

Abstract
Conformational changes in the β2α2 and β6α6 loops in the alpha subunit of tryptophan synthase (αTS) are important for enzyme catalysis and coordinating substrate channeling with the beta subunit (βTS). It was previously shown that disrupting the hydrogen bond interactions between these loops through the T183V substitution on the β6α6 loop decreases catalytic efficiency and impairs substrate channeling. Results presented here also indicate that the T183V substitution decreases catalytic efficiency in Escherchia coli αTS in the absence of the βTS subunit. Nuclear magnetic resonance (NMR) experiments indicate that the T183V substitution leads to local changes in the structural dynamics of the β2α2 and β6α6 loops. We have also used NMR chemical shift covariance analyses (CHESCA) to map amino acid networks in the presence and absence of the T183V substitution. Under conditions of active catalytic turnover, the T183V substitution disrupts long-range networks connecting the catalytic residue Glu49 to the αTS-βTS binding interface, which might be important in the coordination of catalytic activities in the tryptophan synthase complex. The approach that we have developed here will likely find general utility in understanding long-range impacts on protein structure and dynamics of amino acid substitutions generated through protein engineering and directed evolution approaches, and provide insight into disease and drug-resistance mutations.
Keywords: amino acid networks, chemical shift covariance analysis, protein dynamics, tryptophan synthase, nuclear magnetic resonance, enzyme mechanisms
Introduction
Proteins can be described as networks of interacting amino acid residues.1–6 Within this framework, regulatory signals are transmitted through breaking and/or forming new noncovalent interactions, which may result in changes to protein structure and/or dynamics to influence function. Conformational changes, potentially facilitated through binding ligands and/or other macromolecules, would result in substantially altered networks, which may further gear the protein toward additional structural and/or functional changes. Our ability to understand and modulate these amino acid networks would facilitate protein engineering efforts, and/or provide new strategies in drug development aimed towards altering the networks to impact protein function.
One model enzyme to unravel these regulatory networks is tryptophan synthase, a heterotetramer, bienzyme complex in the tryptophan biosynthetic pathway.7 The α-subunit (αTS) forms a (β/α)8-barrel structure, and catalyzes the retro-aldol cleavage of the C3′-C3 bond of indole-3-glycerol phosphate (IGP) to form indole and d-glyceraldehyde 3-phosphate (G3P) (Fig. 1). The indole product is then directly channeled through a 25 Å hydrophobic tunnel to the β-subunit (βTS).8–14 The careful orchestration of αTS and βTS catalytic activities is influenced by the conformation and ligand-bound states of the other subunit.15–21 When IGP is bound, αTS forms a closed state, in which the β2α2 and β6α6 loops fold over the active site. The closed conformation is stabilized by hydrogen bonds between Thr183 from the β6α6 loop and residues on the β2α2, including Ala59 (Thr183-N to Ala59-O), Asp60 (Thr183-Oγ1 to Asp60-Oδ1), and Gly61 (Thr183-Oγ1 to Gly61-N)13,22–26 (Fig. 1).
Figure 1.
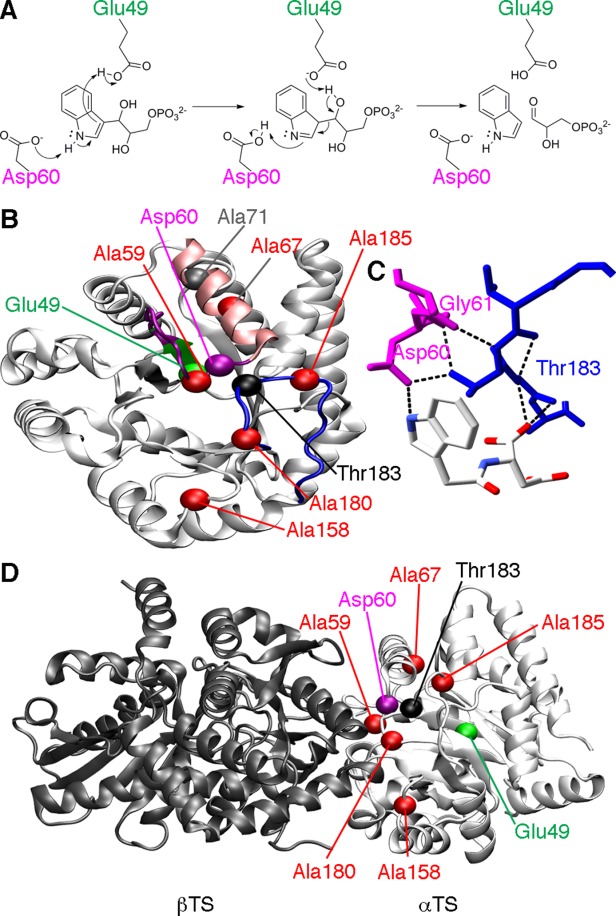
Catalytic function and loop interactions in αTS. (A) Chemical mechanism for αTS, highlighting the roles of Glu49 and Asp60 according to the step-wise mechanism.7 (B) Structure of αTS (PDB: 1K3U), showing the location of important amino acid residues, including Glu49 on the β2 strand (green), Asp60 on the β2α2 loop (magenta), and Thr183 on the β6α6 loop (blue). Also shown are the sites of the perturbations used to characterize the amino acid networks in E. coli αTS, including Ala59, Ala67, Ala158, Ala180, and Ala185 (red). It should be pointed out that in the absence of βTS, the α2' helix (pink) does not form, and instead the β2α2 loop is extended. Ala71 (grey), which is on the same amino acid network as Glu49 under some conditions, is also shown. (C) Close-up of the hydrogen bond interactions between the β2α2 (magenta) and β6α6 (blue) loops involving Thr183. The ligand shown is N-2[1H-indol-3-YL-acetyl]aspartic acid. (D) The locations of important amino acid residues in the context of the α−β TS dimer (PDB: 1K3U). Importantly, all of the NMR and kinetic studies reported here were performed on the αTS subunit alone, in the absence of the βTS subunit.
Thr183 is a key residue to αTS function and the regulation of indole channeling, as indicated by studies on variants of the Salmonella typhimurium enzyme.27,28 Amino acid substitutions at position 183 (e.g. T183A and T183V) lead up to a 100-fold decrease in the catalytic activity of the αTS reaction and severely compromise substrate channeling.27,28 In contrast, the T183S substitution results in only minor changes to TS function,28 suggesting that the hydroxyl group of Thr183 is critical for TS function, likely owing to the hydrogen bond interactions with the β2α2 loop. The β2α2 loop contains one of two residues directly involved in chemical catalysis (i.e. Asp60); the other residue, Glu49, is located on the adjacent β2 strand29–31 (Fig. 1).
