

Abstract
High-throughput omics have proven invaluable in studying human disease, and yet day-to-day clinical practice still relies on physiological, non-omic markers. The metabolic syndrome, for example, is diagnosed and monitored by blood and urine indices such as blood cholesterol levels. Nevertheless, the association between the molecular and the physiological manifestations of the disease, especially in response to treatment, has not been investigated in a systematic manner. To this end, we studied a mouse model of diet-induced dyslipidemia and atherosclerosis that was subject to various drug treatments relevant to the disease in question. Both physiological data and gene expression data (from the liver and white adipose) were analyzed and compared. We find that treatments that restore gene expression patterns to their norm are associated with the successful restoration of physiological markers to their baselines. This holds in a tissue-specific manner—treatments that reverse the transcriptomic signatures of the disease in a particular tissue are associated with positive physiological effects in that tissue. Further, treatments that introduce large non-restorative gene expression alterations are associated with unfavorable physiological outcomes. These results provide a sound basis to in silico methods that rely on omic metrics for drug repurposing and drug discovery by searching for compounds that reverse a disease's omic signatures. Moreover, they highlight the need to develop drugs that restore the global cellular state to its healthy norm rather than rectify particular disease phenotypes.
Keywords: connectivity map, disease reversal, drug repositioning, homeostasis, systems medicine
Introduction
High-throughput gene expression data serve to illuminate the molecular underpinnings of different cellular phenotypes. Such data have proven indispensable to studying the molecular basis of human disease (Cooper-Knock et al, 2012). They were further used to establish risk biomarkers (Scherzer et al, 2007), to predict prognosis and clinical outcome (Sørlie et al, 2001; Pomeroy et al, 2002; Singh et al, 2002; Sotiriou et al, 2003; Willenbrock et al, 2004; Chuang et al, 2007, 2012; Oh et al, 2012), and to examine the effects of drug treatment (Cheok et al, 2003; Baur et al, 2006; Fernald & Altman, 2013). Assays that examine gene expression have already been put to clinical use (McCarthy et al, 2013) and are expected to become a principal element of future personalized medical care.
Yet, present medical diagnosis and decisions concerning clinical interventions rely extensively on measurement of relevant physiological markers such as blood and urine indices that can be readily obtained from patients (Hood & Price, 2014). This is particularly true in the case of the metabolic syndrome and related disorders (Mattix et al, 2002; Huang, 2009; American Diabetes Association, 2013; Imran et al, 2013). A deviation of the physiological markers from their normal levels assists in establishing a diagnosis and indicates elevated disease-associated risk. For example, higher than normal levels of blood glucose and blood triglycerides form two of the criteria that define the metabolic syndrome (Huang, 2009), and are associated with elevated cardiovascular risk. By the same token, clinical interventions are expected to restore these markers to their normal levels, and such a restoration is construed as a sign of reduced disease-associated risk (Qaseem et al, 2007; Moghissi et al, 2009; American Diabetes Association, 2013; Harper et al, 2013).
Thus, the effectiveness of extant treatments is judged not only by their ability to reverse the clinical course of the disease, but also by their ability to restore the patients’ physiological markers to their norm. One might assume that the latter is also indicative of a restoration of disease-affected molecular-level phenotypes to their norm. In other words, it is natural to assume that effective clinical treatments serve, in parallel, to eliminate disease-induced abnormalities both at the organismic level and at the cellular level. Notably, several recent studies have made this implicit assumption by directly searching for drug therapies whose molecular effects anti-correlate with disease molecular signatures (Hu & Agarwal, 2009; Boyle et al, 2010; Chang et al, 2010; Josset et al, 2010; Dudley et al, 2011; Kunkel et al, 2011; Sirota et al, 2011; Qu & Rajpal, 2012; Jahchan et al, 2013; Pacini et al, 2013; Yizhak et al, 2013; Zerbini et al, 2014). The underlying logic is clear: A treatment that eliminates molecular-level abnormalities is assumed to do the same at the physiological level. These studies have already shown considerable success and offer an appealing way to search for novel drug targets, or to repurpose existing drugs rationally. However, to the best of our knowledge, the fundamental relationship between the effects of pharmacological interventions at the physiological, organismic level and their effects at the molecular level has not been rigorously studied up to date.
Here, we address this challenge and ask whether extant treatments that restore physiological indices back to their baseline levels also reverse disease phenotypes at the molecular level. This questions is studied in one case study, a mouse model of dyslipidemia for which pertaining physiological and molecular data have been collected (Radonjic et al, 2013). This model is particularly apt to study the questions at hand because diagnosis and clinical risk assessment in the case of the metabolic syndrome and related disorders depend almost exclusively on physiological markers (Huang, 2009).
Results
Design and methodology
We analyze hepatic and white adipose gene expression, as well as 26 disease-relevant physiological markers, measured in a LDLR−/− mouse model of diet-induced dyslipidemia (Supplementary Table S1). Low-density lipoprotein receptor (LDLR)-deficient mice are genetically predisposed to develop hypercholesterolemia and atherosclerotic lesions. These manifestations appear in mild forms even under a normal diet, but are greatly aggravated when the mice are placed on a high-fat diet (Zadelaar et al, 2007; Getz & Reardon, 2012; Ma et al, 2012). The experimental setting was previously described (Radonjic et al, 2013); it is summarized here and in Box 1A for completeness’ sake.
Box 1: Experimental design and methodology.
(A) The experimental design. Physiological markers and hepatic gene expression were measured in all animals. Adipose gene expression was measured in the experimental groups marked with a white outline. The colors that mark each experimental group are retained throughout the figures in the paper. (B) A schematic illustration of the main research question. Left panel: shows the gene expression space, onto which the gene expression of each subject is projected (Materials and Methods). Blue, red, and green markers represent LFD, untreated HFD, and treated HFD subjects, respectively. The transcriptome deviation index (TDI) of a subject (dashed arrows) is the distance of its gene expression from the mean gene expression of the LFD group (dotted yellow triangle), which represents the healthy baseline. Right panel: shows the physiological space. Organism-level physiological markers are collected from the same subjects and projected onto a physiological space (Materials and Methods). The global physiological deviation index (GPDI) (dashed arrows) is the distance of a sample from the mean of the LFD group in the physiological space. The main research question of the current study translates into asking whether TDIs and GPDIs are correlated.
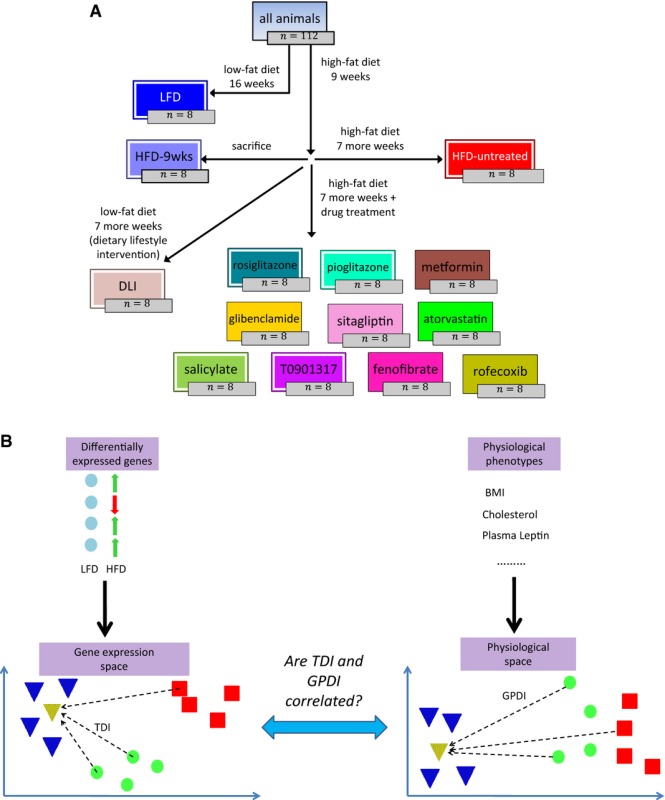
A control group of mice was kept on low-fat maintenance chow diet (LFD) throughout the experiment (16 weeks in total). The other mice were fed high-fat diet (HFD) for 9 weeks to establish dyslipidemia-associated disease markers and were then divided between the following intervention regimens: One group was sacrificed immediately (9-week HFD group) and represents the time-point at which interventions were commenced. The other mice were continued on HFD for 7 more weeks during which they were either left untreated (HFD-untreated) or treated with one of 10 relevant drugs (see below). One group of mice was switched after 9 weeks from HFD to LFD (again, for 7 weeks) and represents a dietary lifestyle intervention (DLI). Hepatic gene expression was measured in all animals. In addition, white adipose gene expression was measured in 4 of the pharmacological intervention groups (rosiglitazone, pioglitazone, T0901317, and salicylate), as well as in the LFD, 9-week HFD, HFD-untreated, and DLI groups.
