

Abstract
Three-helix bundles and coiled-coil motifs are well-established de novo designed scaffolds that have been investigated for their metal-binding and catalytic properties. Satisfaction of the primary coordination sphere for a given metal is sufficient to introduce catalytic activity and a given structure may catalyze different reactions dependent on the identity of the incorporated metal. Here we describe recent contributions in the de novo design of metalloenzymes based on three-helix bundles and coiled-coil motifs, focusing on non-heme systems for hydrolytic and redox chemistry.
Introduction
The relationship between the structure and function of proteins has been a subject of great interest in protein biochemistry since its inception. The interplay between primary, secondary, and tertiary structure has been studied by the likes of chemists such as Linus Pauling, who famously predicted with great accuracy the α helix and β sheet based only on idealized hydrogen bonding situations. Essentially, there are two ways to approach the fundamental question of, “how do proteins work?” One approach is the top-down method of classic protein biochemistry—using mutations, inhibitors, and other tools to perturb or abrogate function to understand key structural features that are required for folding, a specific interaction, or catalysis. The second, bottom-up approach is protein design, where features are incorporated into a design with the goal of understanding to what extent a certain structural element may contribute to a feature found in native proteins.
An estimated one-half to one-third of native proteins require a metal ion for proper folding or catalytic function.[1] Consequently, the field of metalloprotein design has evolved alongside the greater protein design field. Much of this metalloprotein design field has been reviewed extensively in recent articles.[2–6] Protein design can be broadly classified into protein redesign and de novo protein design. Protein redesign uses native proteins as scaffolds and then builds new functions or binding sites into the pre-existing scaffold. De novo protein design seeks to generate a protein “from scratch” and features scaffolds whose primary sequence bears no relation to native proteins, thus identifying the basic, minimal features for function. One significant advantage of de novo design is that a single domain may be explored without the complications of allostery or multiple metal sites. From the perspective of bioinorganic chemistry, these approaches allow researchers to evaluate the extent to which the primary and secondary coordination spheres of a metal affect a metalloprotein’s activity and function. Recent papers from our lab and others have indicated that reproducing the primary coordination sphere alone can confer a certain level of activity to catalytic models.
To date, most established de novo designed protein scaffolds consist of α helical secondary structures, which either self-assemble to form coiled coils or fold as helix-loop-helix motifs into a helical bundle [2], although catalytic metalloenzymes have recently been reported using b-sheet constructs [7]. The α helical regions of the de novo designed a-helical proteins are based on the heptad repeat strategy: where seven amino acids form repeats (with residues a–g see Figure 1a) in which the a and d positions are occupied by hydrophobic residues that face inwards in the coiled coil or helix bundle, driving the folding and association of the peptides. Substitution of a residue in the a or d position with a coordinating amino acid provides a metal-binding site. The TRI family of peptides consists of three heptad repeats with the sequence LKALEEK, which self-assemble into three-stranded coiled-coils above pH 5.5 (Figure 1b).[8,9] A related three-helix bundle protein, designated α3D, was designed by adding loop regions and varying the sequence of the heptads to create a protein that exhibits native-like folding and conformational specificity (Figure 1c).[10,11] Much of the early effort to design non-heme metal centers focused on developing stable scaffolds for transitions metals, heavy metalloids and lanthanides. In this Current Opinion we will outline recent efforts to build efficient non-heme redox and hydrolytic enzymes in de novo designed three-stranded coiled-coils and three-helix bundle proteins.
Figure 1.

a) Helix wheel schematic showing the relative positions of amino acids a–g in a heptad repeat. Residues a and d point in the same direction such that when occupied by hydrophobic residues, they will drive the association of two or more helices. b) The TRI scaffold self assembles into three stranded coiled coils. Substitution of two leucine layers by cysteine and histidine creates a bimetallic construct, HgIISZnIIN(TRIL9CL23H)3, the fasted reported CA mimic for CO2 hydration. Shown is the crystal structure for HgIISZnIIN(CSL9CL23H)3, a crystallographic analogue for the TRI system (PDB: 3PBJ). c) Model of α3DH3 based on the solution structure of α3D (PDB: 2DSX). α3D derivatives fold into an antiparallel three-helix bundle. α3DH3 incorporates three histidine residues for metal binding and has been shown to be capable of CO2 hydration.
