

Abstract
The intracellular enzyme platelet-activating factor acetylhydrolase type-II (PAFAH-II) hydrolyzes platelet-activating factor and oxidatively fragmented phospholipids. PAFAH-II in its resting state is mainly cytoplasmic, and it responds to oxidative stress by becoming increasingly bound to endoplasmic reticulum and Golgi membranes. Numerous studies have indicated that this enzyme is essential for protecting cells from oxidative stress induced apoptosis. However, the regulatory mechanism of the oxidative stress response by PAFAH-II has not been fully resolved. Here, changes to the oligomeric state of human PAFAH-II were investigated as a potential regulatory mechanism toward enzyme trafficking. Native PAGE analysis in vitro and photon counting histogram within live cells showed that PAFAH-II is both monomeric and dimeric. A Gly-2-Ala site-directed mutation of PAFAH-II demonstrated that the N-terminal myristoyl group is required for homodimerization. Additionally, the distribution of oligomeric PAFAH-II is distinct within the cell; homodimers of PAFAH-II were localized to the cytoplasm while monomers were associated to the membranes of the endoplasmic reticulum and Golgi. We propose that the oligomeric state of PAFAH-II drives functional protein trafficking. PAFAH-II localization to the membrane is critical for substrate acquisition and effective oxidative stress protection. It is hypothesized that the balance between monomer and dimer serves as a regulatory mechanism of a PAFAH-II oxidative stress response.
Keywords: Oxidative stress, peripheral membrane protein, phospholipase A2, myristoylation, oligomeric state, trafficking
1. Introduction
PAFAH-II is an intracellular enzyme expressed in a variety of cell types, including human platelets, lymphocytes, neutrophils [1], skin [2], liver and kidney cells [3, 4]. PAFAH-II is a widely conserved protein, found in organisms from S. pombe [5] and C. elegans to higher invertebrates [6], such as mammals. As a member of the phospholipase A2 (PLA2) superfamily, this enzyme hydrolytically cleaves the sn-2 position of a phospholipid, resulting in a free fatty acid and lysophospholipid [7]. PAFAH-II, which is a Ca2+ independent serine hydrolase, was originally named for its ability to hydrolyze and inactivate platelet-activating factor (PAF) [3, 6]. The substrate specificity of PAFAH-II also includes phospholipids structurally resembling PAF, such as oxidatively fragmented phospholipids [4, 8, 9] as depicted in Fig. 1.
Fig. 1.

Model of a phospholipid monolayer depicts an unsaturated phospholipid on the left. The unsaturated chain of this phospholipid can be oxidatively fragmented by a reactive oxygen species (ROS), as shown on the right. Shown on the right is one of many possible “whisker” products. The shortened and more polar chain depicted from this fragmentation is shown pointing toward the aqueous phase, and the ester bond targeted for esterolysis by PAFAH-II is indicated with an arrow.
PAFAH-II has been observed simultaneously distributed in the cytoplasm and localized to intracellular membranes [3, 10, 11]. Upon the addition of oxidative stress, PAFAH-II distribution becomes more heavily membrane associated [2, 11, 12]. Previous research in our laboratory has shown that following oxidative stress, PAFAH-II is specifically localized to the membranes of both the endoplasmic reticulum (ER) and Golgi apparatus [12]. Conversely, when anti-oxidants were added to stressed cells, the distribution of PAFAH-II returned to a pre-stressed distribution with the majority of protein being cytosolic [11].
A number of studies have confirmed the importance of this enzyme in the prevention of oxidative stress induced apoptosis [13–15]. It is believed to function by cleaving the oxidatively fragmented portion of a damaged phospholipid, thereby starting the reparative process [2–4, 6, 8, 11, 16–18]. Its substrate specificity for shortened chains at the sn-2 position prevents PAFAH-II from hydrolyzing intact phospholipids, making this enzyme an essential component of homeostasis [4, 11]. By trafficking from the cytoplasm to the membrane, PAFAH-II can be localized to the areas containing damaged phospholipids.
Previously we constructed a PAFAH-II homology model (Fig. 2) from the plasma PAFAH crystal structure to further understand this enzyme’s structure [12] and interactions at the membrane surface as predicted by the Orientations of Proteins in Membranes (OPM) database computational approach [19, 20]. Specific PAFAH-II regions involved in membrane binding were predicted and then tested using site directed mutagenesis and localization experiments in live HEK293 cells [12]. The location of the active site above the hydrophilic-hydrophobic interface is consistent with the known substrate specificity and model of PAFAH-II targeting “whisker” acyl chains of an oxidized phospholipid [21]. The whisker acyl chain of an oxidatively fragmented phospholipid is shown in Fig. 1 projecting away from the hydrophobic portion and toward the aqueous phase. Furthermore, our previous work demonstrated the role played by both the myristoyl group and the hydrophobic patch of PAFAH-II in directing the enzyme to membrane surfaces [12].
Fig. 2.

The PAFAH-II homology model is shown with the active site Ser, His, Asp catalytic triad in green, hydrophobic patch residues (L76, L79, L327, I328, and F331) in yellow, C-terminus depicted with a red sphere and N-terminus (location of myristoyl group) depicted with a blue sphere [12]. The plane of white spheres represents the hydrophilic-hydrophobic interface of the enzyme’s predicted membrane binding interface [19].
The translocation of myristoylated proteins between the cytoplasm and membrane is regulated by a number of different mechanisms. In general, these select proteins are functionally influenced by the presence of a myristoyl group or by the oligomeric state of the protein. The role of the myristoyl group in protein oligomerization can vary. In Visinin-like protein, the myristoyl group is not directly involved in this protein-protein interaction [22]. For the protein Nef from HIV-1 the myristoyl group prevents formation of higher order oligomers [23, 24]. While for some proteins, the myristoyl group is required to form oligomers, as is the case for NAP-22 [25]. Previous studies had concluded that PAFAH-II purified from bovine liver is monomeric [4]. The oligomeric state of this myristoylated protein has not been studied further, especially for its behavior in a native-like environment. Therefore it is possible that PAFAH-II exists in alternate oligomeric states and that its localization and function may be regulated by changes in this oligomerization.
