

Abstract
Type II R67 dihydrofolate reductase (DHFR) is a bacterial plasmid-encoded enzyme that is intrinsically resistant to the widely-administered antibiotic trimethoprim. R67 DHFR is genetically and structurally unrelated to E. coli chromosomal DHFR and has an unusual architecture, in that four identical protomers form a single symmetrical active site tunnel that allows only one substrate binding/catalytic event at any given time. As a result, substitution of an active-site residue has as many as four distinct consequences on catalysis, constituting an atypical model of enzyme evolution. Although we previously demonstrated that no single residue of the native active site is indispensable for function, library selection here revealed a strong bias toward maintenance of two native protomers per mutated tetramer. A variety of such “half-native” tetramers were shown to procure native-like catalytic activity, with similar KM values but kcat values 5- to 33-fold lower, illustrating a high tolerance for active-site substitutions. The selected variants showed a reduced thermal stability (Tm ∼12°C lower), which appears to result from looser association of the protomers, but generally showed a marked increase in resilience to heat denaturation, recovering activity to a significantly greater extent than the variant with no active-site substitutions. Our results suggest that the presence of two native protomers in the R67 DHFR tetramer is sufficient to provide native-like catalytic rate and thus ensure cellular proliferation.
Keywords: trimethoprim resistance, combinatorial mutations, thermostability, active site engineering, homotetramer
Introduction
Dihydrofolate reductases (DHFRs) play a critical role in proliferation of all living cells. They catalyze the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF), using NADPH as the hydride-donating cofactor. Among its metabolic roles, THF is required for the biosynthesis of purines and thymidylate. As a result, inhibition of DHFR leads to cell death, making this enzyme a main target of antibiotic and anticancer drugs over the past six decades.
Trimethoprim (TMP) is a low-cost and effective bacteriostatic antibiotic that is used worldwide in the prophylaxis and treatment of bacterial infections, both in humans and livestock.1 It is an effective competitive inhibitor of the chromosomally-encoded DHFR in many bacteria and parasites; for example, it inhibits E. coli DHFR with Ki = 0.4 nM.2 Its widespread use has resulted in the development of microbial resistance to TMP, in the form of highly transmissible plasmid-encoded DHFRs. TMP-resistant DHFRs (type I-XVII) are classified into two gene families: dfrA and dfrB.3,4 The dfrA family DHFRs are close genetic variants of the monomeric chromosomally-encoded DHFRs that are 160–190 amino acids long. In contrast, dfrB genes encode shorter sequences, on the order of 78 amino acids, that homotetramerize to form the active DHFR species. They are genetically and structurally unrelated to chromosomal DHFRs. Type II R67 DHFR belongs to this second class. Its single, symmetrical active-site pore characterized by 222 symmetry lies at the heart of a channel formed by the assembly of four protomers that are largely constituted of a SH3-like β-sandwich fold (Fig. 1). Short loops and both termini extend out of each β-sandwich, forming the inter-protomer interfaces.6–8 As R67 DHFR is unrelated to its chromosomal counterparts, it has a different catalytic mechanism4,9–13 as well as a different response to antifolate inhibitors.4 It has been qualified as being “primitive,” in that its catalytic strategy lacks in binding specificity, suggesting that it may not yet be evolutionarily optimized.14 Importantly, as a result of being unrelated to dfrA family DHFRs, it confers resistance to high concentrations of TMP (Ki = 0.15 mM).15
Figure 1.
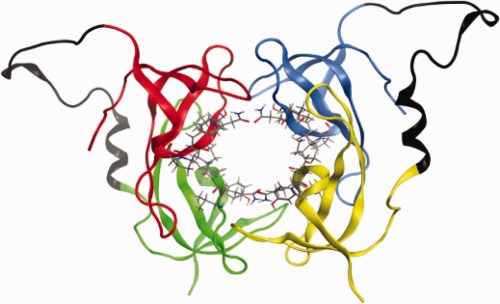
R67 DHFR: The four protomers (A–D) are illustrated in red, green, blue and yellow, respectively. The active site residues 66–69 are shown in sticks representation. The linker introduced for the dimer construct and the N-terminal tail of R67 connecting protomers A,B and C,D are shown in black. The linker conformation was predicted using the loop-modelling tool of the MOE software.5
Here, we investigate the tolerance to active-site modification of this compact reductase. One β-strand per protomer (V66, Q67, I68, and Y69) constitutes the major portion of the active site channel (Fig. 1). The DHF substrate and NADPH cofactor have been proposed to slip into the channel from opposite ends to juxtapose their reactive atoms at the center of the channel, each forming contacts mainly with one protomer, though additional stabilizing contacts are contributed by the three remaining protomers.16,17 For this reason, residues involved in ligand binding can have up to four different consequences within a single binding/catalytic event.9 This makes the active site poorly tolerant to point mutations because of a cumulative effect of each fourfold repeated residue. Despite this observation, we have previously demonstrated that the homotetrameric active site can be heavily modified, including up to four simultaneous mutations per protomer (16 in the tetrameric active site).13 In that selection experiment, the vast majority of modified homotetrameric active sites (99.8% of the variants tested) did not give rise to native-like function, yet this and other studies showed that the simultaneous variation of more than one residue in R67 DHFR can provide compensatory effects, thus allowing multiple mutations to complement each other.13,18–20 This study also revealed that no single residue of the native active site is indispensable for function.
Because our previous active-site mutation efforts using the monomeric species resulted in a low yield of functional variants, here we investigate the tolerance of the active site to more conservative modifications. To reduce the complicating effect of the active-site symmetry, R67 DHFR has previously been genetically linked to provide a covalent dimeric or tetrameric species, using the unstructured N- and C-termini to provide an inter-domain linker (Fig. 1).19,21 This breaks the active-site symmetry by introduction of substitutions on only one (for the covalent tetramer) or two (for the covalent dimer) of the four protomers.18,19,22 In this work, we have modified the active site in the context of the dimeric species so that any substitution is present twice rather than four times. The N-terminal protomer carried multiple active-site substitutions and the C-terminal protomer was either native or carried an independent set of active-site mutations. Screening the library for the native catalytic activity allowed us to further evaluate the tolerance of this antibiotic-resistance enzyme to active-site variation, providing further insight into the potential for this antibiotic-resistance enzyme to adapt its function in response to selective pressure.