Zinc Hydrolytic Enzymes
While enzymes of all classes use zinc as part of the active site or for structural stabilization, perhaps the most well known role for ZnII is in the active site of carbonic anhydrase (CA). Carbonic anhydrase is a hydrolase enzyme that catalyzes the reversible hydration of carbon dioxide. With the exception of cadmium carbonic anhydrase from marine diatoms[12] all carbonic anhydrase enzymes rely on ZnII for their in vivo activity. In α-CAs, the most extensively-studied family of CA, ZnII is tetrahedrally coordinated to three histidine residues and an exogenous water ligand, which is deprotonated to form the catalytically competent species.[13] CA-II of this family is one of the fastest known enzymes, with a catalytic efficiency approaching the diffusion limit in water. The combined simplicity of the primary coordination sphere and the extraordinarily high efficiencies achieved by enzymes make this an attractive design target to understand the relative contributions of the metal site and the protein environment to the rate enhancement of this reaction.
The fastest CA mimic for CO2 hydration was designed in the TRI scaffold as a bifunctional construct with a structurally stabilizing Hg-thiolate site and a catalytic Zn-histidine site.[14] The construct, termed HgII SZnIIN(TRIL9CL23H)3 was characterized by X-ray crystallography and its activity towards the non-native substrate p-nitrophenylacetate and CO2 was evaluated (Figure 1b). The catalytic efficiency at pH 9.5 towards pNPA was 23.3 M−1s−1, or 100-fold slower than CA-II, and its catalytic efficiency towards CO2 hydration was 1.8 × 105 M−1s−1, or 500-fold slower than CA-II (see Table 1). The kinetic pKa was 8.82 as determined by kinetic assays of pNPA hydrolysis. However, native CA has several features, including a hydrogen-bonded threonine near the active site and an extensive hydrogen bond network, which contribute to large rate enhancements and the lowering of the pKa of Zn bound water.[15,16] Removal of Thr199 lowers the catalytic efficiency by ~100-fold for both pNPA hydrolysis and CO2 hydration and raises the pKa of the bound hydroxide from 6.5 to 8.5.[15] Thus, HgIISZnIIN(TRIL9CL23H)3, which only reproduces the primary ZnII coordination sphere, lacks important features found in the native enzyme that are important to achieving similarly high catalytic efficiencies. To improve this design and better understand catalytic site placement in α helical coiled-coils, changes to alter the electrostatics of the catalytic site and substrate access and to incorporate hydrogen-bonding networks should be made.
Table 1.
ZnII affinity and kinetic data for CA isoforms and de novo designed peptides.