In the present study we have looked more closely at the oligomeric state of PAFAH-II in vitro and in live human kidney cells. We followed PAFAH-II oligomerization in living cells using fluorescent fluctuation spectroscopy (FFS), and specifically, we utilized the photon counting histogram (PCH) method. PCH data is collected by monitoring the fluorescence counts of molecules as they move in and out of a specified observation volume [26]. Data for a specific amount of time is compiled as a histogram of photon counts and their frequency. The resulting histogram predicts the probability of finding a number of photons in the observation volume and is used to determine the molecular brightness and concentration that is the result of the collected data [27]. From this statistical analysis, the average counts per second per molecule (CPSM) is determined [26, 27]. Since this is a comparison technique, enhanced green fluorescence protein (eGFP) controls were used to determine the brightness of a monomer and an eGFP-eGFP dimer fusion species [28–30].
The molecular underpinnings of the oxidative stress response of PAFAH-II were explored here to gain a better understanding of the physiological role of this enzyme. The oligomeric state of PAFAH-II was investigated by native PAGE and Western blot analysis using PAFAH-II samples which were purified from mammalian cell culture. We then characterized the oligomeric state of wild-type (WT) and mutant PAFAH-II in live cells and at a low expression level using the FFS technique.
2. Materials and Methods
2.1 Cloning of His-tagged PAFAH-II-YFP, eGFP controls and PAFAH-II-eGFP constructs
To generate a His-tagged PAFAH-II construct, we modified our previous WT-PAFAH-II-YFP-pCEP4 construct [12]. A silent point mutation was made to the endogenous BamHI site in the PAFAH-II DNA sequence, using Quick Change site directed mutagenesis kit (Stratagene) and the following primers: 5′-GCTGCAGGAGGAATGGATTCCTTTCCGTCG-3′ and 5′-CGACGGAAAGGAATCCATTCCTCCTGCAGC-3′. A His-tag was added to the C-terminus of YFP, using a forward primer that conserved the existing XhoI restriction site and a reverse primer that introduced a His-tag and BamHI site: 5′-GGGGCCCCCCACCATCTGTCCAGCCTGCTCGAGATGGTGAGCAAGGGC-3′ and 5′-TTTTTTGGATCCCTAGTGGTGGTGGTGGTGGTGGTGGTGCTTGTACAGCTCGTC CATGCCGAGAGTGATCCCGGCGGCC-3′. WT-PAFAH-II-YFP-pCEP4, with mutated endogenous BamHI site, and PCR amplified YFP with C-terminal His-tag, were digested with XhoI and BamHI (Invitrogen) and ligated using T4 ligase (Invitrogen). To produce unmyristoylated PAFAH-II-YFP-His, Gly-2 was mutated to Ala in the WT-PAFAH-II-YFP-His-pCEP4 construct by site directed mutagenesis using the following primers: 5′-GCTTGCCACCATGGCGGTCAACCAGTC-3′ and 5′-GACTGGTTGACCGCCATGGTGGCAAGC-3′.
To create fluorescent protein fusions, suitable for FFS experiments we used eGFP, which is a fluorophore less prone to photobleaching. The construct pcDNA3-eGFP was purchased from Addgene (Plasmid 13031) originally submitted by Dr. Doug Golenbock. An eGFP-eGFP dimer control with a twelve amino acid linker (GHGTGSTGSGSS) between the two fluorescent proteins was constructed as previously described [28]. This construct was made using a forward primer that introduced a HindIII site, while 3 different reverse primers sequentially introduced the twelve amino acid linker and an XhoI restriction site: 5′-TTTTTTTTAAGCTTGCCACCATGGTGAGCAAGGGC-3′ and (R1) 5′-GCCATGACCCTTGTACAGCTCGTC-3′, (R2) 5′-GGTAGAACCAGTGCCATGACCCTTGTAC-3′ and (R3) 5′-TTTTTTTTCTCGAGCCGGACCCGGTAGAACCAGTGCCATG-3′. This PCR product was cloned into a pCEP4 vector (Invitrogen) as described above. The second eGFP was amplified using a forward primer that introduced an XhoI restriction site and a reverse primer that introduced a double stop codon followed by a BamHI site: 5′-TTTTTTTTCTCGAGCATGGTGAGCAAGGGC-3′ and 5′-TTTTTTTTGGATCCCTATTACTTGTACAGCTCGTCC-3′. The PCR amplified second eGFP was cloned into pCEP4 containing the first eGFP with linker, as described above. DNA sequencing results of this eGFP-eGFP construct confirmed a stop codon directly before the second eGFP. PCR using the following primers was performed to insert an extra nucleotide, thereby removing the stop codon and correcting the frame for proper expression of the eGFP-eGFP dimer fusion. 5′-CGGGTCCGGCTCGAGCATGGTGAGCAAGGGCG-3′ and 5′-CGCCCTTGCTCACCATGCTCGAGCCGGACCCG-3′.
The eGFP-eGFP-pCEP4 construct with the stop codon before the second eGFP, was used as the monomer eGFP control construct. This single subunit control was expressed with the 12 amino acid linker at the C-terminus.
To engineer PAFAH-II with an eGFP tag and no 12 amino acid linker, we utilized mammalian expression constructs that were previously made [12]. PAFAH-II DNA was excised from WT-PAFAH-II-YFP-pCEP4 using HindIII and XhoI restriction enzymes and cloned into eGFP-pCEP4. This construct contained a frame shift between PAFAH-II and eGFP, which was corrected by removing one nucleotide with the following primers: 5′-GTCCAGCCTGCTCGAGATGGTGAGCAAGGGCG-3′ and 5′-CGCCCTTGCTCACCATCTCGAGCAGGCTGGAC-3′. Unmyristoylated PAFAH-II with an eGFP tag was constructed using site directed mutagenesis, as described above.