Results
Selection of TMP-resistant dimeric, doubly-mutated (DDM) R67 DHFR variants yields singly mutated dimers
To introduce asymmetric mutations in the R67 DHFR active site, a library of R67 DHFR protomers mutated at positions 66–69 was linked to a second, similarly mutated library of R67 DHFR protomers to form a library of covalent dimers, doubly mutated (DDM). The DDM dimer library encoded mutated protomer 1 with an N-terminal 6-histidine tag for protein purification and a C-terminal Glu-Leu linker (encoded by an inserted, unique SacI restriction site) connecting to the N-terminus of mutated protomer 2. Residues 66–69 of both protomers were combinatorially mutated according to a previously described strategy for partial randomization.13 Briefly, most mutations were designed to participate in reactivity according to the following mechanisms: (1) The DHF substrate must be protonated at N5 to promote hydride transfer at C6. Contrary to chromosomally-encoded DHFRs, the active site channel of R67 DHFR does not contain an evident proton donor. We thus replaced the central Gln67 by Asp or Glu. (2) The dielectric environment in the E. coli chromosomal DHFR promotes hydride transfer by favoring substrate tautomerization.23 We thus altered the dielectric environment in the active site of R67 DHFR by encoding Val66Asp, Gln67Lys, Ile68Lys, Ile68Arg and Tyr69Phe. (3) The active-site channel is relatively wide; we included bulkier residues in an attempt to increase the proximity between the substrate and cofactor. Because a single primer set was designed to capture all desired features, some additional mutations were also encoded, adding additional sequence diversity.
The library was restricted to eight amino acids encoded at position 66, four at position 67, twelve (and the stop codon) at position 68, and four at position 69 (Supporting Information Table SI). This encoded a maximum of 2.4 × 106 unique, combinatorially mutated dimer variants. The native sequence is not encoded, because no Gln is allowed at codon 67. A silent mutation at codon 74 [CTT (Leu) to TTG (Leu)] was also encoded to detect any potential contamination by native sequence. To confirm that the library construction was successful, 25 clones were picked for DNA sequencing from the obtained library of 106 unselected clones. Slight biases above the expected 1/8 ratio were observed for both protomers at position 66 for Asn and in position 67 a slight bias below the expected 1/4 ratio was observed for Glu. Similarly, position 68 was biased slightly above or below the expected 1/12 ratio for Ser, Asn and Trp; no Leu or Trp was observed among those 25 unselected clones and for protomer 2 no Met or stop codon was observed (Supporting Information Table SI). At position 69 a ratio slightly below the expected 1/4 was observed for Phe. No contamination with the native sequence was observed. In two instances, the C-terminal protomer included unforeseen residues (Supporting Information Table SI: I67 and S69 in DDM clone 14NS; K66 in DDM clone 19NS; compare to amino acids encoded, Supporting Information Table SI), which may result from primer impurity or errors introduced by the polymerase during PCR. Overall, library quality was deemed to be good.
The 106 DDM clones of R67 DHFR were selected for TMP resistance by plating equal aliquots on non-selective and on selective minimal medium plates containing TMP. DNA sequencing of 20 TMP-resistant clones revealed a disparity of the DNA sequence compared to unselected constructs: 25% of the selected clones had only one mutated protomer (Supporting Information Table SI). The Leu74 silent mutation was present in both protomers, demonstrating that this was not the result of contamination with native monomer or with DSM library members. Observation of a non-mutated protomer was unexpected, because the native Gln67 was not encoded in the library; its presence may result from contamination of the mutagenic primers with undesired nucleotides at a level that is too low to be observed in the unselected library, or may result from bacterial mutational activity as a response to strong selective pressure. We note that a single base change can suffice to alter the encoded RAK, to the native CAG.
To evaluate the genetic stability of the selected clones, we re-cloned the 20 individual dimeric inserts into fresh vector and transformed anew. Following two more selection rounds, the majority of the selected variants (14/20) held one protomer with native sequence at residues 66–69 and one mutated protomer; again, the Leu74 silent mutation was present in both protomers (Supporting Information Table SI). Thus, clones with two mutated protomers after the first round of selection acquired a non-mutated protomer in subsequent selection rounds. We note that, in all cases but one, it is the C-terminal protomer that reverted to native sequence. Previous studies of a heavily mutated, covalent tetrameric species of R67 DHFR, incorporating up to 55 mutations, showed long-lasting genetic stability even after 7 days of subculturing under similar conditions.18 In that case, all mutations were positioned in surface or disordered regions of the construct rather than within the central active-site channel. Here, mutagenesis within the active site appears to have triggered molecular mechanisms to “correct” the sequence into a native form, preserving the bacteria from loss of activity of R67 DHFR under selective conditions.
Notwithstanding the mode of its appearance during selection, this indicates that the presence of one protomer with native sequence at residues 66–69 provided a significant survival advantage. This is in good agreement with our previous study of mutated monomers, where only three variants with native-like activity were identified among to 1,536 possible variants screened.13 The selection of the DDM library was repeated in different E. coli strains under similar conditions. Results consistently showed the tendency to enrich clones with one native protomer (data not shown).
Among the non-native protomer sequences that were observed following the rounds of selection, an important bias was observed at positions 66 and 67, which held the non-native Asn66/Lys67 pair in 9/20 variants, one occurrence of which was in a dimer where both protomers remained mutated. We note that the DDM sequences that held no native protomer following three rounds of selection had generally varied throughout the selection rounds.
Selection of TMP-resistant singly mutated dimers (DSM) R67 DHFR variants
On the basis of that preliminary observation, we sought to determine the breadth of sequence diversity that can be functionally accommodated in dimeric variants forming a tetramer with two native and two mutated protomers. To this effect, a library of mutated protomers was genetically linked to a native protomer to form a covalent dimer, singly mutated (DSM) library. As above, residues 66–69 of the N-terminal protomer were combinatorially mutated. To confirm that the library construction was successful, 32 clones were picked for DNA sequencing from the total library of 1.7 × 104 clones obtained. No deviations from the encoded codons were detected and no contamination with the native sequence was observed (Supporting Information Table SI).
Some biases above the expected 1/8 ratio were observed at position 66 for Ile (9/32), Asn (7/32), and Asp (6/32), and no Ala was observed. Similarly, position 68 was slightly biased above the expected 1/12 ratio for Thr (5/32) and the stop codon (4/32); no Phe or Arg were observed among those 32 unselected clones, though they were identified following selection. Position 67 and 69 showed no strong bias towards any residue. Overall, library quality was deemed to be good.
The DSM dimer library was then selected for TMP resistance as described above. A 79% selection rate (number of selected clones/number of unselected) was obtained. DNA sequencing of 32 TMP-resistant clones confirmed that no native sequence had arisen in the mutated N-terminal protomer (Table I). Furthermore, the native C-terminal protomer showed no unexpected mutations. We note that the observed 79% selection rate of the DSM library does not necessarily indicate that nearly four out of five of the possible sequence variations in the N-terminal protomer give rise to an active enzyme species. Bacteria may be multiply transformed, thus harbour more than one variant at a time, which increases the apparent selection rate. Nonetheless, this selection rate is much >0.2% observed upon selection of the same mutated monomeric species.13 As all transformations and selections were undertaken similarly, this indicates that there is a greater fraction of functional variants when the mutated protomer is combined with a native one. This is further supported by the fact that only three variants among that mutated monomeric library had provided TMP resistance upon selection in E. coli,13 while a broad variety of variants were functionally selected here. Among the 32 clones sequenced, 27 unique sequences were identified in the N-terminal protomer (Table I). This represents roughly 2% of all possible combinations. The distribution of codons/amino acids observed for each mutated residue (66–69) was compared for unselected (Supporting Information Table SI) and selected clones (Table I). For the selected clones, a slight (twofold) enrichment of Thr was observed at position 66 and a similar enrichment of Glu was observed at position 67. Only three occurrences of the non-native Asn66/Lys67 pair, which was observed in nearly 50% of the selected DDM variants, were recorded. Interestingly, a new pair emerged at the same position in this selection: the combination of Thr66/Glu67 was observed in nearly 20% of selected DSM variants (Table I). At position 68, a ninefold enrichment of Asn was observed as well as a sixfold enrichment of Phe. The distribution of codons for position 69 was similar for unselected and selected clones. The only unexpected mutation was His66 in mutant 27S, which was not designed into the mutagenic primers. We note that neither the exact sequences of the three functional monomeric variants (SKIH, INRY and GELH13), nor any closely related homolog, were observed among the 32 selected dimers. For those variants, all four protomers were identical such that the mutated sequences needed to be fourfold tolerated in the context of function. Here, the sequence at residues 66–69 for two of the protomers may be complementary, to the native residues on the two other protomers. Thus a greater sequence variation can be expected and, indeed, was observed.