These results are intriguing in the context of our recent NMR results32,33 with the αTS subunit from Escherichia coli, which is 85% identical to the S. typhimurium enzyme. We used the chemical shift covariance analysis (CHESCA) method34–38 to interrogate amino acid networks in αTS, in the absence of the βTS subunit. In the CHESCA approach, correlations between chemical shift changes induced by ligand binding and/or amino acid substitutions are used to map allosteric amino acid networks.34 In our case, we used a series of Ala-to-Gly site mutations to introduce small perturbations to the protein, which resulted in chemical shift changes that were used to map these networks in both the ligand-free resting state and in the ligand-bound working state.33 The working state is a functional state defined by a dynamic chemical equilibrium between IGP and indole with G3P (i.e. a ratio of E:IGP to E:indole:G3P of ∼4:1). Strikingly, the networks were different between the resting and working states. This was especially noticeable in the behavior of Glu49, which changes its network association between the resting and working states.33 Considering that many of our Ala-to-Gly probes reside on either the β2α2 or β6α6 loops, it would suggest that the structure and/or dynamics of these loops greatly influence the underlying amino acid networks. As such, modifying the noncovalent interactions (e.g. through amino acid substitutions at Thr183) between the loops would be predicted to alter the amino acid networks important for protein structure and function.
Our driving hypothesis has been that binding of the βTS subunit influences amino acid networks intrinsic to the αTS subunit, and through these means helps to coordinate structural and functional changes in the TS complex.32,33 Thus, delineation of the amino acid networks in the free αTS subunit can lend insight into the workings of the complex. Indeed, many of the network residues in αTS are found at or near the interface to which the βTS subunit binds.33 Other researchers have also suggested that study of αTS in the presence and absence of βTS can provide deeper insight into how the interactions of the subunits influence the activity of each subunit (e.g. Refs.39–44).
In this article, we have characterized the functional and structural consequences of the T183V substitution on E. coli αTS. Similar to the studies with the S. typhimurium enzyme, the T183V substitution leads to a decrease in the catalytic efficiency of αTS, likely through altering the structural dynamics of the β2α2 and β6α6 active site loops. We mapped the amino acid networks in the presence of the T183V substitution to show that long-range networks are also impacted by this substitution, including those containing residues at the α−β subunit interface. These results suggest that the T183V substitution disrupts multiple interactions that are likely important for subunit communication within the TS complex.
Resuls and Discussion
Kinetic comparison of wild-type and T183V E. coli αTS
Previous studies indicated that the T183V substitution in S. typhimurium αTS results in a large decrease in catalytic activity,27 likely due to the loss of the hydrogen bond interactions between Thr183 on the β6α6 loop and Ala59, Asp60, and Gly61 on the β2α2 loop (Fig. 1). We have now incorporated the T183V substitution into E. coli αTS to determine the functional and structural consequences of this amino acid change. We assayed αTS catalytic activity in the absence of the β-subunit and in the reverse direction (i.e. indole and G3P react to form IGP). It should be noted that the catalytic activity of the free αTS enzyme is lower than when in found in complex with the βTS subunit.40 The T183V substitution also led to a substantial decrease in kcat, but had little effect on the KM for indole or G3P (Table I).
Table I.
Kinetic Analysis of E. coli αTS Variants
| Variant | kcata (s−1) | KMb (indole; mM) | kcat/KM (s−1 M−1) | (kcat)var/(kcat)WT | (kcat/KM)var/(kcat/KM)WT | ΔΔGc (kcat; kcal/mol) | ΔΔGc (kcat/KM; kcal/mol) |
|---|---|---|---|---|---|---|---|
| WT | 0.095 | 0.926 | 103 | — | — | — | — |
| A71G | 0.020 | 1.84 | 10.9 | 0.211 | 0.106 | 0.921 | 1.33 |
| T183V | 0.007 | 1.10 | 6.36 | 0.074 | 0.062 | 1.54 | 1.65 |
| T183V/A71G | 0.003 | 1.70 | 1.76 | 0.032 | 0.017 | 2.04 | 2.41 |
| T183V/A59G | 0.010 | 3.35 | 2.99 | 0.105 | 0.029 | 1.33 | 2.10 |
| T183V/A67G | 0.011 | 0.842 | 13.1 | 0.115 | 0.127 | 1.28 | 1.22 |
| T183V/A158G | 0.007 | 0.804 | 8.71 | 0.074 | 0.085 | 1.54 | 1.46 |
| T183V/A180G | 0.007 | 1.52 | 4.61 | 0.074 | 0.044 | 1.54 | 1.85 |
| T183V/A185G | 0.007 | 0.782 | 8.95 | 0.074 | 0.087 | 1.54 | 1.45 |
Estimated error for kcat is 5 to 10%.
Estimated error for KM is 10 to 25%.
ΔΔG (k) = −RT ln (kvar./kWT), where R is 1.987 × 10−3 kcal K−1 mol−1 and T is 298 K.
Evidence for changes in the αTS structural dynamics induced by the T183V substitution
To gain insight into conformational and/or dynamic changes induced by the T183V substitution, we collected solution-state NMR spectra for the enzyme in the apo-form [i.e. resting state; Fig. 2(A)], bound with G3P [Fig. 2(B)], bound with indole [Fig. 2(C)], and in the working state [Fig. 2(D)]. We note that we collected NMR spectra for both fully 15N labeled protein, and for protein 15N labeled only at the Ala positions; we show the 15N Ala spectra for clarity.
Figure 2.
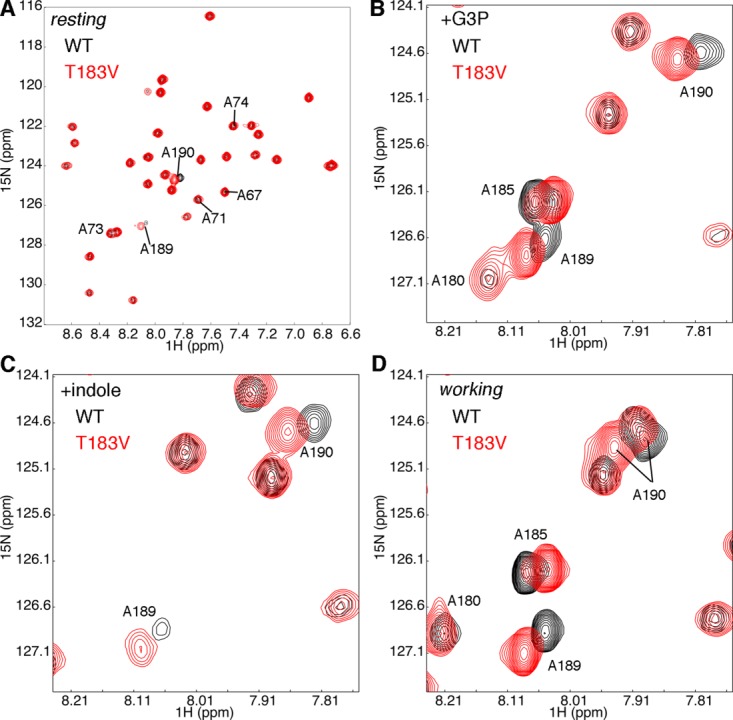
The T183V substitution leads to local changes in the β2α2 and β6α6 loops of αTS. Shown are comparisons of the 1H-15N HSQC spectra for WT (black) and T183V (red) αTS enzymes (A) without ligand, (B) in the presence of 10 mM D-G3P, (C) in the presence of 10 mM indole and (D) under dynamic chemical equilibrium conditions representing a 4:1 ratio of E:IGP to E:indole:G3P forms; these working state conditions are initiated with the addition of 10 mM D-G3P and 10 mM indole. Note that only the Ala residues are 15N labeled. NMR data were collected at 298 K on samples containing 0.5 to 1 mM protein in 50 mM potassium phosphate, pH 7.8, 2 mM DTT, 0.2 mM Na2EDTA, and 10% 2H2O.