DNA sequences for all constructs were confirmed by Genewiz, Inc. DNA for mammalian transfection was purified using Qiagen Maxi Prep Kit to achieve samples with high purity and concentration.
2.2 HEK293 cell culture and transfection
HEK293 cells were maintained and transiently transfected as described previously [12]. For protein purification and native PAGE analysis, cells were transfected with 2.0 μg DNA and 10 μL Lipofectomene2000 (Invitrogen), in a 2 mL Opti-MEM (Invitrogen)/25 cm2 flask (Corning). Proteins were expressed for 24 h. For PCH analysis, HEK293 cells were plated at 50% confluency on 18×18 mm2 high performance cover glasses, D=0.17 mm (Zeiss). The transfection media contained 0.01 μg of DNA and 15 μL Lipofectamine2000 (Invitrogen) in 3 mL Opti-MEM/10 cm petri dish (Fisher Scientific). Proteins were expressed for 40 h. To prepare for imaging, Secure-Seal adhesive spacers (Invitrogen) were used to mount the cover glasses on microscope slides.
2.3 Purification of recombinant PAFAH-II from HEK293 cells
PAFAH-II-YFP-His-pCEP4 constructs expressed in HEK293 cells were partially purified by the following method. Adherent cells were washed from the flasks with phosphate buffered saline (PBS). PBS was removed by centrifugation at 1,900 g, and cells were resuspended in 0.5 mL commercial lysis buffer, MEM-PER (Pierce). Cell suspension was incubated at room temperature for 15 min with rocking to complete lysis. The lysate was clarified by centrifugation at 26,900 g for 15 min at 4 °C. The supernatant was collected, and imidazole (Sigma-Aldrich) was added to a final concentration of 20 mM. This mixture was incubated with gentle rocking with 250 μL bed volume of Ni-Sepharose resin (GE Healthcare) for 1 h at 4 °C to bind PAFAH-II-YFP-His. Unbound impurities were removed by discarding the supernatant and extensively washing the Ni-resin with wash buffer (20 mM Tris-base, pH 8.0, 150 mM NaCl, 10% glycerol and 20 mM imidazole). Bound PAFAH-II-YFP-His was eluted at room temperature by incubation with wash buffer containing 250 mM imidazole for 10 min. Purified PAFAH-II samples were stored at −20 °C.
2.4 Native PAGE and Western blot analysis
To characterize the native state of the PAFAH-II protein purified from HEK293 cells, we resolved protein sample on a 4–12% Tris-glycine native PAGE gel (Invitrogen). Equal amounts of purified protein were loaded, as judged by SDS-PAGE followed by Western blot analysis (Fig. 3B). The protein bands were transferred to a HyBond ECL nitrocellulose membrane (GE Healthcare) in transfer buffer (25 mM Tris, pH 8.3, 192 mM glycine and 20% v/v methanol) at 4 °C overnight at 60 mA. The following steps were carried out at room temperature with gentle shaking. The nitrocellulose membrane was washed in Tris buffered saline with Tween 20 (TBS-T) [20 mM Tris-HCl, pH 7.6, 140 mM NaCl and 0.1% Tween 20 (Sigma-Aldrich)] and blocked in 5% nonfat dry milk in TBS-T for 1 h. The membrane was washed with TBS-T and incubated with GFP-antibody (Abcam, suitable for detecting YFP) 1:2,000 dilution in TBS-T for 1 h. The membrane was washed with TBS-T and incubated with an anti-chicken horseradish peroxidase-bound secondary antibody (Cell Signaling Technology) at a 1:2,000 dilution in TBS-T for 30 min. Finally, the membrane was washed in TBS-T and treated with enhanced chemiluminescence Western blotting substrate (Pierce) for 5 min. Protein bands were imaged with a luminescence filter on a Fluorchem Q using the auto-expose option.
Fig. 3.
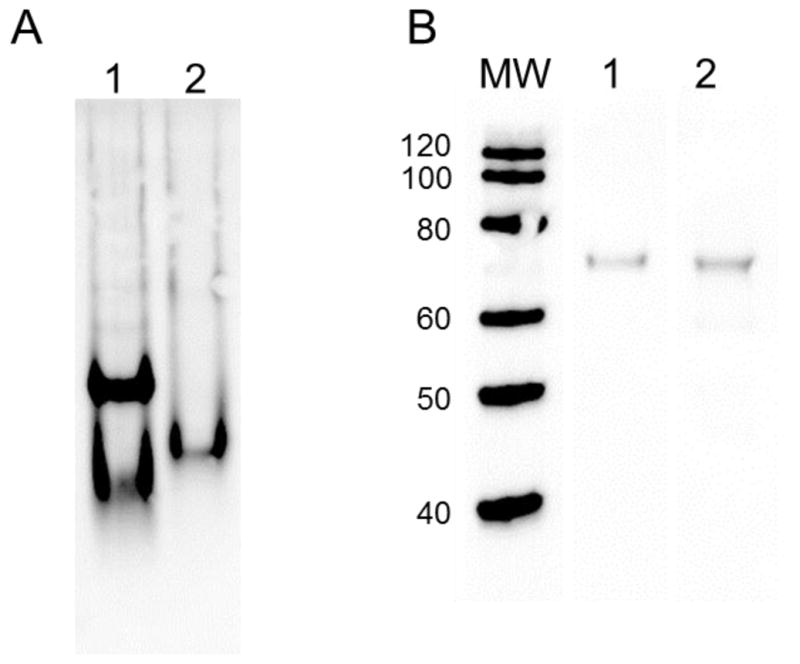
Western blot analysis of PAFAH-II constructs resolved by native PAGE and SDS PAGE. (A) Western blot analysis resolved by native PAGE: lane 1: WT-PAFAH-II-YFP-His, lane 2: G2A mutant-PAFAH-II-YFP-His, and blotted with GFP specific antibodies. WT-PAFAH-II displays monomer and dimer bands, while the myristoyl mutant resolves as a monomer. (B) Western blot analysis resolved by SDS PAGE: molecular weight marker lane (kDa) on left, lane 1: WT-PAFAH-II-YFP-His, lane 2: G2A-PAFAH-II-YFP-His, and blotted with GFP specific antibodies. The wild type and G2A-PAFAH-II fusions with YFP run at their expected molecular weight of 77 kDa.