The ten drugs studied here are all highly relevant to the disease in question (Table1, Supplementary Table S2). Eight of the drugs are FDA-approved and commonly prescribed to treat the metabolic syndrome in human patients. These include oral anti-diabetic compounds that work through diverse mechanisms (metformin, glibenclamide, sitagliptin, rosiglitazone, pioglitazone), lipid-modulating compounds (fenofibrate, atorvastatin), and the anti-inflammatory salicylate; salicylic acid is the main active compound in aspirin, which is prescribed to prevent atherosclerotic complications (Awtry & Loscalzo, 2000; Campbell et al, 2007; American Diabetes Association, 2013). One drug (T0901317) is an experimental compound that has been shown to exert anti-atherogenic effects in the mouse model studied here (Calkin & Tontonoz, 2010), and the last one (rofecoxib) is an anti-inflammatory compound, included because of the critical role of inflammation in atherosclerotic disease (Ross, 1999; Libby et al, 2002; Hansson & Hermansson, 2011; Weber & Noels, 2011; van Diepen et al, 2013).
Table 1.
Drug compounds studied in this work.
| Category | Drug | Status | Notes |
|---|---|---|---|
| Anti-diabetic | Metformin | Approved | Improve glycemic control |
| Glibenclamide | Approved | ||
| Sitagliptin | Approved | ||
| Rosiglitazone | Approved | ||
| Pioglitazone | Approved | ||
| Lipid-modulating | Fenofibrate | Approved | Reduces plasma triglycerides as well as low-density lipoprotein (LDL) cholesterol levels and increases plasma high-density lipoprotein (HDL) cholesterol levels |
| T0901317 | Experimental | A synthetic liver X receptor (LXR) agonist which is known to exert an anti-atherogenic effect in LDLR−/− mice (Calkin & Tontonoz, 2010). LXR modifiers are actively studied as pharmacological targets for treating atherosclerosis (Hong & Tontonoz, 2014) | |
| Atorvastatin | Approved | Lowers plasma LDL cholesterol levels | |
| Anti-inflammatory | Salicylate | Approved | A chronic inflammatory response underlies the metabolic syndrome and the progression of atherosclerotic disease (Libby et al, 2002; Hansson & Hermansson, 2011; Lumeng & Saltiel, 2011; Harper et al, 2013; Van Diepen et al, 2013; McNelis & Olefsky, 2014). Hence, anti-inflammatory drugs were expected to alleviate cardiovascular complications. |
| Rofecoxib | Withdrawn several years after FDA approval due to an increased risk of cardiovascular events (Praticò & Dogné, 2005) | Note that salicylic acid is the main active compound in aspirin, which is commonly prescribed to prevent atherosclerotic complications (Awtry & Loscalzo, 2000; Campbell et al, 2007; American Diabetes Association, 2013) |
Radonjic et al (2013) have recently analyzed a subset of these data and compared the effects of the pharmacological interventions with those of the dietary intervention. They concluded that the pharmacological interventions generally reduced the HFD-induced hyperglycemia, and yet had only a limited effect on other cardiovascular risk markers, whereas the dietary intervention attenuated all risk markers. Transcriptomic and metabolomic analyses of disease pathways confirmed a reversal of HFD-induced disturbances to the liver under DLI. Here, we utilize an extended collection of the data reported by Radonjic et al to rigorously investigate the correlation between the reversal of molecular disease phenotypes and the reversal of physiological disease phenotypes across multiple treatment groups.
We considered the LFD group to be representative of a low-risk baseline (for simplicity of explanation, we refer to these mice as ‘healthy’ despite their genetic deficiency). The untreated 16-week HFD group (henceforth designated ‘untreated HFD’ for brevity) was considered representative of an elevated risk to develop cardiovascular disease. To assess the success of each drug in restoring the normal (LFD) transcriptome, we first identified the genes that were most differentially expressed between the HFD and LFD groups in each of the two tissues, and are thus implicated in diet-induced pathologies and higher cardiovascular risk. These genes were used to define a Euclidean gene expression space for each of the two tissues, onto which we projected the gene expression vector of each animal (Materials and Methods, Discussion). We then computed the (tissue-specific) transcriptome deviation index (TDI) of each animal—the distance in the gene expression space between the animal and the centroid of the healthy (LFD) animals (Box 1B, Materials and Methods). The smaller the TDI, the closer is the animal's transcriptome to the normal, low-risk state. We defined a physiological analog to the TDI based on the aggregate data of the 26 disease-relevant physiological biomarkers measured in the study animals (Supplementary Table S1). These markers represent metabolic abnormalities shown to predispose patients to the development of type 2 diabetes and cardiovascular disease and are instrumental in the clinical management of human patients. We used the aggregated physiological data to compute a global physiological deviation index (GPDI) (Box 1B, Materials and Methods). The smaller is an animal's GPDI, the closer is its physiology to the normal baseline. Our key hypothesis, namely that a restoration of the normal transcriptome following a clinical intervention is correlated with a restoration of the normal physiology, translates into the hypothesis that a direct correlation exists between the TDIs and GPDIs across the studied animals.
The restoration of gene expression correlates with a restoration of physiological markers to their baseline levels
The projection of the animal data into the transcriptomic and physiological spaces allowed us to study their relationship (Fig1, Supplementary Figs S1 and S2). We first examined the post-treatment transcriptomic changes in the hepatic and adipose gene expression spaces separately to obtain a tissue-specific view (Fig1A and B). Many of the treatments altered the gene expression of the treated HFD groups in a way that brought it closer to the gene expression of the LFD group (Supplementary Results S1, Supplementary Fig S1). This observation, which accords with the conceptual image depicted in Box 1B, can also be seen in Fig1A and B: Most dots are closer to the LFD mean (circled in blue) than are the untreated HFD animals. Nonetheless, there exists considerable variability between transcriptomic effects on the animals in each treatment group (Supplementary Results S2, Supplementary Figs S3 and S4). Quite remarkably, the dietary lifestyle intervention was the most effective in reversing the transcriptome back to its healthy state in both tissues, as well as in reinstating the healthy physiology (Fig1, Supplementary Figs S1 and S2), in line with the results of Radonjic et al (2013).
Figure 1.