The NMR spectra suggest that the T183V substitution leads to structural/dynamic changes in the β2α2 and β6α6 loops. As we observed previously,32 resonances associated with residues in the β6α6 loop, including those for Ala180 and Ala185, are broadened out in the absence of G3P, likely due to intermediate exchange (Fig. 2). The T183V substitution leads to both chemical shift changes and peak intensity changes (e.g. A180 when αTS is bound to G3P, A189 when αTS is bound to indole) for residues in the β6α6 loop. The changes in peak intensities might indicate that the T183V substitution changes loop dynamics on the μs-ms timescale. Unfortunately, R2 relaxation dispersion experiments were unable to further reveal and characterize these protein motions, potentially because the timescale of these motions was outside the range of this type of NMR experiment. We also compared steady-state 1H-15N heteronuclear Overhauser effects (hetNOE) for WT and T183V αTS as an indicator of differences in the ps-ns timescale dynamics (Fig. 3); R1 and R2 relaxation rate data were not of sufficient quality for model-free analysis.45,46 The 1H-15N hetNOE data imply that the T183V substitution increases disorder on the ps-ns timescale for some residues in the β2α2 (e.g. Ala67) and β6α6 (e.g. Ala180; statistical testing suggests that there is a difference in the A180 hetNOE data at the 90% confidence interval) loops. These changes in the loop dynamics are not surprising given that the T183V effectively breaks the interactions between the β2α2 and β6α6 loops.
Figure 3.
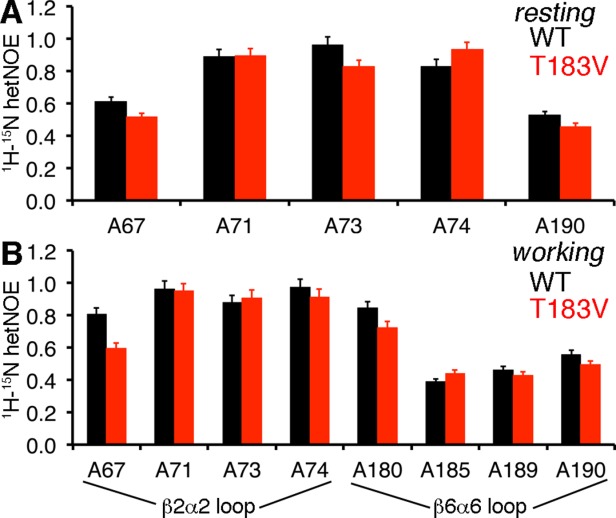
The T183V substitution alters structural dynamics in the β2α2 and β6α6 loops. Comparison of the 1H-15N heteronuclear Overhauser effects (hetNOE) for WT (black) and T183V (red) αTS enzymes for the (A) ligand-free resting state and the (B) working state conditions under dynamic chemical equilibrium. The working state conditions are initiated by the addition of 10 mM indole and 10 mM D-G3P to reach a ratio of E:IGP to E:indole:G3P of ∼4:1.
Network probes only result in minor changes to steady-state kinetic parameters
Besides the local disruption of the hydrogen bond interactions between the β2α2 and β6α6 loops, the T183V substitution may lead to longer-range structure/dynamic changes that would impact αTS function and/or interactions with βTS. We had previously used the CHESCA approach to delineate the amino acid networks in αTS in both the ligand-free resting state and ligand-bound working state.33 Our method depended on perturbing the protein using Ala-to-Gly substitutions, and monitoring chemical shift changes in the fully 15N-labeled proteins. Resonances with strong chemical shift correlations across these perturbations suggest that the corresponding residues respond in a similar fashion to these perturbations, and are thus on the same amino acid network.34 We proposed that the effects of the T183V substitution on the amino acid networks in αTS could be assessed by repeating the analysis but using double-substituted protein containing the previous Ala-to-Gly substitutions together with the T183V substitution itself (i.e. comparing T183V, T183V/A59G, T183V/A67G, T183V/A158G, T183V/A180G and T183V/A185G variants).
Since we were also interested in how ligand-binding impacts the amino acid networks, the amino acid substitutions used to probe the networks should not substantially change ligand binding. Along these lines, we had previously shown that the Ala-to-Gly probes do not substantially alter the steady-state kinetic parameters nor alter the equilibrium ratio between E:IGP and E:indole:G3P in the working state.33 Similarly, the Ala-to-Gly substitutions in the background of the T183V change do not result in any substantial changes to the steady-state kinetic parameters compared with the T183V variant itself (Table I). The largest change is a threefold increase in the KM for indole for the T183V/A59G variant compared with the T183V variant (Table I). Nonetheless, the concentration of indole (i.e. 10 mM) used in the NMR experiments should still be close to saturating for all αTS variants.
The amino acid networks in the working state are unique compared with the resting state and to other ligand-bound states
To begin the CHESCA approach, we collected 1H-15N heteronuclear single quantum coherence (HSQC) spectra for single- and double-substituted protein to compare amino acid networks in the presence and absence of the T183V substitution with and without ligands. As before,33 each resonance was assigned a single, combined chemical shift according to s(x)i = δH + 0.2 δN, where δH and δN are the 1H and 15N chemical shifts, respectively. Uncertainty in these values was accounted for by randomly generating sets of 400 points that followed a Gaussian distribution centered on s(x)i with a standard deviation of σ. The Pearson correlation (R) between two backbone amide resonances was then determined within a series of protein variants using these generated points. While the σ value could be approximated by collecting and comparing spectra, in our hands we have used this mostly as an input value to judge the robustness of the network (i.e. the lowest value we set this to is 0.01 ppm). That is, some of the weaker correlations, or correlations in which the distribution range of the chemical shift data points is small, will essentially disappear at larger values of σ. The robustness of the network can also be gauged by changing the probability or P value at which we determine that the linear correlations are statistically significant. In our view, the P values are a better measure of the statistical significance of the linear correlation than the R value, considering that some chemical shift datasets may not be useable for certain pairwise correlations due to higher than expected chemical shift changes (e.g. due to proximity effects). In this case, the number of data points will change (but will always be larger than four), however, this change in the number of data points is accounted for in the P values. More information about our modified method can be found in our previous manuscript (see Ref.33).
Based on the P values, an agglomerative clustering algorithm using a single-linkage (i.e., nearest-neighbor) model was used to organize the chemical shift correlation matrices (Supporting Information Fig. S1) into dendrograms (Supporting Information Fig. S2), and the clusters identified in the dendrograms were then plotted onto the αTS structure (note that we have used the S. typhimurium structure due to the disordered loops in the E. coli structure; Fig. 4). For simplicity, we will refer to the amino acid networks generated without the T183V substitution as WT networks (i.e. based on the spectra for WT and the single-substituted proteins) and those generated with the T183V substitution as the T183V networks (i.e. based on the spectra for T183V and the double-substituted proteins).
Figure 4.