Table I.
Deduced Amino Acid Sequence of the Active Site Residues 66–69 of the Mutated N-terminal Protomer Within the Dimeric DSM Library Following Selection for TMP Resistance
| Selected clone | Residues 66–69 | Selected clone | Residues 66–69 |
|---|---|---|---|
| 1S | NKSH | 17S | TEFL |
| 2S | TENH | 18S | SDCF |
| 3S | TENH | 19S | GNNL |
| 4S | ANTF | 20S | DDNH |
| 5S | TEFL | 21S | GNNF |
| 6S | NDTF | 22S | SEYF |
| 7S | TNTY | 23S | IETY |
| 8S | SKKY | 24S | TEFL |
| 9S | INSF | 25S | AEFL |
| 10S | DETL | 26S | DDNY |
| 11S | NNRH | 27S | HEYF |
| 12S | TEFY | 28S | NKNY |
| 13S | IKYH | 29S | VDNH |
| 14S | DESH | 30S | NKNY |
| 15S | NDTY | 31S | DDNY |
| 16S | SDWL | 32S | INNF |
The selected mutated dimeric DSM variants 4S, 9S, and 12S were expressed at levels similar to the non-mutated dimer (DNM), whereas variants 3S, 13S, and 30S were expressed about 50% less; variants 5S, 25S, and 27S expressed only in the range of 25% of the DNM level. The molecular weight of DNM and a subset of four selected DSM variants (3S, 5S, 12S, and13S) were verified by mass spectrometry (Supporting Information Table SI), confirming that the dimeric species were expressed with no unexpected modifications.
Kinetic characterization of the selected DSM
The kinetic parameters of nine selected DSM variants were determined and compared to the native-like DNM (Table II). The kcat obtained for DNM was similar to that previously reported for a similar dimer construct (0.28 s−1 vs. 1.2 s−1).21 The KM values obtained for DHF and NADPH (12.9 and 13.3 µM, respectively) were also similar to values reported for the similar dimer construct (6.3 and 2.7 µM, respectively).21 The differences may be attributed to experimental conditions (here, measured at ambient temperature in absence of β-mercaptoethanol rather than at 30°C with β-mercaptoethanol) and the different inter-dimer linker. For the selected DSM variants, the kcat values were similar or 2- to 28-fold lower than for the DNM dimer, except for variant 25S where no activity was detected; this variant was not further analyzed. In contrast, the KM values for DHF and NADPH were generally similar to those of the DNM dimer, except for KMNADPH of variants 5S and 9S, which were significantly increased.
Table II.
Kinetic Parameters for the Non-Mutated DNM Dimer and Selected Mutated DSM Dimer Variants
| Residues 66–69 in N-terminal protomer | kcat (s−1) | KMDHF (µM) | KMNADPH (µM) | kcat/KMNADPH (s−1 µM−1) | kcat/KMDHF (s−1 µM−1) |
|---|---|---|---|---|---|
| DNM: VQIY (native) | 0.28 ± 0.03 | 13 ± 4 | 13 ± 5 | 0.02 | 0.02 |
| DSM: TENH (3S) | 0.18 ± 0.01 | 28 ± 4 | 19 ± 4 | 0.01 | 0.007 |
| ANTF (4S) | 0.28 ± 0.02 | 51 ± 8 | 44 ± 6 | 0.006 | 0.005 |
| TEFL (5S) | 0.03 ± 0.01 | 35 ± 23 | ND | — | 0.001 |
| INSF (9S) | 0.06 ± 0.01 | 16 ± 4 | >200 | < 0.0003 | 0.004 |
| TEFY (12S) | 0.12 ± 0.01 | 18 ± 5 | 84 ± 20 | 0.001 | 0.007 |
| IKYH (13S) | 0.050± 0.004 | 15 ± 4 | 13 ± 2 | 0.004 | 0.003 |
| AEFL (25S) | ND | ND | ND | — | — |
| HEYF (27S) | 0.10 ± 0.01 | 8.5 ± 3.0 | 23 ± 7 | 0.005 | 0.012 |
| NKNY (30S) | 0.11 ± 0.01 | 13 ± 4 | 16 ± 5 | 0.007 | 0.009 |
Functional DSM variants assemble predominantly as “dimers of dimers”
For all subsequent experiments we focused on the selected DSM variants 3S, 9S, 12S, 13S, and 30S, based on their diversity of sequence and kinetic activity. Size exclusion chromatography was carried out to determine the oligomeric state of the native-like DNM dimer as well as the N-terminally mutated DSM variants. For DNM, a single species was observed at ∼36 kDa, confirming the formation of a dimer of the dimeric species, equivalent to the native tetrameric state of R67 DHFR (Table III). Similar results were observed for the variants 9S, 12S, and 13S, where the “dimer of dimers” species was predominant [Fig. 2(b)].
Table III.
Native Molecular Weight Determined by Size Exclusion Chromatography for the Non-Mutated DNM Dimer and Selected Mutated DSM Dimer Variants
| Residues 66–69 in N-terminal protomer | Main peak (kDa) | Second peak (kDa) | Peak ratio (% main/second) |
|---|---|---|---|
| DNM: VQIY (native) | 34.0 | N/Oa | 100 |
| DSM: TENH (3S) | 33.4 | 52.9 | 61.5 |
| INSF (9S) | 33.3 | 45.5 | 92.6 |
| TEFY (12S) | 31.1 | 45.8 | 87.5 |
| IKYH (13S) | 33.3 | N/Oa | 100 |
| NKNY (30S) | 35.3 | 52.9 | 74.1 |
N/O: none observed.
Figure 2.