| Molecule | Kd ZnII [M−1][a] | kinetic pKa | CO2 kcat/Km [M−1s−1][b] | pNPA kcat/Km [M−1s−1][b] |
|---|---|---|---|---|
| CA-II | 4±1 × 10−12 [c] | 6.8 ± 0.1[d] | 9.2±0.4 × 107 [e] | 3640±150[e] |
| CA-III [f] | - | 8.5 ± 0.1 | 2.9±0.5 × 105 | 6 [g] |
| TRIL9CL23H[h] | (2.2±.6) ×10−7 | 9.0 ± 0.1 | 1.58±0.3 × 105 | 23.3±0.3 |
| TRIL2WL23H[i] | (2.4±0.2) × 10−7 | 9.2 ± 0.1 | - | 15.8±1.1 |
| TRILH9L23C[i] | (8±3) × 10−7 | 9.2 ± 0.1 | - | 15.5±0.4 |
| TRIL9CL19H[i] | (4±2) × 10−7 | 9.6 ± 0.1 | - | 13.9±1.1 |
| α3DH3 [j] | (5.9±0.9) × 10−8 | 9.4 | 3.8±0.5 × 104 | - |
More recently, this system was subjected to a systematic study of the position and orientation of the catalytic site along the α helical construct in order to evaluate existing factors in the α helical coiled-coil that might affect future design decisions. The structural site was removed to test how the presence of a negatively charged site (HgS3−) might affect the binding of ZnII and the kinetics of the catalytic site.[17] It was shown that the structural site functions as designed—to stabilize the overall construct—as no change in the Zn Kd, the catalytic efficiency of pNPA hydrolysis, or kinetic pKa was found (Table 1). In a different construct, HgIISZnIIN(TRIL9HL23C)3, the positions of the catalytic site and the structural site were inverted, orienting the bound water/hydroxide towards the bulk solvent rather than towards the hydrophobic interior of the coiled-coil. While this change resulted in 10-fold weaker binding of ZnII to HgIIS(TRIL9HL23C)3, the catalytic efficiency was only slightly decreased (15.8 vs. 23.3 M−1 s−1) (Table 1). Likewise, changing the position of the coordinating histidine from an a site to a d site to produce HgIISZnIIN(TRIL9CL19H)3 only results in a 5-fold decrease in ZnII affinity and a similarly small decrease in catalytic efficiency (13.9 vs. 23.3 M−1 s−1) (Table 1). Overall, this study indicates that faithful reproduction of the primary coordination sphere alone is responsible for the vast majority of the observed rate enhancement. The position and orientation of the catalytic site within the coiled-coil does not have a large effect on the catalytic efficiency, despite altering the substrate and solvent access.[17] Thus, to design constructs with higher rate enhancements, changes to the secondary coordination sphere must be made to induce lower KMs and lower kinetic pKas.
One major drawback to the TRI system is that, due to the parallel, self-assembling nature of the coiled-coils, any change made is necessarily three-fold symmetric. To address this limitation, a new protein was designed based on the α3D scaffold. Leucine residues were replaced with histidines (L18H, L28H, L67H) to create a tris-histidine site and a nearby histidine was changed to a valine (H72V) to prevent competition for ZnII binding (Figure 1c).[18] Adding four extra residues (GSGA) at the C terminus improved expression yields. This construct, α3DH3, is the first reported de novo designed, single-stranded three-helix bundle that functions as a CA mimic. Although the introduction of three bulky residues reduces the stability of the protein relative to α3D (5.1 kcal/mol vs. 3.1 kcal/mol), the protein is still stable enough to assess catalysis up to pH 9.5. α3DH3 was shown to bind ZnII with an affinity of 59 nM at pH 9.0 (Table 1). The ZnII environment is comprised of three histidines at 1.99 Å and one oxygen at 1.90 Å by extended X-Ray absorption fine structure (EXAFS) spectroscopy. This protein catalyses CO2 hydration with a kinetic pKa of 9.4 and a catalytic efficiency of 3.2 × 104 (M−1 s−1), which is 1400-fold slower than CA-II and 2.6-fold slower than HgIISZnIIN(TRIL9CL23H)3 (Table 1).[18] Recall that the kinetic pKa of HgIISZnIIN(TRIL9CL23H)3 is .6 pH units lower than that of α3DH3, which contributes to the observed difference in catalytic efficiency at pH 9.5. This difference is likely affected by the antiparallel orientation of the helices in α3DH3 and the electrostatic environment of the metal site, which are the result of only one round of rational design. Using this existing design, future work will focus on the site-selective incorporation of residues or the design of water channels to lower the kinetic pKa.