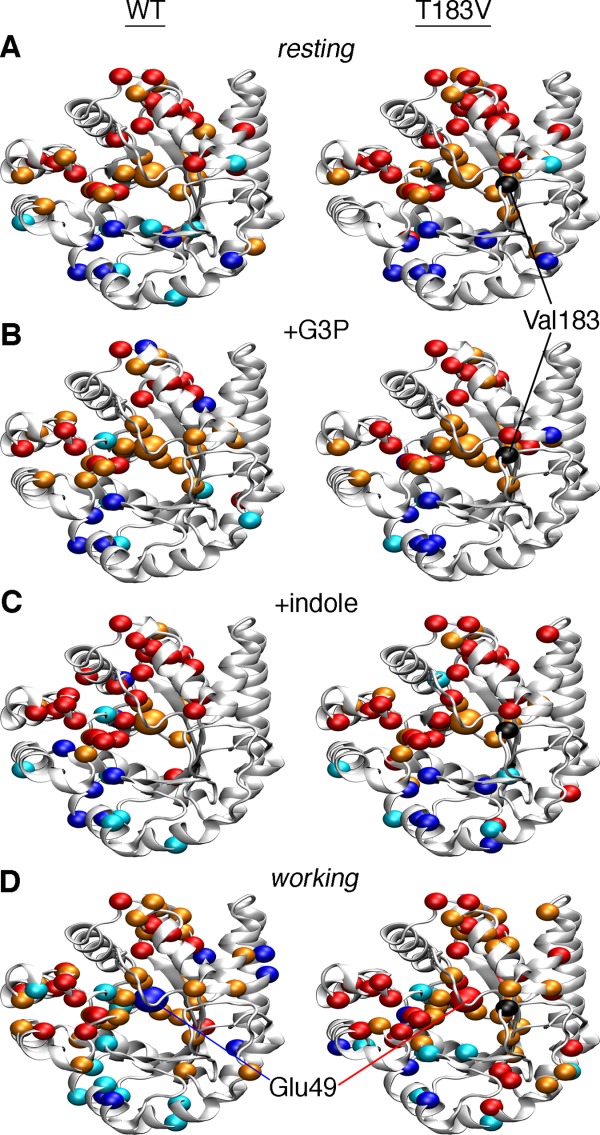
Amino acid networks in E. coli αTS are dependent upon the bound ligand and the presence of the T183V substitution. Agglomerative clustering is used to identify “nearest-neighbor” clusters in αTS (A) in the ligand-free resting state, (B) when bound to G3P, (C) when bound to indole, and (D) in the working state under dynamic chemical equilibrium conditions (i.e. ∼4:1 ratio of E:IGP to E:indole:G3P). To generate the WT networks (left), the CHESCA approach utilized NMR data from WT, A59G, A67G, A158G, A180G, and A185G proteins. To generate the T183V networks (right), the CHESCA approach utilized NMR data from T183V, T183V/A59G, T183V/A67G, T183V/A158G, T183V/A180G, and T183V/A185G proteins. Residues in cluster 1 and cluster 2 are plotted as dark blue/light blue and red/orange spheres onto the αTS structure (PDB: 1K3U). Colors also correspond to the level of statistical significance that these residues are found in their appropriate clusters (P < 0.01, dark blue/red; P < 0.05, light blue/orange). Chemical shift covariance matrices and the associated dendrograms are presented in Supporting Information Figures S1 and S2 respectively.
Our previous studies on the WT resting and working state clusters identified two sets of clusters,33 which we will name cluster 1 (dark blue/light blue in Fig. 4) and cluster 2 (colored red/orange in Fig. 4). For the WT networks, it was noteworthy that Glu49, which is directly involved in chemical catalysis, switches from cluster 2 in the resting state to cluster 1 in the working state.33
We have performed a similar analysis but now in the presence of only one ligand (i.e. G3P or indole alone; Fig. 4). Many of the cluster 2 residues identified are the same between all four states (i.e. resting, bound with G3P, bound with indole, and working states). More changes occur in the cluster 1 residues in the presence of the different ligands. For the resting state, most of the cluster 1 residues are grouped on one face of the active site away from cluster 2. When ligand is present, some cluster 1 residues are interspersed with the cluster 2 residues and remote from the main cluster grouping. The ligands may facilitate interactions that are not present in the apo-enzyme, and in some cases, long-range structural/dynamic changes induced by ligand binding may only be “felt” by a subset of residues. Perhaps the most outstanding finding is that Glu49 remains in cluster 2 when only G3P or indole is bound, indicating that Glu49 only switches clusters when both ligands are present, or when IGP is formed. The cluster 1 residues identified when either G3P or indole are bound are also not just a subset of the cluster 1 residues identified in the working state, suggesting that the working state is comprised of conformational state(s) unique to the active turnover conditions, consistent with our previous findings.32
As another check on these clusters, we have performed singular value decomposition (SVD) analysis. The original article on CHESCA34 performed SVD analysis on the raw chemical shift matrix to provide some sense of function to the clusters that were identified. Here, we have instead performed SVD analysis on the chemical shift correlation matrix. Our rationale is that a network could be described as a linear combination of the correlations within the network, thus each principle component (PC) would represent a network. This analysis would serve as an additional test of the clusters, and identify residues that associate with both clusters, providing potential communication pathways between clusters. We suggest that the SVD analysis performed here is complimentary to the SVD analysis performed in the original CHESCA article.34 Satisfyingly, cluster 1 and cluster 2 residues associated mostly with the PC2 and PC1 axes respectively (Supporting Information Fig. S3). Moreover, Glu49 is near the PC2 axis in the WT working state, but near the PC1 axis in all other tested cases. The residues that scatter away from one axis or another are generally those residues in which we have lower statistical confidence in belonging to a particular cluster (also see Fig. 4). It should be kept in mind, however, that PC1 and PC2 only account for ∼63% of the total variance in the data, which suggests that our clustering analysis might be missing some additional complexity.
The T183V substitution changes the amino acid networks in the working state
We first note that we used chemical shift data for the same residues to generate the WT and T183V networks. As such, differences in the WT and T183V networks must be traced to the T183V substitution, and not to any missing data (e.g. missing or unassigned resonances with the T183V samples). The amino acid networks in the apo-enzyme and when enzyme is bound to indole are very similar in the absence or presence of the T183V substitution (Fig. 4). The T183V substitution induces more substantial changes in the amino acid networks when enzyme is bound to G3P and in the working state. These changes might be due to altered interactions between the β6α6 loop and the phosphate moiety of G3P/IGP. Three clusters of residues are identified by our approach when enzyme is bound to G3P, but only with the T183V substitution. Perhaps most strikingly, Glu49 remains part of cluster 2 in the T183V working state. The long-range interactions involving Glu49 can also be visualized by identifying those residues whose resonances best linearly correlate to the Glu49 resonance (Fig. 5, Supporting Information Fig. S4). Again, the WT working state gives a unique profile compared with the apo- and other ligand-bound states (Supporting Information Fig. S4), and is different from the T183V working state (Fig. 5). These results suggest that the allosteric pathway delineated by the cluster 1 residues depends on the interactions between the β2α2 and β6α6 loops, despite the fact that most of these residues comprising cluster 1 are on the “backside” of the enzyme, away from the active site. This part of the protein has also been termed the “stability face” in the context of TIM-barrel proteins, in contrast to the “catalytic face” that contains the active site.47
Figure 5.