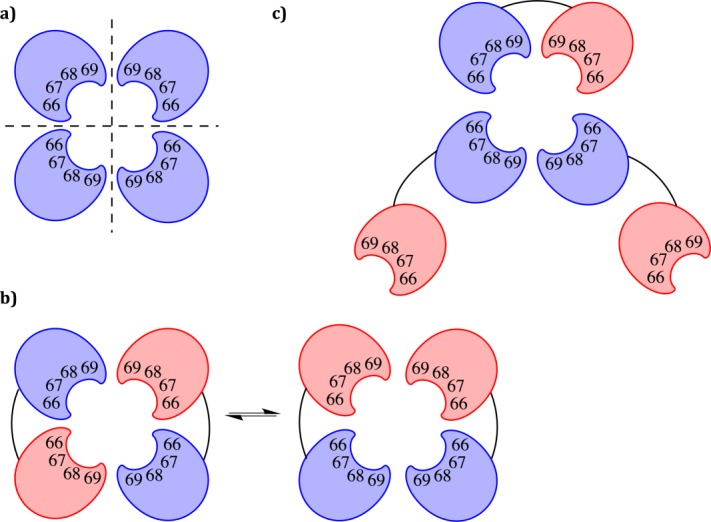
Schematic representation of protomer assembly for R67 DHFR variants. (a) Assembly of the native, monomeric R67 DHFR into a tetramer with a central active-site pore. (b) Assembly of a dimer of dimers, where the red protomers are mutated. (c) Putative assembly of a trimer of dimers to yield an active-site where only one protomer is mutated.
For variants 3S and 30S, the main peak on the chromatogram is consistent with the formation of a “dimer of dimers” yet a shoulder observed at ∼53 kDa is consistent with the formation of a “trimer of dimers.” The formation of “trimers of dimers” opens new possibilities for assembly, which are illustrated in Figure 2. This model is imperfect because the “pending” monomers may not be adequately stabilized; we currently have no direct insight into the mode of assembly. We note that higher-order assembly of R67 DHFR multimers has previously been suggested in relation to the intermolecular association of a covalent tetrameric construct.18
The potential for the dimers to assemble in various configurations to form a functional active site may underlie the high selection rate observed. In a trimeric arrangement, the active site may be constituted of three non-mutated and one mutated protomer, reducing the deleterious effect of some mutations [Fig. 2(c)]. The presence of a non-polar residue at position 66 may favor the formation of a dimer–dimer assembly (9S and 13S). A polar residue at that position may increase the prevalence of the trimeric arrangement up to 37.5% in the most distant variant from the WT sequence, 3S.
Secondary structure is native-like
Circular dichroism (CD) allowed an assessment of the extent of secondary structure formation in functional dimers. The CD spectrum of the native DNM dimer is consistent with that reported for a previously constructed dimer, and is also very similar to that of the native homotetrameric R67 DHFR [see Zhuang et al., Fig. 4(B)].21 The CD scans for DSM variants 3S, 9S, 12S and 30S all yield a CD spectrum that is similar to the non-mutated DNM, and are characteristic of high β-sheet content [Fig. 3(a)]. This is consistent with the native structure of R67 DHFR and indicates that the overall secondary structure was not significantly disrupted by the introduction of the mutations. Only slight differences in steepness were observed among variants.
Figure 4.
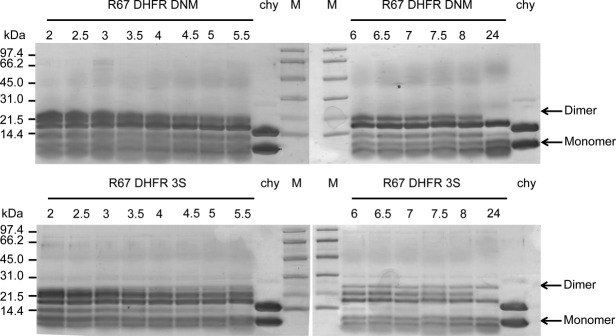
Limited chymotryptic digestion of R67 DHFR DNM (upper panel) and DSM variant 3S (lower panel), observed by tricine SDS PAGE. R67 DHFR DNM or DSM 3S were digested with chymotrypsin (1:100 chymotrypsin : R67 DHFR) for 2–24 h at 4°C, as indicated above each lane. Chy: chymotrypsin reference; M: molecular weight markers. Arrows on the right indicate the bands corresponding to the monomeric and dimeric species.
Figure 3.
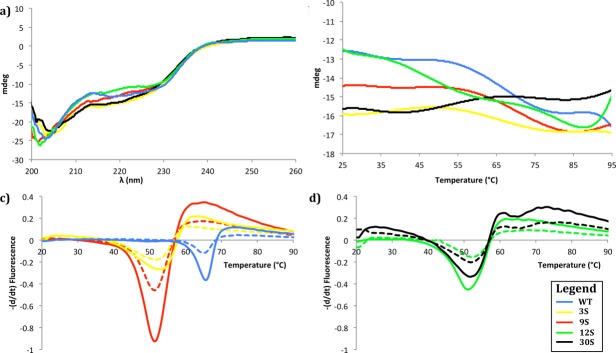
Thermal denaturation of R67 DHFR dimer mutants monitored by CD spectrometry and thermal scanning fluorimetry. (a) Far-UV CD spectra at 25°C of WT, 3S, 9S, 12S, and 30S. (b) Thermal denaturation measured at 215 nm at a rate of 25°C h−1 for WT, 3S, 9S, 12S, and 30S. (c,d) First derivative analysis of representative thermal melting curves observed by fluorescence of SYPRO Orange. Melting curves are shown for a ratio of 3.33 x SYPRO to 8 μM (full line) or 4 μM (dashed line) protein, for (c) WT, 3S, 9S and (d) 12S and 30S.
The thermal stability of the dimeric variants was evaluated with CD between 25 and 95°C, to observe any perturbation of the secondary structure [Fig. 3(b)]. Surprisingly, monitoring the secondary structure showed no clear temperature denaturation for any R67 DHFR variant. Only variants 12S and 30S showed a small increase in ellipticity in the range of 90 to 95°C. This result was in apparent contradiction with the previously published Tm value for native R67 DHFR and a dimer construction,21 determined using differential scanning calorimetry (DSC). DSC reveals not only changes to secondary structure, but also changes to higher order structure. Therefore, we speculated that the previously reported Tm may have reported on changes to the tertiary and quaternary structure, while the secondary structure elements appear to be thermally stable up to at least 90°C.
Protomer assembly into the active structure is weaker in the selected DSM variants
To investigate this possibility we performed a thermal scanning fluorimetry shift assay. We used SYPRO orange as the hydrophobic fluorophore to bind hydrophobic patches of the protein as they become increasingly solvent-exposed upon thermal denaturation.24 Similarly to DSC, this approach can reveal changes in secondary, tertiary, and quaternary structure, as observed by an increase in fluorescence. The resulting melting curve for the native DNM dimer yielded a Tm of 64.5°C, consistent with the value of 61.3°C reported by DSC for a similar native dimer construct [Fig. 3(c,d)].21 A difference of 1–3°C in Tm between the two methods is consistent with previously published data using other proteins.25,26 These results indicate that the secondary structure elements of the native DNM dimer—which is likely to be stabilized as a β-barrel spanning both protomers 6—remain essentially native-like up to ≥90°C, while the quaternary subunit assembly (association of two covalent dimers) breaks down above 60°C. It remains to be investigated whether the high thermostability at the level of secondary structure is an intrinsic property of the native R67 DHFR protomers, or whether it has been increased by linkage of the protomers into a covalent dimer.