Recently, two groups have reported high rates for pNPA hydrolysis in de novo designed esterase catalysts. In general, pNPA hydrolysis has been used to benchmark esterases, as it is a convenient colormetric reaction. In one case the ZnII(His)3(H2O/OH−) site is formed at the interface of two helix-loop-helix monomers, and the rest of the interface helps to form a pocket where a tartrate co-crystallized.[19] This catalyst was capable of catalyzing pNPA hydrolysis with a maximal catalytic efficiency of 630 M−1 s−1, which is only about 4-fold slower than CA-II and makes it the fastest mimic for pNPA hydrolysis to date. Rufo et al reported a β-amyloid design of single, seven residue chains that assemble into fibrils.[7] These fibrils coordinate ZnII with two histidines from one β-strand and a third from an adjacent molecule to form the active site. The incorporation of β-strand-promoting isoleucine residues and a glutamine adjacent to the ZnII-binding histidines gave the most active catalyst with a catalytic efficiency of 62 M−1 s−1. In both cases it seems that the access offered by an interface results in efficient catalysts for pNPA hydrolysis, although whether this larger cavity and increased solvent access is beneficial for CO2 hydration is unknown.
Copper Redox Catalysis
Redox reactions comprise an important class of reactions in both biology and industry. From a design perspective, redox-active metalloproteins have an extra layer of complexity in that the designed construct must bind all redox states of the target metal. In some cases, this means only that multiple bond lengths must be accommodated, but in others, as with copper proteins, different binding geometries must be bound with sufficient affinity. Even when high stability for both oxidation levels is achieved, to obtain the desired reduction potential, the site must have the appropriate relative stabilities for these oxidation states. Copper centers play many roles in known proteins, participating in electron transfer and oxygen transport as well as catalyzing oxygen activation and metabolizing small molecules.[1] Type 2 copper sites are a versatile class of mononuclear copper site that can function as both electron transfer centers and as redox catalysts. The type 2 copper centers in peptidylglycine α-hydroxylating monooxygenase (PHM) and copper nitrite reductase (NiR) exhibit Cu(His)3 ligation, but perform drastically different roles: the type 2 site in PHM transfers an electron to a type 1 copper site[20], while in NiR it accepts an electron from a type 1 copper site and catalyzes the proton-dependent reduction of nitrite to NO and water[21,22]. Protein design is an excellent way to examine the functional switch of these structurally related sites and how the protein environment may mediate this change, particularly since the Cu(His)3 site may be prepared in the absence of the second copper site. The TRI scaffold was utilized to design a functional model for the catalytic site in NiR and was shown to catalyze the reduction of nitrite with ascorbate as a sacrificial electron donor with a pseudo-first order rate constant of 5.2 × 10−4 s−1 at pH 5.8.[23] The copper, when in its reduced state, is bound in a trigonal planar arrangement by the three histidine residues. However, the redox potential of this construct was 402 mV vs. NHE at pH 5.8, which is much higher than that of the type 2 copper center in NiR (218 mV at pH 6.0 in R. sphaeroides [24]). Given that the surrounding environment affects the redox potential of a site, the electrostatics of the site was altered to optimize the redox potential for increased catalytic rates. The previously reported parent peptide, TRI-H (corresponding to TRIL23H) was altered by inverting the position of a lysine above the active site and a glutamate below the active site to give a construct named TRI-EHE27K (corresponding to K22E and E27K), which doesn’t change the overall charge, only the distribution around the active site (Figure 2).[25] This inversion had the greatest effect on the CuI binding affinity, resulting in 100-fold tighter binding regardless of pH (Table 2). To then examine how the charge around the site could be used to alter the CuII stability constant and consequently, the redox potential and activity; the change at K22E (TRI-EH) was kept constant and the residues below the active site (at positions 24 and 27) were varied to produce peptides with overall Δcharge of −3, −6, −9, and −12 (relative to the parent peptide, TRI-H).[25] This series of peptides was studied to examine the pH dependence of CuII binding, affinity for CuI and CuII, CuII EPR spectra, and NiR activities produced by varying the secondary coordination sphere (Table 2).
Figure 2.