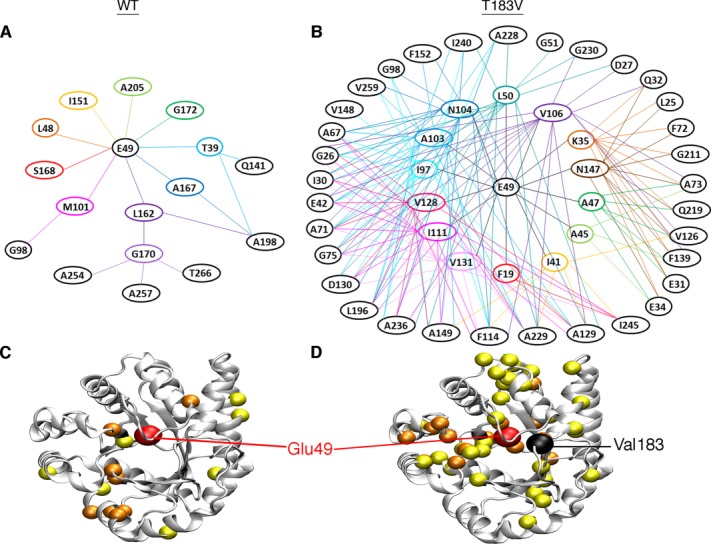
Network associations with Glu49 are different in the presence/absence of the T183V substitution. Shown are the dendrograms for (A) WT and (B) T183V networks under the working state conditions. The WT network utilized NMR data from WT, A59G, A67G, A158G, A180G, and A185G proteins, whereas the T183V network utilized NMR data from T183V, T183V/A59G, T183V/A67G, T183V/A158G, T183V/A180G, and T183V/A185G proteins. Dendrograms are drawn based on the chemical shift covariance matrices in Supporting Information Figure S1. Lines are drawn between residues showing significant (i.e. P < 0.05) linear chemical shift correlations, starting with Glu49 and then to other residues. For improved clarity, connecting lines are not shown between residues within the same “circle” of residues. Residues from these dendrograms are plotted as spheres on the αTS structure for the (C) WT and (D) T183V networks (PDB: 1K3U). Glu49 is plotted as a larger red sphere, the residues showing significant linear chemical shift correlations with Glu49 are plotted as orange spheres, and the residues showing significant linear chemical shift correlations with the first set of residues are plotted as yellow spheres. Analysis of other states is presented in Supporting Information Figure S4.
Probing pathways unique to WT and T183V working states
We have previously used other amino acid substitutions to further probe the unique allosteric pathways in the resting and working states for the WT enzyme.33 For example, Glu49 is only in the same cluster as Ala71 in the resting state; the A71G substitution results in a chemical shift change in the Glu49 resonance in the resting state but not for the working state (Fig. 6). With the T183V amino acid networks, Ala71 is in the same cluster as Glu49 for both the resting and working states (Figs. 4 and 5). We further tested these connections by comparing the chemical shift changes induced by the T183V and T183V/A71G substitutions on the Glu49 resonance (Fig. 6). The T183V substitution led to a more substantial chemical shift change in the working state than the resting state. In contrast to what was previously observed for the A71G variant, the T183V/A71G double substitution led to chemical shift changes to the Glu49 resonance in both the resting and working states compared with what was observed for either WT enzyme or the T183V variant (Fig. 6). These results are consistent with Ala71 and Glu49 being part of the same cluster for both resting and working states when the T183V substitution is present.
Figure 6.
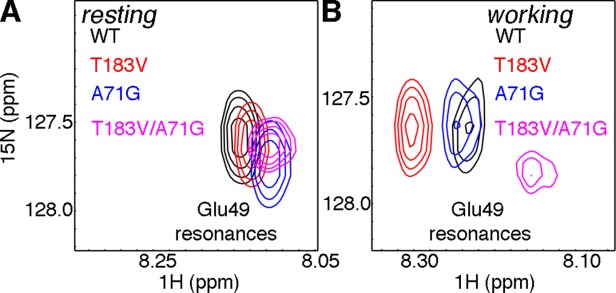
Perturbing unique pathways in the WT and T183V networks of E. coli αTS. Zoomed in pictures of the 1H-15N HSQC spectra showing the Glu49 resonance for WT (black), A71G (blue), T183V (red), and T183V/A71G (magenta) αTS enzymes for the (A) ligand-free resting state and (B) working state under dynamic chemical equilibrium conditions (i.e. a ∼4:1 ratio of E:IGP to E:indole:G3P). The A71G substitution induces a more substantial chemical shift change for the Glu49 resonance in the working state in the presence of the T183V substitution, consistent with Ala71 being in the same cluster as Glu49 in the T183V but not the WT network.
The A71G and T183V/A71G substitutions also led to changes in the steady-state kinetic parameters (Table I). The T183V/A71G double-substitution led to a ∼4-fold decrease in catalytic efficiency (i.e. kcat/KM) compared with the T183V variant, which is less than the ∼9-fold decrease in catalytic efficiency induced by the A71G substitution compared with WT enzyme. However, thermodynamic comparison of the free energy change induced on the catalytic efficiency (i.e. ΔΔG = −RT (ln ((kcat/KM)variant/(kcat/KM)WT) indicated that the sum of the effect of the two single substitutions (i.e. A71G, T183V) was only different by ∼0.5 kcal/mol compared with the double-substitution (i.e. A71G/T183V).
Conclusions
Our results indicate that the T183V substitution substantially decreases the catalytic efficiency of E. coli αTS. These functional impacts likely owe both to changes in the local hydrogen bond interactions between the β2α2 and β6α6 loops and to longer-range changes in the underlying amino acid networks governing the structure and dynamics of αTS. Intriguingly, the switch in the amino acid networks involving Glu49 observed in the working state strictly depend on the presence of both substrates. These unique amino acid networks in the working state are also disrupted by the T183V substitution. We have previously noted that many of the cluster residues are located at or near the interface where βTS binds.33 We have suggested that the amino acid networks intrinsic to αTS might be influenced by the binding of βTS in order to coordinate catalytic activities and indole channeling.32,33 It is interesting to note then that cluster 1 of the T183V working state does not extend to the αTS-βTS interface as it does in the WT working state (Fig. 7). In particular, Phe107, which interacts with β-strands on βTS (i.e. residues 275–279 and 282–289) comprising part of the indole channel, is in the WT but not the T183V working state cluster 1. Phe280 on the turn connecting these β-strands has been shown to block the indole channel in some TS crystal structures.48,49 Cluster 1 in the working state may thus be important in gating the opening/closing of the channel to coordinate indole channeling, and the T183V substitution may disrupt this allosteric pathway to impede indole channeling. NMR studies on the full TS complex should bring additional insights into how the amino acid networks of αTS communicate with those networks intrinsic to βTS; such studies may require the use of solid-state NMR methods that are not inherently size limited, as have been performed previously for TS.50–52
Figure 7.