The melting curves for the four DSM variants yielded Tm values ∼10°C lower than for the DNM dimer, indicative of a perturbation to the quaternary subunit assembly (Table IV). Residues 66–69 are contained in the inner-most β-strand of the active site and do not participate directly in the interface of protein–protein interaction between the two dimers. Nonetheless, their modification, even in only two of the four protomers of the assembled R67 DHFR variants, appears to destabilize the tetrameric core assembly. Interestingly, the Tm value does not differ significantly among the DSM variants, whereas the introduced mutations differ greatly in physical and electrostatic properties.
Table IV.
Tm Values Obtained by Thermal Scanning Fluorimetry for the Non-Mutated DNM Dimer and Selected Mutated DSM Dimer Variants
| Residues 66–69 in N-terminal protomer | Tm (°C) |
|---|---|
| DNM: VQIY (native) | 64.5 |
| DSM: TENH (3S) | 53.0 |
| INSF (9S) | 51.7 |
| TEFY (12S) | 51.8 |
| NKNY (30S) | 52.8 |
To further investigate the high thermal stability of the secondary structure elements, we analyzed the stability of the protomers with respect to controlled proteolysis. Digestion with α-chymostrypsin over 24 h revealed a similar digestion pattern for the native DNM dimer and all DSM variants; variant 3S is shown as a representative example (Fig. 4). We also verified that no significant difference in digestion pattern was observed during the first 2 h (data not shown). α-Chymotrypsin can theoretically recognize up to 22 cleavage sites in the R67 DHFR dimers studied. In all cases, the predominant proteolysis bands observed upon analysis by SDS PAGE following 24 h digestion were equivalent to the molecular weight of a single protomer (monomer) and a slightly shortened dimer, though additional bands indicate further preferred proteolysis sites. Cleavage is most likely to occur in the unstructured N- and C-terminal regions and in the inter-protomer linker as a result of accessibility. We note that the band of highest molecular weight (see “Dimer” arrow, Fig. 4) was digested significantly more rapidly, and to completion, in the DNM sample than for the variants examined, where a significant undigested band remained even after 24 h digestion.
For all variants, the core of the R67 DHFR protomer remained intact and inaccessible to α-chymotrypsin even following 24 h digestion. This indicates that the reduction in Tm by ∼10°C for the selected variants did not destabilize packing to the extent where the core becomes accessible for proteolysis. To verify this, MS/MS analysis was performed on the main band of the DSM 3S dimer remaining following overnight proteolysis, corresponding to a single protomer. Both the non-mutated and the mutated protomers were observed by MS in that sample. Because this method is not quantitative, it was not possible to determine the ratio of non-mutated:mutated protomer resulting from digestion. Nonetheless, this result indicates that the tight fold of each protomer is conserved upon introduction of mutations at the active site, consistent with the maintenance of secondary structure observed by CD upon thermal denaturation.
Recovery of activity following thermal denaturation is enhanced in the selected DSM variants
We further investigated the high thermal stability of the secondary structure elements and the multimeric assembly by verifying the activity of the variants following incubation at 95°C, under conditions mimicking the CD thermal denaturation experiment. After incubating the samples at 95°C for 90 minutes, slow cooling for 45 min allowed partial recovery of the activity (Table V, cf., conditions A and B). All DSM variants showed a high recovery (≥45%) of the initial activity, where the variants 3S and 9S showed the highest recovery (>70%). Interestingly, the recovery of DNM was significantly lower (<25%), consistent with a previous report of 15% activity recovered following heating to 75°C for a similar dimeric construct.21 Shorter incubation at 95°C (conditions C and D) was generally less disruptive to activity. Thus the selected DSM variants have acquired an increased resilience to heat denaturation, despite having reduced Tm values relative to the non-mutated DNM.
Table V.
Recovery of Enzymatic Activity Following High-temperature Incubation
| Condition | DNM (%) | 3S (%) | 9S (%) | 12S (%) | 30S (%) |
|---|---|---|---|---|---|
| A: 95°C, 90 min; RT, 1 min | 11 | 18 | 18 | 26 | 17 |
| B: 95°C, 90 min; RT, 45 min | 23 | 72 | 72 | 46 | 47 |
| C: 95°C, 4 min; RT, 1 min | 78 | 95 | 90 | 50 | 11 |
| D: 95°C, 4 min; RT, 45 min | 71 | >99 | 90 | 53 | 70 |
Samples were incubated at 95°C then at room temperature for the periods indicated, then activity was measured. Activity is expressed as percent of activity for that variant prior to incubation.
Discussion
Overall, we have found that the active, tetrameric form of R67 DHFR is very tolerant to active site variation, particularly in a context where two among the four protomers are native. In most multimeric enzymes, this would not be surprising, since a single protomer can often independently provide catalytic function. This is not the case in R67 DHFR, where the relatively small active site tunnel formed at the center of the tetramer can accommodate a single pair of ligands (DHF substrate + NADPH cofactor) at a time; all four protomers contribute simultaneously to ligand binding.
Through the process of selection, we have obtained DSM variants of R67 DHFR that exhibit a lower Tm. This illustrates that assembly of the dimers into an active structure (generally consisting of 4 protomers, though 6 protomers were also observed according to size-exclusion chromatography) was not as thermally stable in the selected DSM variants as in the non-mutated DNM dimer. Nonetheless, those selected DSM variants showed enhanced recovery of activity following thermal denaturation, suggesting an increased rate of dimer assembly into the active enzyme form. We note that the selection strategy (growth on agar plates at 37°C) was not designed to elicit this trait. We hypothesize that, as 37°C is largely inferior to the Tm of the selected variants (near 52°C), reduced thermal stability was not a selective disadvantage, while the improved dimer assembly into the active tetrameric form appears to have conferred a selective advantage.
Our observation of some selected DDM sequences that contain no native active-site sequence supports our earlier report of active-site modification in R67 DHFR, and confirms that no specific residue in the active-site tunnel formed by residues 66–69 is indispensable for catalytic activity.13 Importantly, we observed no partial enrichment toward native residues with retention of accompanying mutations: the residue 66–69 stretch either became entirely native, or else remained highly variable with only slight sequence biases. There may be a synergy between residues 66–69 that provides one robust solution for catalysis and structural integrity. We do not know if there is a functional or structural significance to the predominance of C-terminal reversion to the native sequence during the selection process; this may reflect different constraints on the two protomers as a result of their non-native fusion and/or the presence of the N-terminal 6-His tag.