2.5 Photon counting histogram data collection and analysis
To measure the oligomeric state of PAFAH-II in live cells, an FFS technique called PCH was used. PCH data collection was carried out on a Zeiss LSM780 confocal microscope using a 40× c-Apochromat (NA = 1.2) water immersion objective. The 488 nm laser was set to 0.2% power, eGFP emission was detected on the BiGaAsP1 detector using a 500–550 nm emission filter. Areas of the cytosol, membranes, and nucleus of healthy cells were observed. We selected cells that displayed low expression levels of eGFP fluorophores and eGFP-PAFAH-II fusions with a brightness between 150 and 300 CPSM. However, studies by others have demonstrated that cells with varying expression levels still have consistent molecular brightness [28]. Five observation volumes per cell were selected and the fluorescent counts of each were measured for 7 s.
Fluorescent counts were organized as histograms of number of photon events per unit time (50 μs bin time). CPSM was calculated using the Zeiss software PCH algorithm for 3-dimensional Gaussian focal volume [27, 31] and incorporating a first order correction [32]. First order correction was not used when measuring the auto fluorescence of untransfected cells.
Monomer and dimer eGFP constructs were used as controls. The average molecular brightness was assigned to each of the two eGFP controls as had been done by Chen et al., where the dimer was approximately double the brightness of the monomer [27].
3. Results
3.1 Western blot analysis demonstrates PAFAH-II dimerization
To examine the oligomeric state of human PAFAH-II, we purified His-tagged PAFAH-II expressed from HEK293 cells by Ni-affinity chromatography. PAFAH-II samples collected by this method displayed 20% purity, as shown by SDS PAGE analysis. Purified proteins were resolved by both native and SDS PAGE. For each gel, PAFAH-II protein bands were detected by Western blot using GFP specific antibodies. Our native PAGE separation and Western blot detection of WT-PAFAH-II-YFP-His fusion protein exhibited two protein bands (Fig. 3). The bands were evenly spaced and demonstrated that WT-PAFAH-II was distributed as both a monomer and dimer. These in vitro results indicate that the WT form of PAFAH-II isolated from mammalian cell culture exists as both a monomer and dimer.
To investigate the role of the myristoyl group in PAFAH-II oligomerization, unmyristoylated G2A-PAFAH-II-YFP-His was also expressed and purified from HEK293 cells. Interestingly, native PAGE and subsequent Western blot analysis of this PAFAH-II myristoyl mutant showed only a single protein band (Fig. 3) and suggested that this construct was a monomer. While the G2A-PAFAH-II-YFP-His band resolved as a single band, this band was positioned between the monomer and dimer bands of the WT-PAFAH-II-YFP-His sample. The variance in gel migration is likely due to the absence of the hydrophobic myristoyl group. This single band suggests that the G2A mutant is a monomer. These results indicate that the myristoyl group is required for PAFAH-II oligomer formation.
3.2 PCH of eGFP monomer and dimer controls
To investigate the oligomeric state of PAFAH-II in live cells, we used a FFS technique called PCH. Prior to each measurement with PAFAH-II constructs, eGFP controls were used to determine the brightness of an eGFP monomer (eGFP-pCEP4) and eGFP dimer mimic (eGFP-eGFP-pCEP4). The brightness of these controls is variable from lab to lab due to laser power, efficiency of detection and expression of fluorophores [26]. Therefore, it was essential to independently determine the molecular brightness of these controls in our lab and during each experiment.
HEK293 cells were transiently transfected with eGFP-pCEP4 or eGFP-eGFP-pCEP4, and proteins were expressed for 40 h. Molecular brightness, in units of CPSM, was measured for each construct from five observation volumes within the nucleus. The average brightness of 31 cells expressing the eGFP monomer was determined to be 6,300 CPSM while the average brightness of 41 cells expressing the dimer was 8,600 CPSM (Fig. 4). The difference between the monomer and dimer brightness is statistically significant. Although the dimer is expected to be nearly twice as bright, measurements by others using this technique were never exactly double [26, 28–30]. Disturbances in protein folding are likely part of the decreased dimer brightness. Brightness of untransfected cells was 890 CPSM. Previous studies have shown that this auto-fluorescence does not affect the brightness of eGFP fluorophores [28].
Fig. 4.

Molecular brightness of eGFP and eGFP-eGFP controls. Error bars show standard deviation, and the asterisk shows statistically significant difference between monomer and dimer brightness (Student’s t-test, p = 0.0057).
3.3 PCH indicates specific cellular distribution of WT-PAFAH-II oligomers
We examined the oligomeric state of PAFAH-II in live cells using PCH. This technique was also used to determine if a particular oligomeric state of PAFAH-II was localized to specific areas of the cell. We accomplished this by separately analyzing the cytoplasm, nucleus, and membrane regions, selecting healthy cells with low expression levels as determined by whole cell imaging.