Potential pathway of communication between the active site of αTS and the βTS binding interface in the (A) WT enzyme that is disrupted in the (B) T183V variant. Working state clusters from Figure 4 are plotted onto αβTS dimer (PDB: 1K3U). αTS and βTS subunits are indicated by white and dark grey ribbons, respectively. Cluster residues are plotted as colored spheres (cluster 1, blue; cluster 2, orange). Green indicates a potential pathway from Glu49 to the αTS-βTS binding interface identified from the cluster analysis in Figure 4, where Phe107 from αTS interacts with β-strands from βTS forming part of the indole channel. These interactions might influence the conformation of Phe280 in βTS, which has been shown to block the indole channel in some TS crystal structures49. The T183V substitution also likely disrupts structural dynamics of the β2α2 and β6α6 loops in αTS that are important for TS catalysis and indole channeling.
Other methods have been important in delineating the amino acid networks in other proteins (e.g. Refs.1, 5–6, and53–56). The CHESCA approach is especially valuable since we have been able to demonstrate that the amino acid networks are dependent on the ligand-bound state of the protein,33 which may suggest means through which conformational changes and/or protein dynamics are controlled through ligand binding.57 Here, we have shown that modifying a key residue impacts both short- and long-range interactions by changing the underlying amino acid networks, but only in specific ligand-bound states. We can envision the approach developed here (i.e. CHESCA with single and double substituted protein) applied to the investigation of other key residues or interactions in proteins to outline how the modification of key residues globally alters the structural dynamics of these proteins, providing new insights into allosteric regulation, molecular evolution and protein engineering.
Materials and Methods
Site-directed mutagenesis of αTS
Single (i.e. T183V) and double mutants (i.e. T183V/A59G, T183V/A67G, T183V/A71G, T183V/A158G, T183V/A180G, T183V/A185G) were generated using the Stratagene QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) and appropriate primers with the pET26 vector. Sequences were confirmed through DNA sequencing (Nucleic Acid Facility, Pennsylvania State University). Other mutants were previously generated according to Refs.32 and33.
Overexpression and purification of WT and variant αTS for kinetic studies
WT and variant αTS proteins were overexpressed in Luria-Bertani media. Cultures of transformed E. coli BL21 (DE3*) were grown at 37°C until an OD600 (optical density at 600 nm) of 0.5 to 0.6 was reached, at which time 1 mM IPTG (final concentration) was added to induce protein expression. Cells were allowed to overexpress αTS for approximately 12 h at 25°C and were then collected by centrifugation at 10,000g for 20 min. WT and variant αTS was purified using the previously described procedure.32
Steady-state kinetic studies of WT and variant αTS
After protein purification, samples were dialyzed into the assay buffer (100 mM potassium phosphate, pH 7.6) for approximately 12 h. Assays were monitored by UV absorbance of IGP at 290 nm with a SpectraMax M2 plate reader (Molecular Devices). Assays were performed in triplicate at 298 K. Initial rate data were fit to the Michaelis-Menten [Eq. (1)] using nonlinear regression with the program Kaleidograph:
| (1) |
where v is the initial reaction velocity, ET is the total amount of enzyme in the assay, and [S] is the substrate concentration. When D-G3P varied (1.5–19 mM), indole was held constant at 1.3 mM, and when indole varied (0.5–3.3 mM), D-G3P was held constant at 10 mM. The concentration of αTS was 7.5 μM for WT enzyme and 37.5 μM for variant proteins carrying the T183V substitution.
Overexpression and purification of WT and T183V TS variants for NMR studies
WT and variant αTS were overexpressed in M9 media, with 15N-labeled ammonium chloride (1 g/L of culture) to achieve full 15N-labeling of the backbone. Cultures of transformed E. coli BL21 (DE3*) were grown at 37°C until an OD600 of 0.5 to 0.6 was reached, at which time 1 mM IPTG (final concentration) was added to induce protein expression. Cells were allowed to overexpress αTS for approximately 12 h at 25°C and were then collected by centrifugation. αTS was purified using the same procedures as described above. Cultures for selective 15N-Ala backbone labeling of WT and T183V αTS for hetNOE studies were overexpressed according to Ref.58.
NMR sample preparation and analysis
Following protein purification and concentration, a ZEBA desalting column (Thermo Scientific) was used to exchange the buffer of all protein samples to NMR buffer (50 mM potassium phosphate, pH 7.8, 2 mM DTT, 0.2 mM Na2EDTA, and 10% 2H2O). The NMR samples generally contained 0.5 to 1 mM protein, and 10 mM indole and/or 10 mM G3P where indicated.
1H-15N hetNOE values were measured by acquiring two spectra, with or without proton presaturation, in an interleaved manner, according to the scheme in Ref.59. A total of two pairs of spectra were collected at 298 K for each of the αTS complexes analyzed. All NMR data was collected on a Bruker Avance III 600 MHz spectrometer equipped with a TCI cryoprobe.
Covariance and cluster analysis
1H-15N HSQC spectra were collected from WT, single variant (i.e. A59G, A67G, A158G, A180G, A185G), and T183V containing double variant αTS (i.e. T183V/A59G, T183V/A67G, T183V/A158G, T183V/A180G, T183V/A185G). The CHESCA analysis was performed using the procedure outlined in Ref.33, which is similar to the original implementation of this method.34
Acknowledgments
The authors thank Dr. Xianrui Yuan for the initial NMR analysis of the T183V variant.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, Venger I, Pietrokovski S. Network analysis of protein structures identifies functional residues. J Mol Biol. 2004;344:1135–1146. doi: 10.1016/j.jmb.2004.10.055. [DOI] [PubMed] [Google Scholar]
- 2.Bode C, Kovacs IA, Szalay MS, Palotai R, Korcsmaros T, Csermely P. Network analysis of protein dynamics. FEBS Lett. 2007;581:2776–2782. doi: 10.1016/j.febslet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 3.Csermely P, Sandhu KS, Hazai E, Hoksza Z, Kiss HJ, Miozzo F, Veres DV, Piazza F, Nussinov R. Disordered proteins and network disorder in network descriptions of protein structure, dynamics and function: hypotheses and a comprehensive review. Curr Protein Pept Sci. 2012;13:19–33. doi: 10.2174/138920312799277992. [DOI] [PubMed] [Google Scholar]
- 4.Lee J, Goodey NM. Catalytic contributions from remote regions of enzyme structure. Chem Rev. 2011;111:7595–7624. doi: 10.1021/cr100042n. [DOI] [PubMed] [Google Scholar]