- A-C The figure shows the first two principal components of (A) the gene expression space of the liver, (B) the gene expression space of white adipose tissue, and (C) the physiological space. In all panels, each dot represents one animal; color codes denote the different experimental groups. Red squares represent the HFD animals, and blue triangles represent the LFD animals. The dashed arrow connects the HFD centroid (yellow square, circled in red) to the LFD centroid (yellow triangle, circled in blue) and denotes the direction of a reversal of the gene expression or physiological state back to the norm.
Gene set enrichment analysis (GSEA; Subramanian et al, 2005) corroborated that pathways that were expected to be modulated by some of the drugs were indeed differentially expressed (except in the case of metformin, which may be due to the lack of statistical power). Interestingly, two particular observations that have been recently reported in other animal models were also detected by this analysis: dysregulation of hepatic ribosomal activity in HFD-fed mice, as in Oie et al (2014), and suppression of the complement and coagulation systems by T0901317, as in Sukardi et al (2012). See Supplementary Results S3 and Supplementary Tables S3 and S4 for details (see also Supplementary Results S4.1 for an alternative definition of TDI via GSEA).
Examining the relationship between the transcriptomic and physiological changes, we find that both the adipose TDI and the hepatic TDI are correlated with the GPDI, as hypothesized. The correlation is particularly strong in the adipose case (Fig2A and B; Pearson rho = 0.91, 0.63 for adipose and liver, respectively; P-values <3e-17, 2e-10; all the results hold when Spearman correlations are used instead of Pearson correlations, see Supplementary Fig S6A and B). This indicates that HFD-induced cardiovascular risk is associated with both transcriptomic and physiological deviations from the norm and that its alleviation by extant treatments is correlated with a restoration of the norm at both levels. The hepatic metabolome was also measured in the animals (Materials and Methods) and represents another dimension of the molecular-level disease state. We projected these data into a metabolome space, and computed metabolome deviation indices (MDI) in a manner analogous to that of the transcriptomic and physiological data (Materials and Methods, Supplementary Fig S7). The hepatic MDI is significantly correlated both with the hepatic TDI (Fig2C, Supplementary Fig S6C; Pearson rho = 0.62, P < 2e-10) and with the GPDI (Pearson rho = 0.46, P < 6e-6). However, it is not correlated with the adipose TDI (Pearson rho = 0.15, P = 0.17).
Figure 2.
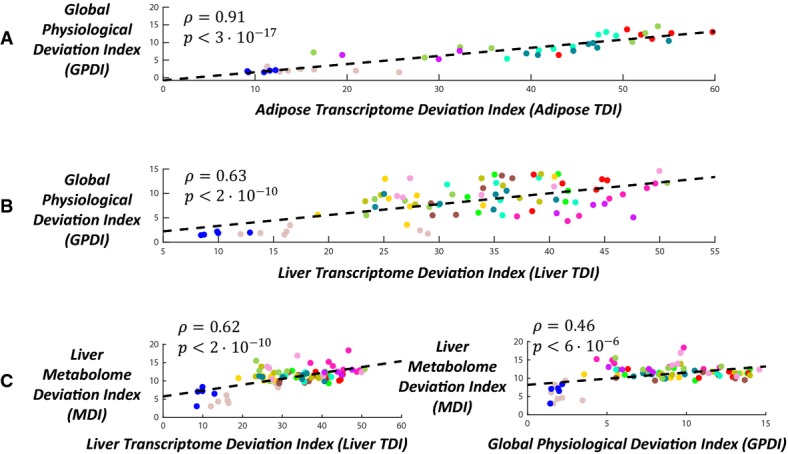
- A, B The correlation between the global physiological deviation index (GPDI) and the transcriptomic deviation index (TDI) in (A) white adipose tissue and (B) liver tissue.
- C Left panel: the correlation between the liver metabolome deviation index (MDI) and the liver TDI. Right panel: the correlation between the liver MDI and the GPDI.
We next turned to examine the correlation between the TDIs of the animals and individual physiological markers; for this purpose, we computed an individual physiological deviation index (PDI) for each of the markers (Materials and Methods). Overall, high correlations were observed and had noticeable tissue-specific trends (Fig3, Supplementary Table S7; Supplementary Figs S8 and S9; P-values were adjusted to multiple hypotheses testing according to the Benjamini–Hochberg FDR method, and false discovery rate was set at α = 5%; Spearman correlations, which are more robust than Pearson correlations in the presence of outliers, were used here because several animals had outlier physiological measurements in particular markers). The white adipose and hepatic TDIs were significantly correlated with 20 and 19 PDIs out of the 26 measured, respectively. The adipose transcriptome was most highly correlated with the animals’ total body weight, total white adipose weight, and plasma leptin levels (Spearman rho = 0.84, 0.85, 0.86, adjusted P < 2.6e-17, 1e-17, 2.6e-17). The latter is a hormone that is produced predominantly by white adipose tissue, and whose blood levels correlate well with total body fat (Considine et al, 1996; Klok et al, 2007; Myers et al, 2008). The hepatic transcriptome was most highly correlated with plasma cholesterol, plasma triglycerides, and liver triglycerides levels (Spearman rho = 0.76, 0.64, 0.61, adjusted P < 2.5e-21, 1.8e-13, 5.5e-12, respectively), reflecting the liver's central role in lipid metabolism (liver MDI was correlated with these markers’ PDIs as well, see Supplementary Table S8 and Supplementary Figs S10 and S11).
Figure 3.
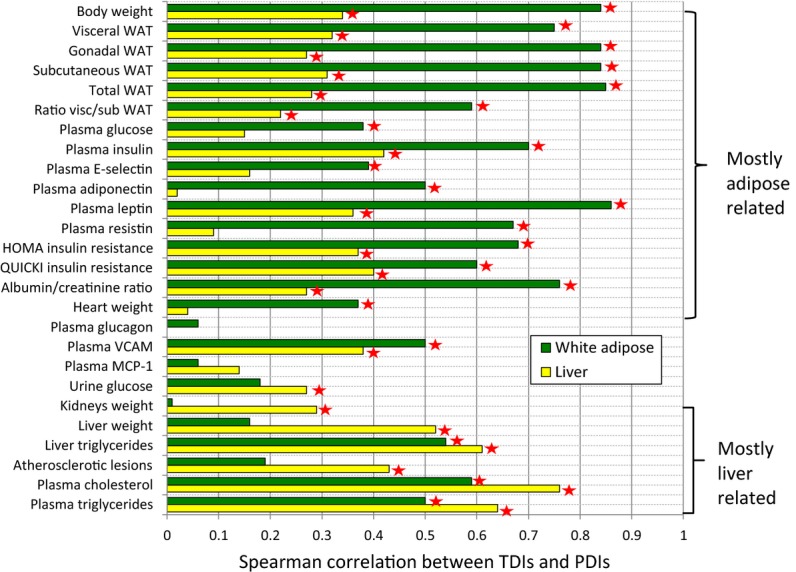
Deviations from the baseline gene expression and deviations from the baseline physiology are correlated in a tissue-specific manner
Deviations from the baseline gene expression (TDIs) in liver and white adipose tissues are correlated with deviations from the normal physiology (PDIs) for markers that are known to be associated with those tissues. Bar lengths represent the Spearman correlations between the TDIs of the two tissues and PDIs of 26 different physiological markers. WAT stands for white adipose tissue, ratio visc/sub WAT for ratio of visceral to subcutaneous WAT. Asterisks mark statistically significant correlations (using the Benjamini–Hochberg correction for multiple hypotheses testing with a 5% FDR level). The top part of the figure displays markers whose PDI is more correlated with white adipose TDI than with hepatic TDI. The bottom part shows markers with a reverse tissue-specific pattern. Source data are available online for this figure.