HEK293 cells expressing WT-PAFAH-II-eGFP-pCEP4 were analyzed in the same fashion as the cells transfected with the eGFP control constructs (section 3.2). Our previously published work indicated that addition of fluorescent proteins to the C-terminus of PAFAH-II did not alter PAFAH-II localization or activity [12]. In a single cell, five observation volumes were selected in the cytoplasm, nucleus, or membrane regions. The average cytoplasmic brightness of 42 cells expressing WT-PAFAH-II was 8,300 CPSM (Fig. 5). The WT cytoplasmic brightness value is comparable to that of the eGFP control dimer and is statistically different from the eGFP monomer control. These results indicate that WT-PAFAH-II exists as a dimer in the cytoplasm of the cell.
Fig. 5.

Molecular brightness of eGFP controls and WT-PAFAH-II-eGFP. Error bars show standard deviation and the asterisks show statistically significant difference between brightness of monomer eGFP and WT-PAFAH-II-eGFP in the cytoplasm (Student’s t test, p = 0.015). A significant difference of brightness was also observed between the dimer eGFP-eGFP and WT-PAFAH-II-eGFP in the nucleus (Student’s t test, p= 0.0068).
The average brightness of WT-PAFAH-II associated to cellular membranes in 22 different cells was 7,800 CPSM and falls between the brightness of both the monomer and dimer eGFP controls (Fig. 5). These results indicated that PAFAH-II proximal to membrane surfaces was a mixture of monomer and dimer species.
Twenty-seven cells analyzed for nuclear WT-PAFAH-II showed an average brightness of 6,300 CPSM, a similar value to the brightness of the eGFP monomer and significantly different from the eGFP dimer (Fig. 3). This data indicates that WT-PAFAH-II exists as a monomer in the nucleus of the cell.
3.4 PCH indicates that G2A-PAFAH-II is a monomer in the cytoplasm
To confirm our in vitro results that showed the unmyristoylated PAFAH-II was monomeric, we analyzed G2A-PAFAH-II in live cells using the PCH technique. Molecular brightness was determined in the cytoplasm, and measurements were not taken in the membranes, as G2A does not localize to these areas [12]. In 21 cells, the average molecular brightness of cytoplasmic G2A-PAFAH-II was 5,900 CPSM. This value corresponds to the brightness of the eGFP monomer and is significantly different from the brightness of the dimer control (Fig. 6).
Fig. 6.

Molecular brightness of eGFP controls and G2A-PAFAH-II-eGFP. Error bars show standard deviation and the asterisk shows a statistically significant difference between brightness of dimer eGFP-eGFP and G2A-PAFAH-II-eGFP in the cytoplasm (Student’s t test, p = 0.0034).
4. Discussion
Research has demonstrated the importance of PAFAH-II in oxidative stress survival, its change in cellular location and membrane binding structures [12–15]. However, little investigation has been done on the regulatory mechanism of this protein’s localization or function. In our present work, we have shown that PAFAH-II forms both monomers and homodimers, and the oligomeric state is linked to its cellular trafficking. Our experiments demonstrate that the myristoyl group is critical for dimerization of PAFAH-II. We propose that cellular localization of PAFAH-II is regulated by the oligomeric state of the protein and the cellular distribution of this protein is critical for proper function as an oxidative stress protector.
Previous research concluded that PAFAH-II purified from bovine liver is monomeric [4]. However, further investigations of in vivo processes of this protein were not completed. Our native PAGE characterization shows that human WT-PAFAH-II, purified from HEK293 cells, resolves as two distinct bands, representing a bimodal population of both monomers and dimers. The samples were analyzed by SDS PAGE and detected by Western, which showed a single band for both WT and G2A constructs. When combined with the native PAGE results, this further strengthens a conclusion that WT exists as a dimer, and G2A exists solely as a monomer. To further understand our in vitro findings, we observed PAFAH-II oligomer formation in live cells, using PCH. This technique allowed us to study the oligomeric state of fluorescently tagged PAFAH-II at a low cellular concentration, which are more closely matched to physiological enzyme abundance than can be obtained using traditional in vitro methods.
We determined the molecular brightness of monomeric and homodimeric species of PAFAH-II by comparison to the molecular brightness of eGFP monomer and dimer controls. Our data indicate that WT-PAFAH-II is a homodimer in the cytoplasm, monomer in the nucleus, and a mixture of monomer and dimer at a position proximal to the membrane. It is important to point out that the PCH method poses a particular challenge at a membrane surface. At best, a window of detection can be defined near a membrane surface, yet the observation likely represents a distribution of membrane surface and the aqueous portion adjacent to the membrane surface. Therefore, results that demonstrate an oligomeric mixture are likely due to detecting both truly membrane and some residual cytoplasmic dimeric protein. The presence of both oligomeric species detected by PCH is consistent with our native PAGE results.
Unlike previous results [12] that suggested WT-PAFAH-II did not localize into the nucleus (Fig. 7), our PCH analysis showed that the WT enzyme is partially nuclear. We believe that nuclear WT-PAFAH-II is only a small portion of the intracellular pool of the enzyme, likely obscured by high cytoplasmic concentrations in prior imaging results (Fig. 7), which were performed with a higher expression level of PAFAH-II [12]. PCH analysis shows that PAFAH-II within the nucleus is solely monomeric. This data provides evidence that homodimeric PAFAH-II dissociates into monomers and these monomers do not remain cytoplasmic. It is plausible that the monomers traffic primarily to the ER and Golgi membranes. However a small population of monomeric PAFAH-II also localizes within the nucleus.
Fig. 7.