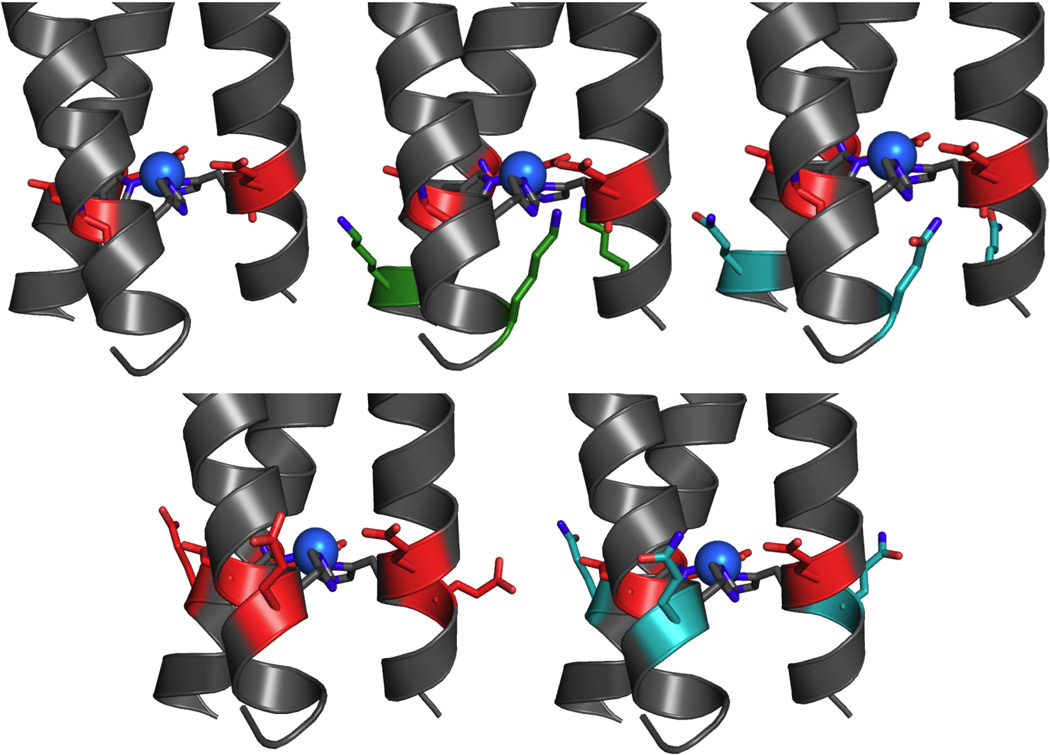
Representation of charge series around copper site in TRI-EH. From top left, clockwise, TRI-EH (corresponds to TRI-HK22E, Δcharge −6), TRI-EHE27K (Δcharge 0), TRI-EHK27Q (Δcharge −3), TRI-EHK24Q (Δcharge −9), TRI-EHK24E (Δcharge −12). Red represents changes to negatively charged residues, green to positively charged residues, and teal to neutrally charged residues. Models are based on the crystal structure of HgIISZnIIN(CSL9CL23H)3 (PDB: 3PBJ).
Table 2.
Spectroscopic and kinetic values for de novo designed NiR mimics based on the TRI scaffold. All values are reported for pH 5.8.