The non-native Asn66/Lys67 residue pair was frequently observed in replacing the native Val66/Gln67 residue pair when the starting point was the doubly mutated dimer library DDM (9/20), but was less frequent when the starting point was the DSM singly mutated dimer library (3/32). In the DSM library the Thr66/Glu67 residue pair was predominant (6/32). This suggests that the Asn66/Lys67 or the Thr66/Glu67 substitution pair confers a selective advantage under the more stringent conditions of selecting for activity in a fully mutated active-site environment (DDM library) relative to the active sites that are part native, part mutated (DSM library). It is tempting to speculate on functional relevance of those pairwise substitutions. However, uncertainty with respect to the “dimer–dimer” arrangement (do the likewise mutated monomers face each other in the tetramer or not?)19 and hence the resulting environment of substrate/co-factor binding opens a number of possible interaction profiles. Further investigation of this question would require the structural resolution of a ligand-bound dimer complex; earlier attempts to resolve the structure of a covalently linked R67 DHFR dimer in our laboratory have not been successful.8
The observation of high sequence diversity at the active site of one protomer in the presence of the second protomer with native sequence suggests that the enzymatic activity of R67 DHFR can be readily supported by an active site that is 50% native. This is supported by the observation of native-like enzyme kinetics for a number of DSM variants. Assembly of the four protomers to form a central active-site channel allows DHF and NADPH to slip in from opposite mouths of the tunnel17; their reactive atoms are juxtaposed near the center of symmetry of the tetramer. Catalysis is proposed to result from proper positioning of the reactive atoms to allow hydride transfer to occur on the pre-protonated DHF molecule. Again, no residue of the enzyme is indispensable for catalysis. Our results suggest that the native 66–69 residues of two protomers are sufficient to provide a native-like catalytic rate enhancement in R67 DHFR.
We also observed an additional structural dimension with the formation of “trimer–dimer.” This evidence for a functional assembly other than the “dimer–dimer” assembly increases the complexity of the system and is likely to reduce the impact of the introduced mutations. Along with the observed reversion of one protomer to the native sequence in the DDM dimers, assembly into trimers can provide an additional route for rapid recovery of trimethoprim resistance in the bacterial cell during selection.
In conclusion, we have found that R67 DHFR can explore a large active-site sequence space and various quaternary structures while maintaining native-like activity. It thus appears that this enzyme, postulated to have recently acquired DHFR activity as a response to selective pressure, is not in a highly specialized, evolutionary dead-end; instead, it appears to have a great potential for continued functional evolution. This finding is highly interesting because newly emerged and still evolving proteins might behave similarly to ancient proteins, and may thus serve as models with which to conduct studies on ancestral proteins.
Our results have direct implications on the threat that R67 DHFR holds with respect to antibiotic resistance. Already natively resistant to trimethoprim as a result of its fold being unrelated to that of DfrA DHFRs, R67 DHFR may continue to evolve even as we direct inhibitors to it,27 and will need to be closely monitored.
Materials and Methods
Materials
All reagents were of the highest available purity. Restriction enzymes and DNA-modifying enzymes were from New England Biolabs and MBI Fermentas. Synthetic oligonucleotides were obtained from Integrated DNA Technologies (Coralville, USA). The infrared dye-labeled sequencing primers were from Li-Cor Biotechnology Division. The Sequenase 2.0 DNA sequencing kit was purchased from GE Healthcare. All aqueous solutions were prepared using water purified with a Millipore BioCell system. The 7,8-dihydrofolate (DHF) was prepared as described previously.28
Bacterial strains and plasmids
E. coli SS320 was used to verify library quality by DNA sequencing. E. coli BL21 containing plasmid pRep4 (Qiagen, Mississauga) was used for expression. Chloramphenicol (Chl) was used at 10 µg mL−1 and kanamycin (Kan) at 50 µg mL−1 for maintenance of plasmids derived from pQE32Chl13 and for pRep4, respectively. Ampicillin (Amp) was used at 100 µg mL−1 for maintenance of plasmids derived from pQE32 (Qiagen). Isopropyl β-d−1-thiogalactopyranoside (IPTG) was used at 1 mM for expression and TMP was used at 70 µg mL−1 during selection.
Construction of the “dimer, doubly mutated” (DDM) and “dimer, singly mutated” (DSM) libraries
Starting from the construction described in Ref. 13, a covalent R67 DHFR dimer mutated at residues 66–69 was created by inserting a second mutated R67 gene between SacI and HindIII restriction sites on pQE32Chl (library DM, where DM stands for “dimer, mutated”). A covalent dimer containing two native (WT) R67 DHFR genes was also created by ligating the BanII-XbaI digested products of the native construct described in Ref. 13 and a construct containing the native R67 DHFR gene in pQE32 (Amp resistant) between SacI and HindIII sites, yielding construct DNM (where DNM stands for “dimer, non-mutated”). Library DM and construct DNM were digested with SacI and XbaI. This generated two fragments of 2500 and 1500 bp for each construct. The 2500 bp fragment of library DM (encoding the N-terminal mutated protomer) and the 1500 bp fragment from the DNM or the DM construct (encoding the C-terminal non-mutated or mutated protomer, respectively) were extracted using a QiaexII extraction kit (Qiagen). The fragments were ligated yielding the dimer libraries, where protomer 1 is mutated at residues 66–69 and protomer 2 is either non-mutated (“dimer, singly mutated,” DSM) or mutated (“dimer, doubly mutated,” DDM). Apart from the mutations, this dimer construct differs slightly from a previously reported dimer, where protomer 1 was immediately connected to protomer 221; the Glu-Leu linker was adopted here because it introduces a SacI restriction site for recombination of protomers into dimers.
The libraries were transformed by electroporation of 0.4–0.5 μg ligated material, into E. coli SS320. The bacteria were plated under non-selective conditions, on 1.2% agar Terrific Broth (TB) containing Chl. Over 2.4 × 106 colonies were obtained for the DDM library and 1.7 × 104 colonies for the DSM library. DNA sequencing of individual clones served to evaluate library quality prior to selection. Sequencing was performed using the Sequenase 2.0 DNA sequencing kit (GE Healthcare) with a Li-Cor IR2 automated system, using dye-labeled primers. Sequences were analyzed using the AlignIR V2.0 software (Li-Cor).
Library selection
The DDM and DSM dimer libraries in E. coli SS320 were harvested, propagated in Terrific Broth (TB), and the DNA retransformed into E. coli BL21/pRep4 for selection. Equal aliquots of transformation mixture were plated on TB plates (+ Chl and Kan) and on M9c minimal medium plates (+ Chl and Kan) containing IPTG and TMP. Colonies were grown at 37°C for 18 h. The plasmid inserts from selected clones were sequenced. Those clones were retransformed into E. coli BL21/pRep4 and subjected to two further rounds of selection and sequencing to confirm that activity was not due to cellular acquisition of TMP resistance or unforeseen changes in their DNA sequences.