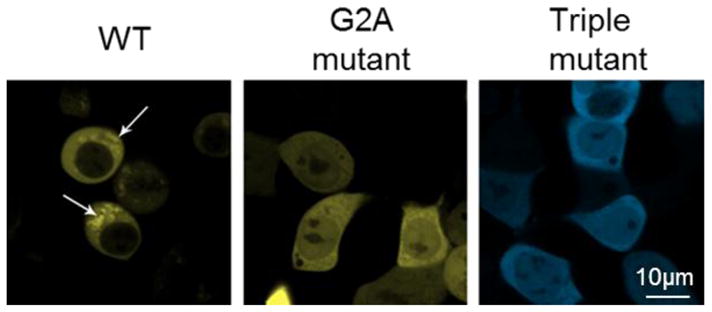
Confocal microscopy images of HEK293 cells expressing PAFAH-II WT, G2A and hydrophobic patch mutant fusions with fluorescent protein. Representative images showing WT-PAFAH-II-YFP, G2A-PAFAH-II-YFP, or Triple-mutant-PAFAH-II-CFP cellular locations. WT-PAFAH-II-YFP was evenly distributed in the cytoplasm and cytoplasmic membranes (shown by white arrows), while PAFAH-II levels were notably reduced inside the nucleus. In contrast, the G2A myristoyl mutant and the hydrophobic patch triple mutant (L327S/I328S/F331S) were evenly distributed in the cytoplasm and nucleus. This image was adapted from a figure of our previous work [12].
Previous research has shown that a protein’s myristoyl group may have a role in protein oligomerization [25]. To study the role of the myristoyl group on the N-terminus of PAFAH-II, we constructed an unmyristoylated form of the enzyme by mutating Gly-2 to Ala. Our native PAGE analysis shows that unmyristoylated PAFAH-II is completely monomeric. PCH analysis of cytoplasmic, unmyristoylated PAFAH-II is consistent with our native PAGE data and shows monomeric species in this cellular location.
We hypothesize that the hydrophobic residues, previously shown to be involved in PAFAH-II membrane binding [12], are also involved in homodimer formation. Native PAGE analysis of a triple hydrophobic patch mutation (L327S/I328S/F331S) showed that this PAFAH-II construct failed to form a dimer (data not shown). Previous whole cell imaging of the triple hydrophobic patch and G2A mutants were completely cytoplasmic and equally localized to the nucleus [12]. Based on the similar native PAGE and nuclear localization behavior of the G2A and hydrophobic patch mutant, it is probable that the triple hydrophobic patch mutant is also monomeric in the cytoplasm of the cell.
The data shown here demonstrates that wild type PAFAH-II forms monomers and homodimers. Additionally, we see that the myristoyl group is required for this protein-protein interaction within the cytoplasm. We have previously shown that the myristoyl group and five hydrophobic residues are required for PAFAH-II membrane localization [12]. Together, these finding suggest that both the myristoyl group and two hydrophobic helices of PAFAH-II are required for monomeric membrane localization or for protein dimerization found in the cytoplasm.
The cellular localization of PAFAH-II is correlated to changes in its oligomeric state. Evidence of this was shown by the distinct localization of homodimers to the cytoplasm and monomers to the membranes. It is likely that the cellular distribution of PAFAH-II is critical for substrate sequestering and proper function as an oxidative stress protector. Therefore, we believe that the regulation of PAFAH-II’s oligomerization state is a crucial part of its oxidative stress response mechanism. This model for PAFAH-II regulation is akin to that of other myristoylated proteins, namely NAP-22 [25]. Myristoylated NAP-22 forms a dimer; the regulation of this enzyme appears to be hinged on competitive binding of the myristoyl group. The myristoyl group drives NAP-22 to form a homodimer or to bind to the membrane where it functions [25].
We propose that PAFAH-II oxidative stress trafficking is mediated by changes in its oligomeric state. In unstressed situations, PAFAH-II remains cytosolic and exists as a dimer. Fig. 8 shows a possible model where PAFAH-II dimerizes in a head to tail orientation, and the membrane binding regions of the two monomers serve as the protein-protein interface of this homodimer. The hydrophobic residues from one protein conceal the hydrophobic residues of the other protein, while the myristoyl groups bury into the opposing protein. The PAFAH-II homodimer dissociates into a monomer when exposed to oxidative stress. It is possible that a post-translational modification, such as cysteine nitrosylation, lysine alkylation or tyrosine nitration, linked to oxidative stress facilitates the exposure of the myristoyl group, as described recently for the myristoyl switch reported for cAMP dependent protein kinase [33]. As a monomer, the hydrophobic patch of PAFAH-II including its myristoyl group, previously hidden as a dimer, is now exposed and able to bind to the membranes of the Golgi and ER. The association of PAFAH-II to the membrane is understood to be dynamic and thermodynamically balanced, as even a single residue mutation in the hydrophobic patch was enough to prevent membrane binding [12]. Work is underway to test this speculative model with experiments aimed at uncovering the molecular details of the homodimer formation as well as the identification of post-translational modifications tied to its regulation.
Fig. 8.
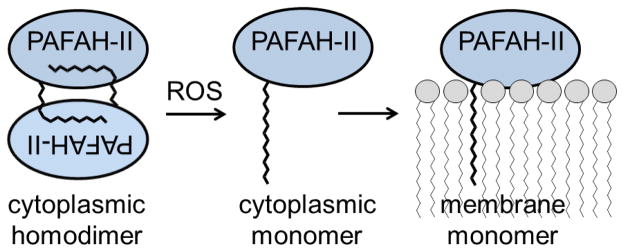
Proposed PAFAH-II trafficking model. A model of oxidative stress induced trafficking consistent with our observation begins with WT PAFAH-II existing as a homodimer in an unstressed state. The myristoyl group of one subunit packs into the hydrophobic pocket of the dimer partner. Upon ROS oxidative stress, post-translational modification of PAFAH-II disrupts the homodimer, thereby driving the PAFAH-II monomer to seek out a membrane surface for myristoyl group and hydrophobic patch mediated membrane surface interaction.
5. Conclusion
We have shown that PAFAH-II oligomerization is correlated to its localization. The oligomeric forms of this enzyme are distinctly positioned within the cell, where dimeric PAFAH-II is cytoplasmic and membrane associated PAFAH-II is monomeric. Additionally, we see that the myristoyl group of PAFAH-II, required for membrane binding [12], is also required for protein dimerization. We also predict that the five hydrophobic residues observed to be critical for membrane binding [12], are also important for protein dimerization. Finally, we propose that the oxidative stress post-translational response of PAFAH-II, by which it becomes more membrane bound, is regulated via the oligomeric state of the protein.