Significant correlations were also identified between TDIs and PDIs of known hallmark markers of cardiovascular risk: Albuminuria is correlated with adipose TDI, and atherosclerotic lesion size is correlated with hepatic TDI. Albuminuria is an established predictor for cardiovascular morbidity and mortality, as well as for poor renal outcome, which is often diagnosed via the albumin/creatinine ratio in the urine (Gerstein et al, 2001; Isomaa et al, 2001; Mattix et al, 2002; Bahrami et al, 2008; Basi et al, 2008). Interestingly, there exists in our data a strong correlation between the PDI of the albumin/creatinine ratio and the adipose TDI (Spearman rho = 0.76, adjusted P < 2.4e-10; only a weaker correlation was obtained with the hepatic TDI). This result is in accordance with recent findings concerning the role of adipose-derived hormones, and particularly adiponectin, in the development of albuminuria and renal dysfunction (Tsioufis et al, 2005; Sharma et al, 2008; Sharma, 2009; Ix & Sharma, 2010; Meyvis et al, 2013; Christou & Kiortsis, 2014). Liver TDI was significantly correlated with the PDI of atherosclerosis (Spearman rho = 0.43, adjusted P < 1.2e-5), which in turn triggers myocardial infarction and stroke (Weber & Noels, 2011). This reflects the causative role of dyslipidemia and liver inflammation in the formation of atherosclerotic lesions (Tannock et al, 2005; Kleemann et al, 2007; Harper et al, 2013; Van Diepen et al, 2013).
Notably, the associations between TDIs and PDIs hold across all the experimental groups: animals in different phases of disease progression, treated and untreated animals, and treatments that work through different mechanisms (Fig4A–D; four of the physiological markers for which particularly high correlations with TDIs were obtained are presented in this figure; the complete data are provided in Supplementary Table S7 and Supplementary Figs S8 and S9). This indicates that effective pharmacological interventions, which work through diverse mechanisms, ultimately serve to reverse the transcriptomic deviations from the healthy baseline, and that these effects are associated with a restoration of the normal physiology. The same holds also for the non-pharmacological, dietary intervention. Exceptions do occur, however. For example, the drug T090137 does not adhere to the general correlation trend between the adipose TDI and the plasma cholesterol and triglyceride PDIs (Fig4E). This probably stems from this drug's particular mechanism of action—it is an agonist of a transcription factor that has a pivotal role in lipid metabolism (Schultz et al, 2000; Steffensen & Gustafsson, 2004; Ulven et al, 2005; Zhao & Dahlman-Wright, 2010). This drug also has particularly high hepatic TDIs compared with other treatment groups, which again point to an interference with lipid metabolism (Fig1A, Supplementary Figs S1A and S12A, and see below).
Figure 4.

- A-D White adipose (A, B) and liver (C, D) TDIs (deviations from the baseline gene expression) are correlated with PDIs (deviations from the baseline physiology) across different experimental groups and treatment regimens. PDIs of four physiological markers are shown, two that are highly correlated with adipose transcriptome (A, B) and two that are highly correlated with hepatic transcriptome (C, D).
- E Adipose TDIs and plasma cholesterol PDIs are generally correlated, but this does not hold for animals treated with T090137 (purple dots at the top left corner of the figure), probably due to this drug's particular mechanism of action (see main text). The correlation displayed in (E) increases from 0.59 to 0.8 (P < 1.3e-13) when excluding the T090137 animals.
Non-restorative alterations to the gene expression are associated with unfavorable outcomes
Four drugs in our data introduced large transcriptomic alterations that are incongruent with the direction of the disease reversal axis that goes from the HFD centroid to the LFD centroid (such alterations will be henceforth referred to in short as “non-restorative”): pioglitazone and rosiglitazone in white adipose, and fenofibrate and T090137 in the liver (Fig1A and B; Supplementary Figs S12 and S13A–C). This seems to stem from their respective mechanisms of action—each of these drugs modulates a master transcription regulator, which is highly expressed in the tissue in which it causes the major non-restorative transcriptomic shifts (Supplementary Table S2). In contrast, the dietary intervention, which was the most successful one in reversing disease gene expression patterns, was also characterized by small non-restorative transcriptomic effects in both tissues when compared with the other intervention groups. We hypothesized that non-restorative shifts in gene expression are associated with unfavorable physiological outcomes.
A comprehensive test of this hypothesis is impossible in the current dataset: The physiological indices collected in the study animals were selected in the first place because of their relevance to the disease model, and this precludes us from studying unfavorable effects that arise solely as a side effect of the drugs rather than outcomes of the HFD feeding (Supplementary Table S9). Therefore, we resorted to considering only a limited scope of unfavorable drug effects that are detectable in the set of 26 physiological markers measured in the study animals (the same ones that had been analyzed throughout this paper, Supplementary Table S1). We considered a marker to indicate the presence of an unfavorable treatment outcome if it fulfilled two conditions: (i) Its levels in the HFD group were higher than in the LFD group, and (ii) its levels in any of the treatment groups were even higher than in the HFD group in a statistically significant manner (one-sided t-test; a parallel criterion was used for markers in which the HFD levels were lower than the LFD levels; P-values were adjusted to multiple hypotheses testing according to the Benjamini–Hochberg method, and false discovery rate was set at α = 5%). In other words, a marker was considered as manifesting an unfavorable outcome of a certain treatment if its values in the treated animals were even farther from the baseline than its values in the untreated HFD animals (Supplementary Fig S13D). We found that according to this definition, four drugs were associated with statistically significant unfavorable physiological outcomes: pioglitazone, rosiglitazone, fenofibrate, and T090137 (Supplementary Fig S14). Notably, these are the same drugs that were previously shown to introduce the highest non-restorative transcriptomic alterations.
The unfavorable outcomes ascribed to these drugs by our analysis were all previously described in the medical literature. Moreover, the unfavorable outcomes ascribed to each drug concern physiological markers that are known to be associated with the tissue in which it causes major non-restorative transcriptional shifts. T0901317 and fenofibrate introduced large non-restorative alterations to hepatic gene expression. Indeed, our analysis implicated T0901317 in hepatomegaly (i.e., liver enlargement), atherogenesis, and abnormally high plasma cholesterol and plasma triglyceride levels (Chisholm et al, 2003; Jung et al, 2011), all indicative of an underlying liver pathology. The presence of drug-induced hepatotoxicity is further corroborated by the observation that T0901317 treatment also led to significant up-regulation, compared with the untreated HFD animals, of several pro-inflammatory genes (Supplementary Results S5). Fenofibrate was implicated in hepatomegaly and elevated plasma cholesterol (but not triglyceride) levels (Toda et al, 2003; Backes et al, 2007). Treatment with the two TZDs, pioglitazone and rosiglitazone, led to large non-restorative alterations in adipose gene expression. These treatments also led to abnormally high levels of the hormone adiponectin, which is secreted exclusively by adipocytes (Chandran et al, 2003; Lihn et al, 2005; Ye & Scherer, 2013). While increased adiponectin levels are associated with reversal of insulin resistance (Yamauchi et al, 2001; Gao et al, 2013) and are thus considered a beneficial effect of TZD administration (Maeda et al, 2001; Hirose et al, 2002; Yang et al, 2002), the data studied here suggest that TZDs increase them far beyond (3–6 fold) normal levels. Notably, irregularly high adiponectin levels have been reported to correlate with increased cardiovascular disease and all-cause mortality (Laughlin et al, 2007; Jorsal et al, 2008; Ohashi et al, 2008). We conclude that in the animal model studied here, treatments that induce non-restorative gene expression alterations tend to be associated with unfavorable physiological outcomes.
