Endlich N, Bek MJ, Witherden I, Mathieson PW, Saleem MA, Pavenstadt H, Fischer KG. Functional expression of the renin-angiotensin system in human podocytes. Am J Physiol Renal Physiol. 2006;290:F710–719. doi: 10.1152/ajprenal.00475.2004. [DOI] [PubMed] [Google Scholar]
- 48.Schulz WW, Hagler HK, Buja LM, Erdos EG. Ultrastructural localization of angiotensin I-converting enzyme (EC 3.4.15.1) and neutral metalloendopeptidase (EC 3.4.24.11) in the proximal tubule of the human kidney. Lab Invest. 1988;59:789–797. [PubMed] [Google Scholar]
- 49.Casarini DE, Boim MA, Stella RC, Krieger-Azzolini MH, Krieger JE, Schor N. Angiotensin I-converting enzyme activity in tubular fluid along the rat nephron. Am J Physiol. 1997;272:F405–409. doi: 10.1152/ajprenal.1997.272.3.F405. [DOI] [PubMed] [Google Scholar]
- 50.Redublo Quinto BM, Camargo de Andrade MC, Ronchi FA, Santos EL, Alves Correa SA, Shimuta SI, Pesquero JB, Mortara RA, Casarini DE. Expression of angiotensin I-converting enzymes and bradykinin B2 receptors in mouse inner medullary-collecting duct cells. Int Immunopharmacol. 2008;8:254–260. doi: 10.1016/j.intimp.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 51.Metzger R, Bohle RM, Pauls K, Eichner G, Alhenc-Gelas F, Danilov SM, Franke FE. Angiotensin-converting enzyme in non-neoplastic kidney diseases. Kidney Int. 1999;56:1442–1454. doi: 10.1046/j.1523-1755.1999.00660.x. [DOI] [PubMed] [Google Scholar]
- 52.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension. 2011;57:355–362. doi: 10.1161/HYPERTENSIONAHA.110.163519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prieto MC, Gonzalez-Villalobos RA, Botros FT, Martin VL, Pagan J, Satou R, Lara LS, Feng Y, Fernandes FB, Kobori H, et al. Reciprocal changes in renal ACE/ANG II and ACE2/ANG 1–7 are associated with enhanced collecting duct renin in Goldblatt hypertensive rats. Am J Physiol Renal Physiol. 2011;300:F749–755. doi: 10.1152/ajprenal.00383.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54**.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, et al. The absence of intrarenal ACE protects against hypertension. J Clin Invest. 2013;123:2011–2023. doi: 10.1172/JCI65460. This manuscript demonstrates a key role of renal ACE in the development of angiotensin II-induced hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55**.Giani JF, Janjulia T, Kamat N, Blackwell WL, Shah K, Shen XZ, Fuchs S, Delpire E, Bernstein KE, McDonough AA, et al. Renal ACE is essential for the hypertension induced by nitric oxide synthesis inhibition. J Am Soc Nephrol. 2014;25:2752–2763. doi: 10.1681/ASN.2013091030. This study demonstrates that renal ACE-derived angiotensin II accumulation plays a key role in the development of L-NAME hypertension. A complete analysis of sodium transporters along the nephon is offered. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 57.Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D. Glomerular localization and expression of Angiotensin-converting enzyme 2 and Angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol. 2006;17:3067–3075. doi: 10.1681/ASN.2006050423. [DOI] [PubMed] [Google Scholar]
- 58.Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1–7): an evolving story in cardiovascular regulation. Hypertension. 2006;47:515–521. doi: 10.1161/01.HYP.0000196268.08909.fb. [DOI] [PubMed] [Google Scholar]
- 59.Atlas SA. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm. 2007;13:9–20. doi: 10.18553/jmcp.2007.13.s8-b.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension. 2002;39:316–322. doi: 10.1161/hy0202.103821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Norwood VF, Garmey M, Wolford J, Carey RM, Gomez RA. Novel expression and regulation of the renin-angiotensin system in metanephric organ culture. Am J Physiol Regul Integr Comp Physiol. 2000;279:R522–530. doi: 10.1152/ajpregu.2000.279.2.R522. [DOI] [PubMed] [Google Scholar]