Highlights.
Membrane associated PAFAH-II is monomeric while cytoplasmic protein forms homodimers.
PAFAH-II’s myristoyl group is required for dimerization within the cytoplasm.
The oligomeric state of PAFAH-II drives protein localization and membrane association.
PAFAH-II’s functional trafficking is mediated by oxidative stress and its oligomeric state.
Acknowledgments
We are thankful to Dr. John Koh and Dr. Daniel Simmons for use of their lab space and equipment. We thank the imaging team at the DBI imaging facility for assistance. This work was supported by National Institutes of Health (NIH) grants 8P30GM103519 from the National Institute of General Medical Sciences and 5R01HL084366 from the National Heart, Lung, and Blood Institute.
Abbreviations
- CPSM
counts per second per molecule
- eGFP
enhanced green fluorescent protein
- eGFP-pCEP4
enhanced green fluorescent protein monomer
- eGFP-eGFP-pCEP4
enhanced green fluorescent protein dimer mimic
- ER
endoplasmic reticulum
- FFS
fluorescent fluctuation spectroscopy
- G2A
glycine-2 to alanine mutant
- OPM
Orientations of Proteins in Membranes
- PAGE
polyacrylamide gel electrophoresis
- PAF
platelet-activating factor
- PAFAH-II
platelet-activating factor acetylhydrolase type II
- PCH
photon counting histogram
- PLA2
phospholipase A2
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulfate
- TBS-T
Tris buffered saline with Tween 20
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rice SQ, Southan C, Boyd HF, Terrett JA, MacPhee CH, Moores K, Gloger IS, Tew DG. Expression, Purification and Characterization of a Human Serine-Dependent Phospholipase A2 with High Specificity for Oxidized Phospholipids and Platelet Activating Factor. Biochem J. 1998;330(Pt 3):1309–1315. doi: 10.1042/bj3301309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marques M, Pei Y, Southall MD, Johnston JM, Arai H, Aoki J, Inoue T, Seltmann H, Zouboulis CC, Travers JB. Identification of Platelet-Activating Factor Acetylhydrolase II in Human Skin. J Invest Dermatol. 2002;119:913–919. doi: 10.1046/j.1523-1747.2002.01859.x. [DOI] [PubMed] [Google Scholar]
- 3.Hattori K, Adachi H, Matsuzawa A, Yamamoto K, Tsujimoto M, Aoki J, Hattori M, Arai H, Inoue K. cDNA Cloning and Expression of Intracellular Platelet-Activating Factor (PAF) Acetylhydrolase II. Its Homology with Plasma PAF Acetylhydrolase. J Biol Chem. 1996;271:33032–33038. doi: 10.1074/jbc.271.51.33032. [DOI] [PubMed] [Google Scholar]
- 4.Hattori K, Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Purification and Characterization of Platelet-Activating Factor Acetylhydrolase II from Bovine Liver Cytosol. J Biol Chem. 1995;270:22308–22313. doi: 10.1074/jbc.270.38.22308. [DOI] [PubMed] [Google Scholar]
- 5.Foulks JM, Weyrich AS, Zimmerman GA, McIntyre TM. A Yeast PAF Acetylhydrolase Ortholog Suppresses Oxidative Death. Free Radic Biol Med. 2008;45:434–442. doi: 10.1016/j.freeradbiomed.2008.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inoue T, Sugimoto A, Suzuki Y, Yamamoto M, Tsujimoto M, Inoue K, Aoki J, Arai H. Type II Platelet-Activating Factor-Acetylhydrolase Is Essential for Epithelial Morphogenesis in Caenorhabditis Elegans. Proc Natl Acad Sci USA. 2004;101:13233–13238. doi: 10.1073/pnas.0405507101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaloske RH, Dennis EA. The Phospholipase A2 Superfamily and Its Group Numbering System. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Stafforini DM, McIntyre TM, Zimmerman GA, Prescott SM. Platelet-Activating Factor Acetylhydrolases. J Biol Chem. 1997;272:17895–17898. doi: 10.1074/jbc.272.29.17895. [DOI] [PubMed] [Google Scholar]
- 9.Chakraborty S, Asgeirsson B, Rao BJ. A Measure of the Broad Substrate Specificity of Enzymes Based on ‘Duplicate’ Catalytic Residues. PloS One. 2012;7:e49313. doi: 10.1371/journal.pone.0049313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kono N, Inoue T, Yoshida Y, Sato H, Matsusue T, Itabe H, Niki E, Aoki J, Arai H. Protection against Oxidative Stress-Induced Hepatic Injury by Intracellular Type II Platelet-Activating Factor Acetylhydrolase by Metabolism of Oxidized Phospholipids in Vivo. J Biol Chem. 2008;283:1628–1636. doi: 10.1074/jbc.M708622200. [DOI] [PubMed] [Google Scholar]
- 11.Matsuzawa A, Hattori K, Aoki J, Arai H, Inoue K. Protection against Oxidative Stress-Induced Cell Death by Intracellular Platelet-Activating Factor-Acetylhydrolase II. J Biol Chem. 1997;272:32315–32320. doi: 10.1074/jbc.272.51.32315. [DOI] [PubMed] [Google Scholar]