Discussion
In this paper, we examined whether effective drug treatments, which restore various organism-level physiological indices to their norm, also serve to reverse molecular-level, disease-induced gene expression deviations from the norm. This assumption has profound implications to clinical practice. First, because restoration of disease-relevant physiological markers to baseline levels is construed as a sign of reduced risk, and hence as a sign of the treatment's efficacy. Second, because this assumption has already been made implicitly by successful studies that sought to identify novel drug therapies by searching for drug candidates whose molecular effects anti-correlate with disease molecular signatures (Hu & Agarwal, 2009; Boyle et al, 2010; Chang et al, 2010; Josset et al, 2010; Dudley et al, 2011; Kunkel et al, 2011; Sirota et al, 2011; Qu & Rajpal, 2012; Jahchan et al, 2013; Pacini et al, 2013; Yizhak et al, 2013). While this assumption seems reasonable enough, it is important to test it directly in a quantitative, systematic manner.
To this end, we studied data from a mouse model of diet-induced dyslipidemia and atherosclerosis that was subject to various drug treatments; both gene expression and physiological data were analyzed and compared. Overall, the data provide strong support to this hypothesis: Treatments that restore gene expression patterns to their norm are also more successful in restoring the physiological markers to their baselines. This observation holds in a tissue-specific manner, at least with respect to the two tissues studied here: Treatments that reverse the transcriptomic signatures of the disease in a particular tissue were associated with positive physiological effects in that tissue. For example, the LXR ligand T0901317 induced major transcriptional shifts in both liver and adipose. The shifts were mostly disease-reversing in adipose tissue, but not in the liver. Accordingly, mice treated with T0901317 were characterized by favorable adipose-related effects, including reduced body weight and normal levels of the adipose-derived hormones leptin, resistin, and adiponectin. However, they also exhibited adverse phenotypes consistent with drug-related impaired liver functions, such as elevated liver weight and blood cholesterol levels, which were both higher than those of untreated high-fat diet animals. A related and particularly troubling observation was that the T0901317 group had significantly larger atherosclerotic plaques than the untreated high-fat diet group.
One should note, however, that our findings are based only on the animal model studied here. While we hypothesize that they are more generally applicable, it remains for future work: (i) to extend them to other diseases and other organs than those studied here, (ii) to test them in human subjects, and (iii) to extend them to other omic data types that capture other facets of the molecular-level drug effects, as well as to other clinically relevant physiological indices. Using more sophisticated dimensionality reduction methods than the one employed here may prove illuminating. In addition, one notes that this study established only an association between treatment effects at the molecular and physiological levels; any causative relationship between the two levels is probably complex and multifaceted.
A major challenge left for future work is the incorporation of adverse drug side effects into the methodological framework employed in this study. We have shown that drugs that induce large non-restorative shifts in gene expression are associated, to some extent, with unfavorable physiological outcomes. We hypothesize that, more generally, the non-restorative gene expression alterations brought about by the drug may be correlated with the severity of the drug's side effects. The limited scope of the present data does not allow testing this hypothesis directly: Adverse drug side effects may manifest in biomarkers that are unaffected by the disease itself, whereas in the experiment analyzed here, only disease-relevant biomarkers were measured. Similarly, a study of side effects should consider genes that are unaffected by the disease itself but may still be affected by the drug treatment, while here we reduced the data's dimensionality by focusing only on disease-associated genes that were differentially expressed between the LFD and untreated HFD groups. Dimensionality reduction was necessary because distance metrics become unstable in high dimension (Beyer et al, 1999; Hinneburg et al, 2000; Aggarwal et al, 2001). Future studies that wish to consider adverse drug side effects may overcome the dimensionality challenge by augmenting the data-driven approach presented here with knowledge-based elements; that is, incorporating into the gene expression spaces known pathways and gene modules that are not disrupted by the disease, but might be related to adverse drug side effects.
In conclusion, the contribution of this study is twofold: First, it provides a sound basis to in silico methods that rely on omic metrics for drug repurposing and drug discovery; such methods are expected to develop into a cornerstone of future pharmacological research (Hurle et al, 2013; Iorio et al, 2013). Second, our results imply that drugs that are most successful in reversing the transcriptomic signatures of the disease across multiple tissues should also exert the most favorable outcomes. This highlights the need to develop drugs that restore the global cellular state of multiple tissues back to their healthy norms, rather than rectify particular disease phenotypes.
Materials and Methods
Data collection and processing
A detailed description of the experimental design, the pharmacological intervention regimens, and methods of data collection was given by Radonjic et al (2013). In summary, the experiment consisted of 132 animals (15 animals in the HFD-untreated group and 9 in all the other groups), but hepatic mRNA was collected only from 112 animals (8 per group). The other 20 animals were excluded from the current analysis. Out of these 112 animals, white adipose mRNA was collected only from 62 animals from 7 experimental groups as detailed in the main text (and see also Box 1A). White adipose was taken from the gonadal depot. In addition, 168 liver metabolites were measured for each animal, refer to Radonjic et al (2013) for details.
Microarray data were background-corrected, log2-transformed, and quantile-normalized by the lumi package of R/Bioconductor (Du et al, 2008). Statistical tests for differentially expressed genes were run through the limma package (Smyth, 2005). Liver and adipose data are publicly available at the ArrayExpress (Accession Number E-MTAB-1063) and GEO (Accession Number GSE57659) repositories, respectively.
The gene expression space and the transcriptome deviation index
The top N = 200 differentially expressed probes between the untreated 16-week HFD group and the LFD group were selected (Supplementary Tables S5 and S6; we verified that our results were not sensitive to the choice of N within an order of magnitude). Z-scores of their respective normalized expression (across all the study animals) were transformed by principal components analysis (PCA), with feature weights as described below, to form the gene expression space. All distance computations in this study used all the dimensions of the PCA space; that is, we did not use only the top principal components. We verified that using only the top principal components did not significantly alter the results.
In order to account for the different power that the selected N = 200 probes might have to separate the LFD and the untreated HFD groups, we weighted the features when computing the PCA by their Fisher scores, which are defined as follows:
with k = 1,2 being indices for the two groups (LFD and untreated HFD). F(x) is the Fisher score of a particular feature x whose mean across both groups is μ. μk and σk denote the mean and standard deviation, respectively, of the feature in group k. nk denotes the number of observations in group k. We note that the conclusions presented in this paper are robust to changes in the method by which features are weighted, and uphold even when assigning equal weights to all features.
Once the gene expression space has been defined, we project the gene expression data of all animals onto it. We then compute the centroid of all the LFD animals in this space, which represents the healthy baseline. The transcriptome deviation index (TDI) associated with a particular animal is the Euclidean distance between its representation in the gene expression space and the LFD centroid. We emphasize that the computation of the gene expression space and TDIs was done separately for each of the studied tissues.
Physiological and metabolome deviation indices
The physiological space was defined in an analogous manner to the gene expression space. Z-scores were computed for each of the 26 physiological markers collected from the animals (Supplementary Table S1), and PCA-transformed with features weighting as described above. Again, all principal components, rather than only the top ones, were used to define distances in the PCA space. The global physiological deviation index (GPDI) of a particular animal is the Euclidean distance between its representation in the physiological space and the LFD centroid. Animals for which not all of the 26 markers were available (due to failing the quality control of at least one assay) were excluded from GPDI analyses (29 out of 112). PDI is a special case of the GPDI that pertains to only one physiological marker. It is defined as the absolute value of the difference between the value of this marker in a particular animal and the mean of the marker's values in the LFD animals. The metabolome space and metabolome deviation indices were computed by the same procedure as the gene expression and physiological ones. Metabolites whose values were not available for all animals (31 out of 168) were excluded from MDI analyses.