- 5.Reynolds KA, Russ WP, Socolich M, Ranganathan R. Evolution-based design of proteins. Methods Enzymol. 2013;523:213–235. doi: 10.1016/B978-0-12-394292-0.00010-2. [DOI] [PubMed] [Google Scholar]
- 6.van den Bedem H, Bhabha G, Yang K, Wright PE, Fraser JS. Automated identification of functional dynamic contact networks from X-ray crystallography. Nat Methods. 2013;10:896–902. doi: 10.1038/nmeth.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunn MF. Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch Biochem Biophys. 2012;519:154–166. doi: 10.1016/j.abb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson KS, Miles EW, Johnson KA. Serine modulates substrate channeling in tryptophan synthase. A novel intersubunit triggering mechanism. J Biol Chem. 1991;266:8020–8033. [PubMed] [Google Scholar]
- 9.Brzovic PS, Hyde CC, Miles EW, Dunn MF. Characterization of the functional role of a flexible loop in the alpha-subunit of tryptophan synthase from Salmonella typhimurium by rapid-scanning, stopped-flow spectroscopy and site-directed mutagenesis. Biochemistry. 1993;32:10404–10413. doi: 10.1021/bi00090a016. [DOI] [PubMed] [Google Scholar]
- 10.Brzovic PS, Sawa Y, Hyde CC, Miles EW, Dunn MF. Evidence that mutations in a loop region of the alpha-subunit inhibit the transition from an open to a closed conformation in the tryptophan synthase bienzyme complex. J Biol Chem. 1992;267:13028–13038. [PubMed] [Google Scholar]
- 11.Dunn MF, Aguilar V, Brzovic P, Drewe WF, Jr, Houben KF, Leja CA, Roy M. The tryptophan synthase bienzyme complex transfers indole between the alpha- and beta-sites via a 25-30 A long tunnel. Biochemistry. 1990;29:8598–8607. doi: 10.1021/bi00489a015. [DOI] [PubMed] [Google Scholar]
- 12.Houben KF, Dunn MF. Allosteric effects acting over a distance of 20-25 A in the Escherichia coli tryptophan synthase bienzyme complex increase ligand affinity and cause redistribution of covalent intermediates. Biochemistry. 1990;29:2421–2429. doi: 10.1021/bi00461a028. [DOI] [PubMed] [Google Scholar]
- 13.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 14.Schlichting I, Yang XJ, Miles EW, Kim AY, Anderson KS. Structural and kinetic analysis of a channel-impaired mutant of tryptophan synthase. J Biol Chem. 1994;269:26591–26593. [PubMed] [Google Scholar]
- 15.Fan YX, McPhie P, Miles EW. Regulation of tryptophan synthase by temperature, monovalent cations, and an allosteric ligand. Evidence from Arrhenius plots, absorption spectra, and primary kinetic isotope effects. Biochemistry. 2000;39:4692–4703. doi: 10.1021/bi9921586. [DOI] [PubMed] [Google Scholar]
- 16.Harris RM, Dunn MF. Intermediate trapping via a conformational switch in the Na(+)-activated tryptophan synthase bienzyme complex. Biochemistry. 2002;41:9982–9990. doi: 10.1021/bi0255672. [DOI] [PubMed] [Google Scholar]
- 17.Harris RM, Ngo H, Dunn MF. Synergistic effects on escape of a ligand from the closed tryptophan synthase bienzyme complex. Biochemistry. 2005;44:16886–16895. doi: 10.1021/bi0516881. [DOI] [PubMed] [Google Scholar]
- 18.Leja CA, Woehl EU, Dunn MF. Allosteric linkages between beta-site covalent transformations and alpha-site activation and deactivation in the tryptophan synthase bienzyme complex. Biochemistry. 1995;34:6552–6561. doi: 10.1021/bi00019a037. [DOI] [PubMed] [Google Scholar]
- 19.Ngo H, Harris R, Kimmich N, Casino P, Niks D, Blumenstein L, Barends TR, Kulik V, Weyand M, Schlichting I, Dunn MF. Synthesis and characterization of allosteric probes of substrate channeling in the tryptophan synthase bienzyme complex. Biochemistry. 2007;46:7713–7727. doi: 10.1021/bi700385f. [DOI] [PubMed] [Google Scholar]
- 20.Peracchi A, Bettati S, Mozzarelli A, Rossi GL, Miles EW, Dunn MF. Allosteric regulation of tryptophan synthase: effects of pH, temperature, and alpha-subunit ligands on the equilibrium distribution of pyridoxal 5′-phosphate-l-serine intermediates. Biochemistry. 1996;35:1872–1880. doi: 10.1021/bi951889c. [DOI] [PubMed] [Google Scholar]
- 21.Phillips RS, McPhie P, Miles EW, Marchal S, Lange R. Quantitative effects of allosteric ligands and mutations on conformational equilibria in Salmonella typhimurium tryptophan synthase. Arch Biochem Biophys. 2008;470:8–19. doi: 10.1016/j.abb.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 22.Barends TR, Domratcheva T, Kulik V, Blumenstein L, Niks D, Dunn MF, Schlichting I. Structure and mechanistic implications of a tryptophan synthase quinonoid intermediate. Chembiochem. 2008;9:1024–1028. doi: 10.1002/cbic.200700703. [DOI] [PubMed] [Google Scholar]
- 23.Miles EW, Kawasaki H, Ahmed SA, Morita H, Nagata S. The beta subunit of tryptophan synthase. Clarification of the roles of histidine 86, lysine 87, arginine 148, cysteine 170, and cysteine 230. J Biol Chem. 1989;264:6280–6287. [PubMed] [Google Scholar]
- 24.Sachpatzidis A, Dealwis C, Lubetsky JB, Liang PH, Anderson KS, Lolis E. Crystallographic studies of phosphonate-based alpha-reaction transition-state analogues complexed to tryptophan synthase. Biochemistry. 1999;38:12665–12674. doi: 10.1021/bi9907734. [DOI] [PubMed] [Google Scholar]
- 25.Weyand M, Schlichting I. Crystal structure of wild-type tryptophan synthase complexed with the natural substrate indole-3-glycerol phosphate. Biochemistry. 1999;38:16469–16480. doi: 10.1021/bi9920533. [DOI] [PubMed] [Google Scholar]
- 26.Weyand M, Schlichting I, Marabotti A, Mozzarelli A. Crystal structures of a new class of allosteric effectors complexed to tryptophan synthase. J Biol Chem. 2002;277:10647–10652. doi: 10.1074/jbc.M111285200. [DOI] [PubMed] [Google Scholar]
- 27.Kulik V, Weyand M, Seidel R, Niks D, Arac D, Dunn MF, Schlichting I. On the role of alphaThr183 in the allosteric regulation and catalytic mechanism of tryptophan synthase. J Mol Biol. 2002;324:677–690. doi: 10.1016/s0022-2836(02)01109-9. [DOI] [PubMed] [Google Scholar]
- 28.Yang XJ, Miles EW. Threonine 183 and adjacent flexible loop residues in the tryptophan synthase alpha subunit have critical roles in modulating the enzymatic activities of the beta subunit in the alpha 2 beta 2 complex. J Biol Chem. 1992;267:7520–7528. [PubMed] [Google Scholar]
- 29.Lim WK, Sarkar SK, Hardman JK. Enzymatic properties of mutant Escherichia coli tryptophan synthase alpha-subunits. J Biol Chem. 1991;266:20205–20212. [PubMed] [Google Scholar]
- 30.Nagata S, Hyde CC, Miles EW. The alpha subunit of tryptophan synthase. Evidence that aspartic acid 60 is a catalytic residue and that the double alteration of residues 175 and 211 in a second-site revertant restores the proper geometry of the substrate binding site. J Biol Chem. 1989;264:6288–6296. [PubMed] [Google Scholar]