- 12.Thevenin AF, Monillas ES, Winget JM, Czymmek K, Bahnson BJ. Trafficking of Platelet-Activating Factor Acetylhydrolase Type II in Response to Oxidative Stress. Biochemistry. 2011;50:8417–8426. doi: 10.1021/bi200802w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foulks JM, Weyrich AS, Zimmerman GA, McIntyre TM. A Yeast PAF Acetylhydrolase Ortholog Suppresses Oxidative Death. Free Radic Biol Med. 2008;45:434–442. doi: 10.1016/j.freeradbiomed.2008.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kono N, Inoue T, Yoshida Y, Sato H, Matsusue T, Itabe H, Niki E, Aoki J, Arai H. Protection against Oxidative Stress-Induced Hepatic Injury by Intracellular Type II Platelet-Activating Factor Acetylhydrolase by Metabolism of Oxidized Phospholipids in Vivo. J Biol Chem. 2008;283:1628–1636. doi: 10.1074/jbc.M708622200. [DOI] [PubMed] [Google Scholar]
- 15.Matsuzawa A, Hattori K, Aoki J, Arai H, Inoue K. Protection against Oxidative Stress-Induced Cell Death by Intracellular Platelet-Activating Factor-Acetylhydrolase II. J Biol Chem. 1997;272:32315–32320. doi: 10.1074/jbc.272.51.32315. [DOI] [PubMed] [Google Scholar]
- 16.Arai H. Platelet-Activating Factor Acetylhydrolase. Prostaglandins Other Lipid Mediat. 2002;68–69:83–94. doi: 10.1016/s0090-6980(02)00023-0. [DOI] [PubMed] [Google Scholar]
- 17.Tjoelker LW, Stafforini DM. Platelet-Activating Factor Acetylhydrolases in Health and Disease. Biochim Biophys Acta. 2000;1488:102–123. doi: 10.1016/s1388-1981(00)00114-1. [DOI] [PubMed] [Google Scholar]
- 18.Stafforini DM, McIntyre TM. Determination of Phospholipase Activity of PAF Acetylhydrolase. Free Radic Biol Med. 2013;59:100–107. doi: 10.1016/j.freeradbiomed.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI. Positioning of Proteins in Membranes: A Computational Approach. Protein Sci. 2006;15:1318–1333. doi: 10.1110/ps.062126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: Orientations of Proteins in Membranes Database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 21.Greenberg ME, Li XM, Gugiu BG, Gu X, Qin J, Salomon RG, Hazen SL. The Lipid Whisker Model of the Structure of Oxidized Cell Membranes. J Biol Chem. 2008;283:2385–2396. doi: 10.1074/jbc.M707348200. [DOI] [PubMed] [Google Scholar]
- 22.Li C, Pan W, Braunewell KH, Ames JB. Structural Analysis of Mg2+ and Ca2+ Binding, Myristoylation, and Dimerization of the Neuronal Calcium Sensor and Visinin-Like Protein 1 (Vilip-1) J Biol Chem. 2011;286:6354–6366. doi: 10.1074/jbc.M110.173724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breuer S, Gerlach H, Kolaric B, Urbanke C, Opitz N, Geyer M. Biochemical Indication for Myristoylation-Dependent Conformational Changes in HIV-1 Nef. Biochemistry. 2006;45:2339–2349. doi: 10.1021/bi052052c. [DOI] [PubMed] [Google Scholar]
- 24.Dennis CA, Baron A, Grossmann JG, Mazaleyrat S, Harris M, Jaeger J. Co-Translational Myristoylation Alters the Quaternary Structure of HIV-1 Nef in Solution. Proteins. 2005;60:658–669. doi: 10.1002/prot.20544. [DOI] [PubMed] [Google Scholar]
- 25.Epand RM, Braswell EH, Yip CM, Epand RF, Maekawa S. Quaternary Structure of the Neuronal Protein Nap-22 in Aqueous Solution. Biochim Biophys Acta. 2003;1650:50–58. doi: 10.1016/s1570-9639(03)00191-2. [DOI] [PubMed] [Google Scholar]
- 26.Slaughter BD, Li R. Toward Quantitative “in Vivo Biochemistry” with Fluorescence Fluctuation Spectroscopy. Mol Biol Cell. 2010;21:4306–4311. doi: 10.1091/mbc.E10-05-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Muller JD, So PT, Gratton E. The Photon Counting Histogram in Fluorescence Fluctuation Spectroscopy. Biophys J. 1999;77:553–567. doi: 10.1016/S0006-3495(99)76912-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Wei LN, Muller JD. Probing Protein Oligomerization in Living Cells with Fluorescence Fluctuation Spectroscopy. Proc Natl Acad Sci USA. 2003;100:15492–15497. doi: 10.1073/pnas.2533045100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slaughter BD, Huff JM, Wiegraebe W, Schwartz JW, Li R. Sam Domain-Based Protein Oligomerization Observed by Live-Cell Fluorescence Fluctuation Spectroscopy. PLoS One. 2008;3:e1931. doi: 10.1371/journal.pone.0001931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slaughter BD, Schwartz JW, Li R. Mapping Dynamic Protein Interactions in Map Kinase Signaling Using Live-Cell Fluorescence Fluctuation Spectroscopy and Imaging. Proc Natl Acad Sci USA. 2007;104:20320–20325. doi: 10.1073/pnas.0710336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, Muller JD, Ruan Q, Gratton E. Molecular Brightness Characterization of eGFP in Vivo by Fluorescence Fluctuation Spectroscopy. Biophys J. 2002;82:133–144. doi: 10.1016/S0006-3495(02)75380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang B, Perroud TD, Zare RN. Photon Counting Histogram: One-Photon Excitation. Chemphyschem. 2004;5:1523–1531. doi: 10.1002/cphc.200400176. [DOI] [PubMed] [Google Scholar]
- 33.Gaffarogullari EC, Masterson LR, Metcalfe EE, Traaseth NJ, Balatri E, Musa MM, Mullen D, Distefano MD, Veglia G. A Myristoyl/Phosphoserine Switch Controls cAMP-Dependent Protein Kinase Association to Membranes. J Mol Biol. 2011;411:823–836. doi: 10.1016/j.jmb.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]