Non-restorative gene expression alterations
Let vLFD, vHFD be the centroids of the LFD and untreated HFD groups, respectively, in the gene expression space. The direction vLFD − vHFD can be considered the desired effect of the treatment, which corresponds to a reversal of the disease gene expression phenotypes; effects that occur in orthogonal directions are termed non-restorative. The gene expression vector v of a treated animal can be thus decomposed into two orthogonal components: v's projection onto vLFD − vHFD, which we denote v1, and v2 = v − v1. ||v1||2 and ||v2||2 are the magnitudes of the disease-reversal (i.e., restorative) and non-restorative alterations, respectively, brought about by the treatment (see also Supplementary Fig S13A).
Implementation
The analysis was implemented in Matlab version 8.1.0.604 (R2013a). The source code used to compute the gene expression spaces, TDIs, and their correlations with PDIs can be downloaded from http://www.cs.tau.ac.il/~allonwag/LDLR_paper/LDLR_paper_code.zip. The source code is also available, along with the gene expression, physiological, and metabolome data, in Supplementary Dataset S1.
Acknowledgments
The authors thank Kevin G. Becker, Rafael de Cabo, Simon Kasif, Mary-Elizabeth Patti, Gideon Stein, William H. Wood III, and Keren Yizhak for their assistance in various phases of this study. AW was supported by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel Aviv University, and by an excellence scholarship in memory of Ms. Moran Tzafrir. ER acknowledges the generous support of grants from the Israeli Science Foundation (ISF), the Israeli Cancer Research Fund (ICRF) and the Israeli Centers for Research Excellence (I-CORE) program (Grant No 41/11).
Author contributions
AW, NC, and ER designed the study. AW and NC performed research. AW, NC, TK, UA, EL, DMS, and MR analyzed data. DMS, MR, and ER supervised the research. AW and ER wrote the paper. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Supplementary Dataset S1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
References
- Aggarwal CC, Hinneburg A, Keim DA. Proceedings of the 8th. London, UK: Springer-Verlag; 2001. On the Surprising Behavior of Distance Metrics in High Dimensional Spaces; pp. 420–434. International Conference on Database Theory. [Google Scholar]
- American Diabetes Association. Standards of Medical Care in Diabetes—2013. Diabetes Care. 2013;36:S11–S66. doi: 10.2337/dc13-S011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awtry EH, Loscalzo J. Aspirin. Circulation. 2000;101:1206–1218. doi: 10.1161/01.cir.101.10.1206. [DOI] [PubMed] [Google Scholar]
- Backes JM, Gibson CA, Ruisinger JF, Moriarty PM. Fibrates: what have we learned in the past 40 years? Pharmacother J Hum Pharmacol Drug Ther. 2007;27:412–424. doi: 10.1592/phco.27.3.412. [DOI] [PubMed] [Google Scholar]
- Bahrami H, Bluemke DA, Kronmal R, Bertoni AG, Lloyd-Jones DM, Shahar E, Szklo M, Lima JAC. Novel metabolic risk factors for incident heart failure and their relationship with obesity: the MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;51:1775–1783. doi: 10.1016/j.jacc.2007.12.048. [DOI] [PubMed] [Google Scholar]
- Basi S, Fesler P, Mimran A, Lewis JB. Microalbuminuria in Type 2 Diabetes and Hypertension: a marker, treatment target, or innocent bystander? Diabetes Care. 2008;31:S194–S201. doi: 10.2337/dc08-s249. [DOI] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer KS, Goldstein J, Ramakrishnan R, Shaft U. Proceedings of the 7th. London, UK: Springer-Verlag; 1999. When Is Nearest Neighbor’’ Meaningful? pp. 217–235. International Conference on Database Theory. [Google Scholar]
- Boyle JO, Gümüş ZH, Kacker A, Choksi VL, Bocker JM, Zhou XK, Yantiss RK, Hughes DB, Du B, Judson BL, Subbaramaiah K, Dannenberg AJ. Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev Res. 2010;3:266–278. doi: 10.1158/1940-6207.CAPR-09-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkin AC, Tontonoz P. Liver X Receptor Signaling Pathways and Atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1513–1518. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, Smyth S, Montalescot G, Steinhubl S. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA. 2007;297:2018–2024. doi: 10.1001/jama.297.18.2018. [DOI] [PubMed] [Google Scholar]
- Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care. 2003;26:2442–2450. doi: 10.2337/diacare.26.8.2442. [DOI] [PubMed] [Google Scholar]
- Chang M, Smith S, Thorpe A, Barratt MJ, Karim F. Evaluation of phenoxybenzamine in the CFA model of pain following gene expression studies and connectivity mapping. Mol Pain. 2010;6:56. doi: 10.1186/1744-8069-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheok MH, Yang W, Pui C-H, Downing JR, Cheng C, Naeve CW, Relling MV, Evans WE. Treatment-specific changes in gene expression discriminate in vivo drug response in human leukemia cells. Nat Genet. 2003;34:85–90. doi: 10.1038/ng1151. [DOI] [PubMed] [Google Scholar]
- Chisholm JW, Hong J, Mills SA, Lawn RM. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res. 2003;44:2039–2048. doi: 10.1194/jlr.M300135-JLR200. [DOI] [PubMed] [Google Scholar]
- Christou GA, Kiortsis DN. The role of adiponectin in renal physiology and development of albuminuria. J Endocrinol. 2014;221:R49–R61. doi: 10.1530/JOE-13-0578. [DOI] [PubMed] [Google Scholar]
- Chuang H, Lee E, Liu Y, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol Syst Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang H-Y, Rassenti L, Salcedo M, Licon K, Kohlmann A, Haferlach T, Foà R, Ideker T, Kipps TJ. Subnetwork-based analysis of chronic lymphocytic leukemia identifies pathways that associate with disease progression. Blood. 2012;120:2639–2649. doi: 10.1182/blood-2012-03-416461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- Cooper-Knock J, Kirby J, Ferraiuolo L, Heath PR, Rattray M, Shaw PJ. Gene expression profiling in human neurodegenerative disease. Nat Rev Neurol. 2012;8:518–530. doi: 10.1038/nrneurol.2012.156. [DOI] [PubMed] [Google Scholar]
- Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, Morgan AA, Sarwal MM, Pasricha PJ, Butte AJ. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med. 2011;3:96ra76. doi: 10.1126/scitranslmed.3002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernald GH, Altman RB. Using molecular features of Xenobiotics to predict hepatic gene expression response. J Chem Inf Model. 2013;53:2765–2773. doi: 10.1021/ci3005868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Fall T, van Dam RM, Flyvbjerg A, Zethelius B, Ingelsson E, Hägg S. Evidence of a causal relationship between adiponectin levels and insulin sensitivity: a mendelian randomization study. Diabetes. 2013;62:1338–1344. doi: 10.2337/db12-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein HC, Mann JFE, Yi Q, Zinman B, Dinneen SF, Hoogwerf B, Hallé JP, Young J, Rashkow A, Joyce C, Nawaz S, Yusuf S the HOPE Study Investigators. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA. 2001;286:421–426. doi: 10.1001/jama.286.4.421. [DOI] [PubMed] [Google Scholar]
- Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1104–1115. doi: 10.1161/ATVBAHA.111.237693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- Harper W, Clement M, Goldenberg R, Hanna A, Main A, Retnakaran R, Sherifali D, Woo V, Yale J-F. Pharmacologic management of Type 2 diabetes. Can J Diabetes. 2013;37:S61–S68. doi: 10.1016/j.jcjd.2013.01.021. [DOI] [PubMed] [Google Scholar]
- Hinneburg A, Aggarwal CC, Keim DA. Proceedings of the 26th. San Francisco, CA: Morgan Kaufmann Publishers Inc; 2000. What Is the Nearest Neighbor in High Dimensional Spaces? pp. 506–515. International Conference on Very Large Data Bases. [Google Scholar]
- Hirose H, Kawai T, Yamamoto Y, Taniyama M, Tomita M, Matsubara K, Okazaki Y, Ishii T, Oguma Y, Takei I, Saruta T. Effects of pioglitazone on metabolic parameters, body fat distribution, and serum adiponectin levels in Japanese male patients with type 2 diabetes. Metabolism. 2002;51:314–317. doi: 10.1053/meta.2002.30506. [DOI] [PubMed] [Google Scholar]
- Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov. 2014;13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- Hood L, Price ND. Demystifying disease, democratizing health care. Sci Transl Med. 2014;6:225ed5. doi: 10.1126/scitranslmed.3008665. [DOI] [PubMed] [Google Scholar]
- Hu G, Agarwal P. Human disease-drug network based on genomic expression profiles. PLoS ONE. 2009;4:e6536. doi: 10.1371/journal.pone.0006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. 2009;2:231–237. doi: 10.1242/dmm.001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurle MR, Yang L, Xie Q, Rajpal DK, Sanseau P, Agarwal P. Computational drug repositioning: from data to therapeutics. Clin Pharmacol Ther. 2013;93:335–341. doi: 10.1038/clpt.2013.1. [DOI] [PubMed] [Google Scholar]
- Imran SA, Rabasa-Lhoret R, Ross S. Targets for Glycemic control. Can J Diabetes. 2013;37:S31–S34. doi: 10.1016/j.jcjd.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Iorio F, Rittman T, Ge H, Menden M, Saez-Rodriguez J. Transcriptional data: a new gateway to drug repositioning? Drug Discov Today. 2013;18:350–357. doi: 10.1016/j.drudis.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomaa B, Almgren P, Tuomi T, Forsén B, Lahti K, Nissén M, Taskinen M-R, Groop L. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24:683–689. doi: 10.2337/diacare.24.4.683. [DOI] [PubMed] [Google Scholar]
- Ix JH, Sharma K. Mechanisms linking obesity, chronic kidney disease, and fatty liver disease: the roles of fetuin-A, adiponectin, and AMPK. J Am Soc Nephrol. 2010;21:406–412. doi: 10.1681/ASN.2009080820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahchan NS, Dudley JT, Mazur PK, Flores N, Yang D, Palmerton A, Zmoos A-F, Vaka D, Tran KQT, Zhou M, Krasinska K, Riess JW, Neal JW, Khatri P, Park KS, Butte AJ, Sage J. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 2013;3:1364–1377. doi: 10.1158/2159-8290.CD-13-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorsal A, Tarnow L, Frystyk J, Lajer M, Flyvbjerg A, Parving H-H, Vionnet N, Rossing P. Serum adiponectin predicts all-cause mortality and end stage renal disease in patients with type I diabetes and diabetic nephropathy. Kidney Int. 2008;74:649–654. doi: 10.1038/ki.2008.201. [DOI] [PubMed] [Google Scholar]
- Josset L, Textoris J, Loriod B, Ferraris O, Moules V, Lina B, N'Guyen C, Diaz J-J, Rosa-Calatrava M. Gene expression signature-based screening identifies new broadly effective influenza a antivirals. PLoS ONE. 2010;5:e13169. doi: 10.1371/journal.pone.0013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung UJ, Millman PN, Tall AR, Deckelbaum RJ. n−3 Fatty acids ameliorate hepatic steatosis and dysfunction after LXR agonist ingestion in mice. Biochim Biophys Acta – Mol Cell Biol Lipids. 2011;1811:491–497. doi: 10.1016/j.bbalip.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleemann R, Verschuren L, van Erk M, Nikolsky Y, Cnubben N, Verheij E, Smilde A, Hendriks H, Zadelaar S, Smith G, Kaznacheev V, Nikolskaya T, Melnikov A, Hurt-Camejo E, van der Greef J, van Ommen B, Kooistra T. Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: a combined transcriptomics and metabolomics analysis. Genome Biol. 2007;8:R200. doi: 10.1186/gb-2007-8-9-r200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klok MD, Jakobsdottir S, Drent ML. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: a review. Obes Rev. 2007;8:21–34. doi: 10.1111/j.1467-789X.2006.00270.x. [DOI] [PubMed] [Google Scholar]
- Kunkel SD, Suneja M, Ebert SM, Bongers KS, Fox DK, Malmberg SE, Alipour F, Shields RK, Adams CM. mRNA expression signatures of human skeletal muscle atrophy identify a natural compound that increases muscle mass. Cell Metab. 2011;13:627–638. doi: 10.1016/j.cmet.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin GA, Barrett-Connor E, May S, Langenberg C. Association of adiponectin with coronary heart disease and mortality: the rancho bernardo study. Am J Epidemiol. 2007;165:164–174. doi: 10.1093/aje/kwk001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, Tang A, Gabriel G, Ly C, Adamjee S, Dame ZT, Han B, Zhou Y, Wishart DS. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Maseri A. Inflammation and Atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- Lihn AS, Pedersen SB, Richelsen B. Adiponectin: action, regulation and association to insulin sensitivity. Obes Rev. 2005;6:13–21. doi: 10.1111/j.1467-789X.2005.00159.x. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Wang W, Zhang J, Lu Y, Wu W, Yan H, Wang Y. Hyperlipidemia and atherosclerotic lesion development in Ldlr-deficient mice on a long-term high-fat diet. PLoS ONE. 2012;7:e35835. doi: 10.1371/journal.pone.0035835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, Kuriyama H, Hotta K, Nakamura T, Shimomura I, Matsuzawa Y. PPARγ ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–2099. doi: 10.2337/diabetes.50.9.2094. [DOI] [PubMed] [Google Scholar]
- Mattix HJ, Hsu C, Shaykevich S, Curhan G. Use of the Albumin/Creatinine ratio to detect Microalbuminuria: implications of sex and race. J Am Soc Nephrol. 2002;13:1034–1039. doi: 10.1681/ASN.V1341034. [DOI] [PubMed] [Google Scholar]
- McCarthy JJ, McLeod HL, Ginsburg GS. Genomic medicine: a decade of successes, challenges, and opportunities. Sci Transl Med. 2013;5:189sr4. doi: 10.1126/scitranslmed.3005785. [DOI] [PubMed] [Google Scholar]
- McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48. doi: 10.1016/j.immuni.2014.05.010. [DOI] [PubMed] [Google Scholar]
- Meyvis K, Verrijken A, Wouters K, Van Gaal L. Plasma adiponectin level is inversely correlated with albuminuria in overweight and obese nondiabetic individuals. Metabolism. 2013;62:1570–1576. doi: 10.1016/j.metabol.2013.05.023. [DOI] [PubMed] [Google Scholar]
- Moghissi ES, Korytkowski MT, DiNardo M, Einhorn D, Hellman R, Hirsch IB, Inzucchi SE, Ismail-Beigi F, Kirkman MS, Umpierrez GE. American association of clinical endocrinologists and American diabetes association consensus statement on inpatient glycemic control. Diabetes Care. 2009;32:1119–1131. doi: 10.2337/dc09-9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Cowley MA, Münzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- Oh SC, Park Y-Y, Park ES, Lim JY, Kim SM, Kim S-B, Kim J, Kim SC, Chu I-S, Smith JJ, Beauchamp RD, Yeatman TJ, Kopetz S, Lee J-S. Prognostic gene expression signature associated with two molecularly distinct subtypes of colorectal cancer. Gut. 2012;61:1291–1298. doi: 10.1136/gutjnl-2011-300812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi N, Kato A, Misaki T, Sakakima M, Fujigaki Y, Yamamoto T, Hishida A. Association of serum Adiponectin levels with all-cause mortality in Hemodialysis patients. Intern Med. 2008;47:485–491. doi: 10.2169/internalmedicine.47.0614. [DOI] [PubMed] [Google Scholar]