| peptide | Δcharge | d-d λmax (ε/M−1cm−1) |
reduction potential |
CuI Kd, [M−1] | CuII Kd, [M−1] |
rate [M−1min−1] | pKaE | pKaW |
|---|---|---|---|---|---|---|---|---|
| TRI-H | 0 | 644 (143) | 402±8 | (3.1±0.7) × 10−12 | (4.0±0.8) × 10−8 | (2.2±0.2) × 10−6 | 8.53±0.02 | |
| TRI-EHE27K | 0 | 671 (80) | 600±10 | (1.4±0.6) × 10−14 | (3±1) × 10−7 | (1.1±0.7) × 10−6 | 9.6±0.2 | |
| TRI-EHE27Q | −3 | 560±10 | (2±1) × 10−14 | (1.3±0.3) × 10−7 | (1.3±0.9) × 10−6 | |||
| TRI-EH | −6 | 659 (110) | 590±10 | (4.2±0.2) × 10−14 | (7±3) × 10−7 | (2.0±0.3) × 10−6 | 6.33±0.04 | 9.86±0.05 |
| TRI-EHK24Q | −9 | 490±20 | (5±3) × 10−13 | (2±1) × 10−7 | (3.8±0.8) × 10−6 | |||
| TRI-EHK24E | −12 | 665 (101) | 520±12 | (3.52±0.09) × 10−14 | (5±2) × 10−7 | (3.6±0.6) × 10−6 | 6.76±0.06 | 9.8±0.2 |
From ref [25]
Careful examination of the pH dependence of the d-d visible band and EPR signals revealed that the presence of K22E introduces a hydrogen bond to the Cu-coordinating imidazole and raises the pKa of an exogenous water bound to the copper center. The increase in local negative charge across the series resulted in less positive reduction potentials, as expected, however this was due to a destabilization of CuI binding, rather than a higher stability constant for CuII.[25] This study showed that, by careful choice of the pH conditions and the peptide sequence, one could vary the reduction potential of this site by nearly 200 mV. Future efforts will focus on the redesign of this construct to incorporate other features to perturb reactivity, such as cavities for substrate binding or modification of the electronics of the coordinating ligand.
Given the success of incorporating carbonic anhydrase activity into the three-helix bundle scaffold, α3D, a natural extension to this work is to incorporate copper into α3DH3. The folding and electrostatics of α3DH3 are known to be different from histidine-substitute TRI derivatives. The protein scaffold has been shown to affect the carbonic anhydrase activity of ZnII-α3DH3 relative to TRI. It is likely that copper substitution in this peptide to give CuI/II-α3DH3 would result in drastically different properties than what is observed in TRI-H and the TRI-EH series given the demonstrated sensitivity to charge for this system. Unpublished work indicates that α3DH3 is capable of binding to Cu(I/II) and that this system behaves as a type 2 copper center.
Conclusion
The same fundamental sequence can support different activities based on the identity of the metal, as long as the primary coordination sphere has been satisfied; thus TRI-H with ZnII is capable of CO2 hydration and with Cu(I/II) is capable of nitrite reductase activity. While some baseline level of activity is present, these results emphasize the effect that millions of years of evolutionary pressure has had on the surrounding protein environment to optimize activity. These scaffolds provide the template with which to understand the individual effects of a given structural element and what cooperative effect multiple elements may have.
Beyond what can be learned from comparison within a series of peptides based on the same scaffold, comparisons made between different scaffolds allow us to understand how more basic questions in protein biochemistry may affect the intended function of a protein. For example, the extent to which native-life folding and the antiparallel or parallel nature of α helices and β sheets play a role in native enzyme rates is not know. Similarly, the preference of certain types and classes of enzymes for structural motifs has been observed. While some instances (like CA) appear to be the result of convergent evolution, the underlying factors driving this selective pressure are not well understood. By building up a “toolbox” of functions and scaffolds we understand well, we hope to not only answer some of these fundamental questions, but to reach a point where novel enzymes and reactions can be designed and tailored to a particular application.
Highlights.