- 62.Carey RM, Wang ZQ, Siragy HM. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension. 2000;35:155–163. doi: 10.1161/01.hyp.35.1.155. [DOI] [PubMed] [Google Scholar]
- 63.Siragy HM, Carey RM. Protective role of the angiotensin AT2 receptor in a renal wrap hypertension model. Hypertension. 1999;33:1237–1242. doi: 10.1161/01.hyp.33.5.1237. [DOI] [PubMed] [Google Scholar]
- 64.Siragy HM, Inagami T, Ichiki T, Carey RM. Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proc Natl Acad Sci U S A. 1999;96:6506–6510. doi: 10.1073/pnas.96.11.6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Doan TN, Gletsu N, Cole J, Bernstein KE. Genetic manipulation of the renin-angiotensin system. Curr Opin Nephrol Hypertens. 2001;10:483–491. doi: 10.1097/00041552-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 66.Gribouval O, Moriniere V, Pawtowski A, Arrondel C, Sallinen SL, Saloranta C, Clericuzio C, Viot G, Tantau J, Blesson S, et al. Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum Mutat. 2012;33:316–326. doi: 10.1002/humu.21661. [DOI] [PubMed] [Google Scholar]
- 67.Michaud A, Acharya KR, Masuyer G, Quenech’du N, Gribouval O, Moriniere V, Gubler MC, Corvol P. Absence of cell surface expression of human ACE leads to perinatal death. Hum Mol Genet. 2014;23:1479–1491. doi: 10.1093/hmg/ddt535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen XZ, Li P, Weiss D, Fuchs S, Xiao HD, Adams JA, Williams IR, Capecchi MR, Taylor WR, Bernstein KE. Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am J Pathol. 2007;170:2122–2134. doi: 10.2353/ajpath.2007.061205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cole J, Quach DL, Sundaram K, Corvol P, Capecchi MR, Bernstein KE. Mice lacking endothelial angiotensin-converting enzyme have a normal blood pressure. Circ Res. 2002;90:87–92. doi: 10.1161/hh0102.102360. [DOI] [PubMed] [Google Scholar]
- 70.Pollock DM, Polakowski JS, Divish BJ, Opgenorth TJ. Angiotensin blockade reverses hypertension during long-term nitric oxide synthase inhibition. Hypertension. 1993;21:660–666. doi: 10.1161/01.hyp.21.5.660. [DOI] [PubMed] [Google Scholar]
- 71.Johnson RA, Freeman RH. Renin release in rats during blockade of nitric oxide synthesis. Am J Physiol. 1994;266:R1723–1729. doi: 10.1152/ajpregu.1994.266.6.R1723. [DOI] [PubMed] [Google Scholar]
- 72*.Gonzalez-Villalobos RA, Billet S, Kim C, Satou R, Fuchs S, Bernstein KE, Navar LG. Intrarenal angiotensin-converting enzyme induces hypertension in response to angiotensin I infusion. J Am Soc Nephrol. 2011;22:449–459. doi: 10.1681/ASN.2010060624. This paper demonstrates that intrarenal ACE-derived angiotensin II formation, even in the absence of systemic ACE, increases kidney angiotensin II levels and promotes the development of hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73*.Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, et al. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–475. doi: 10.1016/j.cmet.2011.03.001. This paper demonstrate that abrogation of AT1a receptor signaling in the proximal tubule alone is sufficient to lower blood pressure, despite intact vascular responses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, et al. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci U S A. 2009;106:4384–4389. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem. 2012;287:660–671. doi: 10.1074/jbc.M111.298919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Campbell DJ. Clinical relevance of local Renin Angiotensin systems. Front Endocrinol (Lausanne) 2014;5:113. doi: 10.3389/fendo.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodriguez-Iturbe B. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N Engl J Med. 2002;346:913–923. doi: 10.1056/NEJMra011078. [DOI] [PubMed] [Google Scholar]