- Oie S, Matsuzaki K, Yokoyama W, Tokunaga S, Waku T, Han S-I, Iwasaki N, Mikogai A, Yasuzawa-Tanaka K, Kishimoto H, Hiyoshi H, Nakajima Y, Araki T, Kimura K, Yanagisawa J, Murayama A. Hepatic rRNA transcription regulates high-fat-diet-induced obesity. Cell Rep. 2014;7:807–820. doi: 10.1016/j.celrep.2014.03.038. [DOI] [PubMed] [Google Scholar]
- Pacini C, Iorio F, Gonçalves E, Iskar M, Klabunde T, Bork P, Saez-Rodriguez J. DvD: an R/Cytoscape pipeline for drug repurposing using public repositories of gene expression data. Bioinformatics. 2013;29:132–134. doi: 10.1093/bioinformatics/bts656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JYH, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- Praticò D, Dogné J-M. Selective Cyclooxygenase-2 inhibitors development in cardiovascular medicine. Circulation. 2005;112:1073–1079. doi: 10.1161/CIRCULATIONAHA.104.524231. [DOI] [PubMed] [Google Scholar]
- Qaseem A, Vijan S, Snow V, Cross JT, Weiss KB, Owens DK. Glycemic control and type 2 diabetes mellitus: the optimal hemoglobin A1c targets. a guidance statement from the American college of physicians. Ann Intern Med. 2007;147:417–422. doi: 10.7326/0003-4819-147-6-200709180-00012. [DOI] [PubMed] [Google Scholar]
- Radonjic M, Wielinga PY, Wopereis S, Kelder T, Goelela VS, Verschuren L, Toet K, van Duyvenvoorde W, van der Werff van der Vat B, Stroeve JHM, Cnubben N, Kooistra T, van Ommen B, Kleemann R. Differential effects of drug interventions and dietary lifestyle in developing type 2 diabetes and complications: a systems biology analysis in LDLr−/− mice. PLoS ONE. 2013;8:e56122. doi: 10.1371/journal.pone.0056122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis — an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Scherzer CR, Eklund AC, Morse LJ, Liao Z, Locascio JJ, Fefer D, Schwarzschild MA, Schlossmacher MG, Hauser MA, Vance JM, Sudarsky LR, Standaert DG, Growdon JH, Jensen RV, Gullans SR. Molecular markers of early Parkinson's disease based on gene expression in blood. Proc Natl Acad Sci. 2007;104:955–960. doi: 10.1073/pnas.0610204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, RamachandraRao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, Falkner B, Goldstein BJ. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest. 2008;118:1645–1656. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K. The link between obesity and albuminuria: adiponectin and podocyte dysfunction. Kidney Int. 2009;76:145–148. doi: 10.1038/ki.2009.137. [DOI] [PubMed] [Google Scholar]
- Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D'Amico AV, Richie JP, Lander ES, Loda M, Kantoff PW, Golub TR, Sellers WR. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–209. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- Sirota M, Dudley JT, Kim J, Chiang AP, Morgan AA, Sweet-Cordero A, Sage J, Butte AJ. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med. 2011;3:96ra77. doi: 10.1126/scitranslmed.3001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lønning PE, Børresen-Dale A-L. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C, Neo S-Y, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci. 2003;100:10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen KR, Gustafsson J-Å. Putative metabolic effects of the liver X receptor (LXR) Diabetes. 2004;53:S36–S42. doi: 10.2337/diabetes.53.2007.s36. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukardi H, Zhang X, Lui EY, Ung CY, Mathavan S, Gong Z, Lam SH. Liver X receptor agonist T0901317 induced liver perturbation in zebrafish: histological, gene set enrichment and expression analyses. Biochim Biophys Acta – Gen Subj. 2012;1820:33–43. doi: 10.1016/j.bbagen.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Tannock LR, O'Brien KD, Knopp RH, Retzlaff B, Fish B, Wener MH, Kahn SE, Chait A. Cholesterol feeding increases C-Reactive protein and serum Amyloid a levels in lean insulin-sensitive subjects. Circulation. 2005;111:3058–3062. doi: 10.1161/CIRCULATIONAHA.104.506188. [DOI] [PubMed] [Google Scholar]
- Toda K, Okada T, Miyaura C, Saibara T. Fenofibrate, a ligand for PPARα, inhibits aromatase cytochrome P450 expression in the ovary of mouse. J Lipid Res. 2003;44:265–270. doi: 10.1194/jlr.M200327-JLR200. [DOI] [PubMed] [Google Scholar]
- Tsioufis C, Dimitriadis K, Chatzis D, Vasiliadou C, Tousoulis D, Papademetriou V, Toutouzas P, Stefanadis C, Kallikazaros I. Relation of microalbuminuria to adiponectin and augmented C-reactive protein levels in men with essential hypertension. Am J Cardiol. 2005;96:946–951. doi: 10.1016/j.amjcard.2005.05.052. [DOI] [PubMed] [Google Scholar]
- Ulven SM, Dalen KT, Gustafsson J-Å, Nebb HI. LXR is crucial in lipid metabolism. Prostaglandins Leukot Essent Fatty Acids. 2005;73:59–63. doi: 10.1016/j.plefa.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Van Diepen JA, Berbée JFP, Havekes LM, Rensen PCN. Interactions between inflammation and lipid metabolism: relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis. 2013;228:306–315. doi: 10.1016/j.atherosclerosis.2013.02.028. [DOI] [PubMed] [Google Scholar]
- Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- Willenbrock H, Juncker AS, Schmiegelow K, Knudsen S, Ryder LP. Prediction of immunophenotype, treatment response, and relapse in childhood acute lymphoblastic leukemia using DNA microarrays. Leukemia. 2004;18:1270–1277. doi: 10.1038/sj.leu.2403392. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- Yang W-S, Jeng C-Y, Wu T-J, Tanaka S, Funahashi T, Matsuzawa Y, Wang J-P, Chen C-L, Tai T-Y, Chuang L-M. Synthetic peroxisome proliferator-activated receptor-γ agonist, rosiglitazone, increases plasma levels of adiponectin in type 2 diabetic patients. Diabetes Care. 2002;25:376–380. doi: 10.2337/diacare.25.2.376. [DOI] [PubMed] [Google Scholar]
- Ye R, Scherer PE. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol Metab. 2013;2:133–141. doi: 10.1016/j.molmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K, Gabay O, Cohen H, Ruppin E. Model-based identification of drug targets that revert disrupted metabolism and its application to ageing. Nat Commun. 2013;4:2632. doi: 10.1038/ncomms3632. [DOI] [PubMed] [Google Scholar]
- Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27:1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- Zerbini LF, Bhasin MK, de Vasconcellos JF, Paccez JD, Gu X, Kung AL, Libermann TA. Computational repositioning and preclinical validation of pentamidine for renal cell cancer. Mol Cancer Ther. 2014;13:1929–1941. doi: 10.1158/1535-7163.MCT-13-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Dahlman-Wright K. Liver X receptor in cholesterol metabolism. J Endocrinol. 2010;204:233–240. doi: 10.1677/JOE-09-0271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Dataset S1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4