-
-
α-helical de novo designed proteins are versatile scaffolds
-
-
The primary coordination sphere confers significant hydrolytic or redox catalytic activity
-
-
secondary coordination sphere engineering is required for enhancing reactivity
Acknowledgements
The authors would like to thank the National Institutes of Health grant R01ES012236. AGT acknowledges training grant support from the University of Michigan Chemistry-Biology Interface (CBI) training program (NIH grant 5T32GM008597) and the Chateaubriand International Fellowship. VLP acknowledges the Blaise Pascal International Chair for funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holm RH, Kennepohl P, Solomon EI. Structural and Functional Aspects of Metal Sites in Biology. Chem. Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 2.Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL. Protein design: toward functional metalloenzymes. Chem. Rev. 2014;114:3495–3578. doi: 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peacock AF. Incorporating metals into de novo proteins. Current Opinion in Chemical Biology. 2013;17:934–939. doi: 10.1016/j.cbpa.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 4.Zastrow ML, Pecoraro VL. Designing functional metalloproteins: from structural to catalytic metal sites. Coordination Chemistry Reviews. 2013;257:2565–2588. doi: 10.1016/j.ccr.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zastrow ML, Pecoraro VL. Designing hydrolytic zinc metalloenzymes. Biochemistry. 2014;53:957–978. doi: 10.1021/bi4016617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plegaria JS, Pecoraro VL. Sculpting Metal Binding Environments in De Novo Designed Three-Helix Bundles. Israel Journal of Chemistry. doi: 10.1002/ijch.201400146. [no date], [no volume]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rufo CM, Moroz YS, Moroz OV, Stöhr J, Smith TA, Hu X, DeGrado WF, Korendovych IV. Short peptides self-assemble to produce catalytic amyloids. Nat Chem. 2014;6:303–309. doi: 10.1038/nchem.1894. This paper describes the synthesis and characterization of the catalytic activity of short 7-residue peptides that self-assemble to form fibrils, chelating metals between adjacent β strands.
- 8.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O'Halloran TV, Penner-Hahn JE, DeGrado WF, Pecoraro VL. De novo design of mercury-binding two-and three-helical bundles. J. Am. Chem. Soc. 1997;119:6195–6196. [Google Scholar]
- 9.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. The role of protonation and metal chelation preferences in defining the properties of mercury-binding coiled coils. J. Mol. Biol. 1998;280:897–912. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 10.Bryson JW, Desjarlais JR, Handel TM, DeGrado WF. From coiled coils to small globular proteins: design of a native-like three-helix bundle. Protein Sci. 1998;7:1404–1414. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh ST, Cheng H, Bryson JW, Roder H, DeGrado WF. Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Feng L, Jeffrey PD, Shi Y, Morel FMM. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature. 2008;452:56–61. doi: 10.1038/nature06636. [DOI] [PubMed] [Google Scholar]
- 13.Imtaiyaz Hassan M, Shajee B, Waheed A, Ahmad F, Sly WS. Structure, function and applications of carbonic anhydrase isozymes. Bioorg. Med. Chem. 2013;21:1570–1582. doi: 10.1016/j.bmc.2012.04.044. [DOI] [PubMed] [Google Scholar]
- 14. Zastrow ML, Peacock AFA, Stuckey JA, Pecoraro VL. Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat Chem. 2012;4:118–123. doi: 10.1038/nchem.1201. The synthesis and characterization of the first bifunctional, bimetallic de novo designed protein, which has the fastest rates for CO2 hydration of any CA mimic.
- 15.Liang Z, Xue Y, Behravan G, Jonsson BH, Lindskog S. Importance of the conserved active-site residues Tyr7, Glu106 and Thr199 for the catalytic function of human carbonic anhydrase II. Eur. J. Biochem. 1993;211:821–827. doi: 10.1111/j.1432-1033.1993.tb17614.x. [DOI] [PubMed] [Google Scholar]
- 16.Krebs JF, Ippolito JA, Christianson DW, Fierke CA. Structural and functional importance of a conserved hydrogen bond network in human carbonic anhydrase II. J. Biol. Chem. 1993;268:27458–27466. [PubMed] [Google Scholar]
- 17. Zastrow ML, Pecoraro VL. Influence of active site location on catalytic activity in de novo-designed zinc metalloenzymes. J. Am. Chem. Soc. 2013;135:5895–5903. doi: 10.1021/ja401537t. This paper describes the systematic variation of location and orientation in a bifunctional de novo designed metalloenzyme.