- 31.Yutani K, Ogasahara K, Tsujita T, Kanemoto K, Matsumoto M, Tanaka S, Miyashita T, Matsushiro A, Sugino Y, Miles EW. Tryptophan synthase alpha subunit glutamic acid 49 is essential for activity. Studies with 19 mutants at position 49. J Biol Chem. 1987;262:13429–13433. [PubMed] [Google Scholar]
- 32.Axe JM, Boehr DD. Long-range interactions in the alpha subunit of tryptophan synthase help to coordinate ligand binding, catalysis, and substrate channeling. J Mol Biol. 2013;425:1527–1545. doi: 10.1016/j.jmb.2013.01.030. [DOI] [PubMed] [Google Scholar]
- 33.Axe JM, Yezdimer EM, O'Rourke KF, Kerstetter NE, You W, Chang CE, Boehr DD. Amino acid networks in a (beta/alpha)(8) barrel enzyme change during catalytic turnover. J Am Chem Soc. 2014;136:6818–6821. doi: 10.1021/ja501602t. [DOI] [PubMed] [Google Scholar]
- 34.Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc Natl Acad Sci USA. 2011;108:6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akimoto M, Selvaratnam R, McNicholl ET, Verma G, Taylor SS, Melacini G. Signaling through dynamic linkers as revealed by PKA. Proc Natl Acad Sci USA. 2013;110:14231–14236. doi: 10.1073/pnas.1312644110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dawson JE, Farber PJ, Forman-Kay JD. Allosteric coupling between the intracellular coupling helix 4 and regulatory sites of the first nucleotide-binding domain of CFTR. PLoS One. 2013;8:e74347. doi: 10.1371/journal.pone.0074347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Selvaratnam R, Mazhab-Jafari MT, Das R, Melacini G. The auto-inhibitory role of the EPAC hinge helix as mapped by NMR. PLoS One. 2012;7:e48707. doi: 10.1371/journal.pone.0048707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cembran A, Kim J, Gao J, Veglia G. NMR mapping of protein conformational landscapes using coordinated behavior of chemical shifts upon ligand binding. Phys Chem Chem Phys. 2014;16:6508–6518. doi: 10.1039/c4cp00110a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fatmi MQ, Chang CE. The role of oligomerization and cooperative regulation in protein function: the case of tryptophan synthase. PLoS Comput Biol. 2010;6:e1000994. doi: 10.1371/journal.pcbi.1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weischet WO, Kirschner K. Steady-state kinetic studies of the synthesis of indoleglycerol phosphate catalyzed by the alpha subunit of tryptophan synthase from Escherichia coli. Comparison with the alpha2 beta2-complex. Eur J Biochem. 1976;65:375–385. doi: 10.1111/j.1432-1033.1976.tb10351.x. [DOI] [PubMed] [Google Scholar]
- 41.Kim JW, Kim EY, Park HH, Jung JE, Kim HD, Shin HJ, Lim WK. Homodimers of mutant tryptophan synthase alpha-subunits in Escherichia coli. Biochem Biophys Res Commun. 2001;289:568–572. doi: 10.1006/bbrc.2001.6022. [DOI] [PubMed] [Google Scholar]
- 42.Jeong MS, Jeong JK, Lim WK, Jang SB. Structures of wild-type and P28L/Y173F tryptophan synthase alpha-subunits from Escherichia coli. Biochem Biophys Res Commun. 2004;323:1257–1264. doi: 10.1016/j.bbrc.2004.08.222. [DOI] [PubMed] [Google Scholar]
- 43.Nishio K, Morimoto Y, Ishizuka M, Ogasahara K, Tsukihara T, Yutani K. Conformational changes in the alpha-subunit coupled to binding of the beta 2-subunit of tryptophan synthase from Escherichia coli: crystal structure of the tryptophan synthase alpha-subunit alone. Biochemistry. 2005;44:1184–1192. doi: 10.1021/bi047927m. [DOI] [PubMed] [Google Scholar]
- 44.Nishio K, Ogasahara K, Morimoto Y, Tsukihara T, Lee SJ, Yutani K. Large conformational changes in the Escherichia coli tryptophan synthase beta(2) subunit upon pyridoxal 5'-phosphate binding. FEBS J. 2010;277:2157–2170. doi: 10.1111/j.1742-4658.2010.07631.x. [DOI] [PubMed] [Google Scholar]
- 45.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 46.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc. 1982;104:4559–4570. [Google Scholar]
- 47.Sterner R, Hocker B. Catalytic versatility, stability, and evolution of the (betaalpha)8-barrel enzyme fold. Chem Rev. 2005;105:4038–4055. doi: 10.1021/cr030191z. [DOI] [PubMed] [Google Scholar]
- 48.Rhee S, Parris KD, Ahmed SA, Miles EW, Davies DR. Exchange of K+ or Cs+ for Na+ induces local and long-range changes in the three-dimensional structure of the tryptophan synthase alpha2beta2 complex. Biochemistry. 1996;35:4211–4221. doi: 10.1021/bi952506d. [DOI] [PubMed] [Google Scholar]
- 49.Miles EW, Rhee S, Davies DR. The molecular basis of substrate channeling. J Biol Chem. 1999;274:12193–12196. doi: 10.1074/jbc.274.18.12193. [DOI] [PubMed] [Google Scholar]
- 50.Lai J, Niks D, Wang Y, Domratcheva T, Barends TR, Schwarz F, Olsen RA, Elliott DW, Fatmi MQ, Chang CE, Schlichting I, Dunn MF, Mueller LJ. X-ray and NMR crystallography in an enzyme active site: the indoline quinonoid intermediate in tryptophan synthase. J Am Chem Soc. 2011;133:4–7. doi: 10.1021/ja106555c. [DOI] [PubMed] [Google Scholar]
- 51.Mueller LJ, Dunn MF. NMR crystallography of enzyme active sites: probing chemically detailed, three-dimensional structure in tryptophan synthase. Acc Chem Res. 2013;46:2008–2017. doi: 10.1021/ar3003333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caulkins BG, Bastin B, Yang C, Neubauer TJ, Young RP, Hilario E, Huang YM, Chang CE, Fan L, Dunn MF, Marsella MJ, Mueller LJ. Protonation states of the tryptophan synthase internal aldimine active site from solid-state NMR spectroscopy: direct observation of the protonated schiff base linkage to pyridoxal-5′-phosphate. J Am Chem Soc. 2014;136:12824–12827. doi: 10.1021/ja506267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Livesay DR, Kreth KE, Fodor AA. A critical evaluation of correlated mutation algorithms and coevolution within allosteric mechanisms. Methods Mol Biol. 2012;796:385–398. doi: 10.1007/978-1-61779-334-9_21. [DOI] [PubMed] [Google Scholar]
- 54.Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 55.Ichiye T, Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991;11:205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 56.Rivalta I, Sultan MM, Lee NS, Manley GA, Loria JP, Batista VS. Allosteric pathways in imidazole glycerol phosphate synthase. Proc Natl Acad Sci USA. 2012;109:E1428–E1436. doi: 10.1073/pnas.1120536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 58.Muchmore DC, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW. Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods Enzymol. 1989;177:44–73. doi: 10.1016/0076-6879(89)77005-1. [DOI] [PubMed] [Google Scholar]
- 59.Ferrage F, Cowburn D, Ghose R. Accurate sampling of high-frequency motions in proteins by steady-state (15)N-{(1)H} nuclear Overhauser effect measurements in the presence of cross-correlated relaxation. J Am Chem Soc. 2009;131:6048–6049. doi: 10.1021/ja809526q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information