Small-scale protein overexpression and purification
An overnight culture (0.2 mL) was used to inoculate 20 mL TB (+ Kan, and Chl or Amp for DNM). Cultures were propagated at 37°C with shaking until OD600 reached 0.7. Protein expression was induced for 3 h following the addition of IPTG (1 mM final concentration). The cells were harvested by centrifugation (30 min, 3000g, 4°C). The cell pellet was resuspended in 4 mL of lysis buffer (100 mM potassium phosphate, pH adjusted to 8.0) and was disrupted by sonication on ice for 3 × 30 s with 1 min intervals. Following centrifugation at 3000g for 30 min at 4°C, 1 mL of Ni-NTA agarose resin was added to the supernatant. The mixture was gently shaken for 30 min at 4°C and transferred into a gravitation column. The resin was washed twice with 5 mL lysis buffer, and twice with the same containing 10 and 20 mM imidazole, respectively. The enzyme was eluted with elution buffer 100 mM potassium phosphate buffer, pH 8 containing 250 mM imidazole.
Large-scale protein over-expression and purification
Induction of protein expression was undertaken as above, in 500 mL of TB. The cell pellet was resuspended in 100 mM Tris-Cl buffer pH 8, and lysed by one passage through a cell disrupter (Constant Systems) adjusted to 27 kpsi. Following centrifugation (30 min, 3800g, 4°C), the supernatant was filtered over a 0.2-µm filter. Purification was performed following a two-step purification protocol on an Äkta FPLC (GE Healthcare, Piscataway, NJ) at 4°C. The supernatant was applied to a 5 mL His-trap column pre-equilibrated with 100 mM Tris-Cl, pH 8. The protein was eluted with a gradient of imidazole (0–300 mM). Fractions were analyzed for DHFR activity13 and by tricine SDS-PAGE.29 Those containing the most target protein were pooled and concentrated using an Ultra Amicon Ultracel 3.5K (Millipore). The concentrate was applied to a 1.5 × 28 cm2 Superose 12 column equilibrated in 100 mM TrisCl, pH 8 and eluted in the same buffer. The fractions were collected and analyzed for DHFR activity, by tricine SDS-PAGE and using the Bio-Rad protein assay.
Mass spectral characterization of R67 DHFR variants
Mass spectral analysis of R67 variants was achieved by HPLC-MS. Separations were performed on an 1100 LC system coupled to an ESI-TOF mass spectrometer (Agilent Technologies, Santa Clara, CA). The chromatographic column was a Poroshell 300SB-C8, 75 × 2.1 mm2, 5μ particle size (Agilent Technologies) operated at 0.4 mL min−1. The eluents used were 0.1% formic acid in water and 0.1% formic acid in acetonitrile. A linear acetonitrile gradient from 20 to 95% was used for elution. The mass spectrometer was operated in positive electrospray mode and mass spectra were acquired from m/z 110 to 3200. Deconvolution on the multiply charged species mass spectrum was performed using BioConfirm, an integrated program from MassHunter Qualitative Analysis.
Determination of multimerization state
Size exclusion chromatography was carried out at 4°C using a 1.5 × 28 cm2 Superose 12 column equilibrated in 100 mM Tris-Cl buffer pH 8. Ribonuclease A (13.7 kDa ± 15%, 10 mg mL−1), chymotrypsinogen A (25.0 kDa ± 25%; 3 mg mL−1), ovalbumin (43.0 kDa ± 15%, 7 mg mL−1) and albumin (67.0 kDa ± 10%; 7 mg mL−1) were used as standards to determine the oligomeric state of the R67 DHFR variants. The flow rate was 1.5 mg mL−1. Standard curves were produced by plotting the logarithm of the molecular mass of standards versus Kav. Kav = (VE − Vv)/(VB − Vv) where VE is the elution volume, Vv is the void volume and VB is the bed volume of the column matrix.
Kinetic characterization
Spectrophotometric enzyme activity assays were carried using a Varian Cary 100 Bio spectrophotometer. The assay buffer was MTA (50 mM MES, 100 mM Tris-Cl, 50 mM acetic acid), pH 7. Assays were performed in the presence of 50 nM TMP to inhibit any residual E. coli chromosomal DHFR. The extinction coefficients used were 28,400 M−1 cm−1 at 282 nm for DHF, 6230 M−1 cm−1 at 340 nm for NADPH and 12,300 M−1 cm−1 at 340 nm to determine product formation.13 To determine KMDHF, DHF was held at 100 µM while NADPH was varied from 0 to 100 µM. Similarly, NADPH was fixed at 100 µM to determine KMNADPH while DHF was varied from 0 to 100 µM. The data were fitted to the Michaelis–Menten equation using non-linear regression analysis with the GraphPad software. Protein concentration was determined with the BioRad assay.
Circular dichroism
Circular dichroism (CD) spectra were recorded on a Jasco J-815 spectropolarimeter with a Peltier element for temperature control. Protein solutions were prepared at a final concentration of 0.25 µg µL−1 in 50 mM potassium phosphate, pH 7. The path length of the quartz cell was 0.2 cm. Spectra were acquired at 20°C over the range 190–280 nm at a scan rate of 20 nm min−1. Three scans were averaged. Buffer background recorded under the same conditions was subtracted. A single protein preparation was tested for all proteins except for DNM, the non-mutated R67 DHFR dimer (two preparations). Thermal denaturation experiments were performed by monitoring changes in ellipticity at 215 nm, at a scan rate of 25°C h−1. The Savitzky–Golay method was used for curve smoothing.30 The Tm values were determined using the derivatives function of the Jasco Spectra Manager software using the Savitzky–Golay algorithm for determination of first order derivative, where Tm is given as the maximum of this derivative.
Thermal scanning fluorimetry shift assays
Tm values were also determined by thermally induced incorporation of SYPRO Orange into the unfolding protein, with analysis using a Q-PCR thermal cycler as previously described.24 Briefly, each combination of 10×, 5×, and 3.33× SYPRO Orange solution (Invitrogen) with 4 μM or 8 μM test protein was probed in a 96-well LightCycler plate (Sarstedt). SYPRO Orange and the protein were diluted with 50 mM potassium phosphate, pH 7, to a final volume of 20 μL per well. Controls contained SYPRO Orange in buffer. The plates were sealed using Optically Clear Sealing Tape (Sarstedt) and heated from 20 to 95°C in a LightCycler 480 apparatus (Roche) with a ramp speed of 0.04°C s−1 and 10 acquisitions/°C. Fluorescence was monitored with a CCD camera, using λexc = 483 nm and λem = 568 nm and a 1 s exposure time. Any curve showing a maximum fluorescence plateau during denaturation was excluded from the Tm calculation.
For the Tm calculations, both temperature and fluorescence data were smoothed.30 The first derivatives dFluo or dTemp were calculated using the cubic spline interpolation. The preliminary maximum was determined to obtain the half-values to the left and right of it. The linear fit for the curve outside the half-values was calculated, followed by the calculation of the average deviation from the fit. If the maximum was below the detection limit (fit value + 3 × deviation), the Tm determination was considered uncertain. The quadratic fit around the maximum was then calculated as follows to obtain Tm. The first derivative of the quadratic fit function (y-value) was set to 0 and the x-axis value (temperature) was resolved. Then, the average deviation of the curve points around the maximum from the quadratic fit was calculated. If the relative deviation was >5%, the Tm values were rejected if the corresponding maximum was below the detection limit. However, Tm values with a maximum above the detection but a relative deviation >5% were defined as uncertain.