- 18. Cangelosi VM, Deb A, Penner-Hahn JE, Pecoraro VL. A de novo designed metalloenzyme for the hydration of CO2. Angew. Chem. Int. Ed. Engl. 2014;53:7900–7903. doi: 10.1002/anie.201404925. This paper describes the first de novo designed, single-stranded, three-helix bundle capable of catalyzing CO2 hydration.
- 19. Der BS, Edwards DR, Kuhlman B. Catalysis by a De Novo Zinc-Mediated Protein Interface: Implications for Natural Enzyme Evolution and Rational Enzyme Engineering. Biochemistry. 2012;51:3933–3940. doi: 10.1021/bi201881p. The design of metal sites in interfaces between designed proteins yielded the fastest catalyst for pNPA hydrolysis.
- 20.Chufán EE, Prigge ST, Siebert X, Eipper BA, Mains RE, Amzel LM. Differential reactivity between two copper sites in peptidylglycine α-hydroxylating monooxygenase. J. Am. Chem. Soc. 2010;132:15565–15572. doi: 10.1021/ja103117r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Libby E, Averill BA. Evidence that the type 2 copper centers are the site of nitrite reduction by Achromobacter cycloclastes nitrite reductase. Biochemical and Biophysical Research Communications. 1992;187:1529–1535. doi: 10.1016/0006-291x(92)90476-2. [DOI] [PubMed] [Google Scholar]
- 22.Tocheva EI, Rosell FI, Mauk AG, Murphy MEP. Side-on copper-nitrosyl coordination by nitrite reductase. Science. 2004;304:867–870. doi: 10.1126/science.1095109. [DOI] [PubMed] [Google Scholar]
- 23. Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, Pecoraro VL. Designing a functional type 2 copper center that has nitrite reductase activity within α-helical coiled coils. Proceedings of the National Academy of Sciences. 2012;109:21234–21239. doi: 10.1073/pnas.1212893110. This paper reports the first de novo designed nitrite reductase mimic.
- 24.Jacobson F, Pistorius A, Farkas D, De Grip W, Hansson O, Sjolin L, Neutze R. pH Dependence of Copper Geometry, Reduction Potential, and Nitrite Affinity in Nitrite Reductase. Journal of Biological Chemistry. 2006;282:6347–6355. doi: 10.1074/jbc.M605746200. [DOI] [PubMed] [Google Scholar]
- 25. Yu F, Penner-Hahn JE, Pecoraro VL. De novo-designed metallopeptides with type 2 copper centers: modulation of reduction potentials and nitrite reductase activities. J. Am. Chem. Soc. 2013;135:18096–18107. doi: 10.1021/ja406648n. Careful engineering of charge and its distribution around the active site of a nitrite reductase catalyst reveals the importance of secondary coordination sphere as CuI binding was destabilized, leading to less positive reduction potentials.
- 26.Kiefer LL, Krebs JF, Paterno SA, Fierke CA. Engineering a cysteine ligand into the zinc binding site of human carbonic anhydrase II. Biochemistry. 1993;32:9896–9900. doi: 10.1021/bi00089a004. [DOI] [PubMed] [Google Scholar]
- 27.Fierke CA, Calderone TL, Krebs JF. Functional consequences of engineering the hydrophobic pocket of carbonic anhydrase II. Biochemistry. 1991;30:11054–11063. doi: 10.1021/bi00110a007. [DOI] [PubMed] [Google Scholar]
- 28.Jackman JE, Merz KM, Fierke CA. Disruption of the active site solvent network in carbonic anhydrase II decreases the efficiency of proton transfer. Biochemistry. 1996;35:16421–16428. doi: 10.1021/bi961786+. [DOI] [PubMed] [Google Scholar]
- 29.Jewell DA, Tu C, Paranawithana SR, Tanhauser SM, LoGrasso PV, Laipis PJ, Silverman DN. Enhancement of the catalytic properties of human carbonic anhydrase III by site-directed mutagenesis. Biochemistry. 1991;30:1484–1490. doi: 10.1021/bi00220a006. [DOI] [PubMed] [Google Scholar]