Thermal denaturation and refolding assay
To simulate the thermal denaturation conditions of the CD experiment, the same temperature ramp was programmed into a BioRad PTC-200 PCR cycler. The total volume in each PCR tube was 70 µL with a protein concentration at 1 µg µL−1 in 50 mM potassium phosphate, pH 7. The activity of the R67 DHFR dimer variants was measured as described above for the different conditions. Condition A: Ramp to 95°C, hold for 90 min, keep at RT for 1 min and determine activity. B: Ramp to 95°C, hold for 90 min, keep at RT for 45 min and determine activity. C: Ramp to 95°C, hold for 4 min, keep at RT for 1 min and determine activity. D: Ramp to 95°C, hold for 4 min, keep at RT for 45 min and determine activity. Control: immediately determine activity. Specific activity was determined as described above.
Proteolysis by chymotrypsin
The packing of the mutated protomers was assessed by digesting with α-chymotrypsin (MP Biomedicals). Each R67 DHFR species was purified and quantitated as described above. The protein concentration was 3 µg µL−1 and chymotrypsin was added in a mass ratio of 1%. Incubation was at 4°C and samples were taken at various time points up to 24 h. The progress of the proteolysis was analyzed by tricine SDS-PAGE.
Acknowledgments
The authors thank the Regional Center for Mass Spectrometry at Université de Montréal for their help with the MS/MS experiments. They also thank Professors Joanne Turnbull and Judith Kornblatt from Concordia University for access and training for CD experiments.
Glossary
- CD
circular dichroism
- DDM
R67 DHFR mutant library, doubly mutated
- DHF
dihydrofolate
- DHFR
dihydrofolate reductase
- DNM
R67 DHFR dimer, non-mutated
- DSM
R67 DHFR mutant library, singly mutated
- THF
tetrahydrofolate
- TMP
trimethoprim.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- 1.Threlfall EJ, Ward LR, Frost J, Willshaw G. The emergence and spread of antibiotic resistance in food-borne bacteria. Int J Food Microbiol. 2000;62:1–5. doi: 10.1016/s0168-1605(00)00351-2. [DOI] [PubMed] [Google Scholar]
- 2.Matthews D, Smith S, Baccanari D, Burchall J, Oatley S, Kraut J. Crystal structure of a novel trimethoprim-resistant dihydrofolate reductase specified in Escherichia coli by R-plasmid R67. Biochemistry. 1986;15:4194–4204. doi: 10.1021/bi00363a005. [DOI] [PubMed] [Google Scholar]
- 3.White P, Rawlinson W. Current status of the aadA and dfr gene cassette families. J Antimicrob Chemother. 2001;47:495–496. doi: 10.1093/jac/47.4.495. [DOI] [PubMed] [Google Scholar]
- 4.Howell EE. Searching sequence space: two different approaches to dihydrofolate reductase catalysis. Chembiochem. 2005;6:590–600. doi: 10.1002/cbic.200400237. [DOI] [PubMed] [Google Scholar]
- 5. CCG. Inc. (2014) Molecular operating environment (MOE), 2013.08. 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7.
- 6.Stone D, Smith S. The amino acid sequence of the trimethoprim-resistant dihydrofolate reductase specified in Escherichia coli by R-plasmid R67. J Biol Chem. 1979;254:10857–10861. [PubMed] [Google Scholar]
- 7.Narayana N, Matthews D, Howell EE, Nguyen-huu X. A plasmid-encoded dihydrofolate reductase from trimethoprim-resistant bacteria has a novel D2-symmetric active site. Nat Struct Biol. 1995;2:1018–1025. doi: 10.1038/nsb1195-1018. [DOI] [PubMed] [Google Scholar]
- 8.Yachnin BJ, Colin DY, Volpato JP, Ebert M, Pelletier JN, Berghuis AM. Novel crystallization conditions for tandem variant R67 DHFR yield a wild-type crystal structure. Acta Cryst. 2011;F67:1316–1322. doi: 10.1107/S1744309111030417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strader MB, Smiley RD, Stinnett LG, VerBerkmoes NC, Howell EE. Role of S65, Q67, I68, and Y69 residues in homotetrameric R67 dihydrofolate reductase. Biochemistry. 2001;40:11344–11352. doi: 10.1021/bi0110544. [DOI] [PubMed] [Google Scholar]
- 10.Nichols R, Weaver C, Eisenstein E, Blakley RL, Appleman J, Huang TH, Howell EE. Titration of histidine 62 in R67 dihydrofolate reductase is linked to a tetramer <-> two-dimer equilibrium. Biochemistry. 1993;32:1695–1706. doi: 10.1021/bi00058a002. [DOI] [PubMed] [Google Scholar]
- 11.Park H, Bradrick TD, Howell EE. A glutamine 67–> histidine mutation in homotetrameric R67 dihydrofolate reductase results in four mutations per single active site pore and causes substantial substrate and cofactor inhibition. Protein Eng. 1997;10:1415–1424. doi: 10.1093/protein/10.12.1415. [DOI] [PubMed] [Google Scholar]
- 12.Bradrick TD, Beechem JM, Howell EE. Unusual binding stoichiometries and cooperativity are observed during binary and ternary complex formation in the single active pore of R67 dihydrofolate reductase, a D2 symmetric protein. Biochemistry. 1996;35:11414–11424. doi: 10.1021/bi960205d. [DOI] [PubMed] [Google Scholar]
- 13.Schmitzer AR, Lépine F, Pelletier JN. Combinatorial exploration of the catalytic site of a drug-resistant dihydrofolate reductase: creating alternative functional configurations. Protein Eng Des Sel. 2004;17:809–819. doi: 10.1093/protein/gzh090. [DOI] [PubMed] [Google Scholar]
- 14.Strader MB, Chopra S, Jackson M, Smiley RD, Stinnett L, Wu J, Howell EE. Defining the binding site of homotetrameric R67 dihydrofolate reductase and correlating binding enthalpy with catalysis. Biochemistry. 2004;43:7403–7412. doi: 10.1021/bi049646k. [DOI] [PubMed] [Google Scholar]
- 15.Amyes SG, Smith JT. The purification and properties of the trimethoprim-resistant dihydrofolate reductase mediated by the R-factor, R388. Eur J Biochem. 1976;61:597–603. doi: 10.1111/j.1432-1033.1976.tb10055.x. [DOI] [PubMed] [Google Scholar]
- 16.Deng H, Callender R, Howell EE. Vibrational structure of dihydrofolate bound to R67 dihydrofolate reductase. J Biol Chem. 2001;276:48956–48960. doi: 10.1074/jbc.M105107200. [DOI] [PubMed] [Google Scholar]
- 17.Krahn JM, Jackson MR, DeRose EF, Howell EE, London RE. Crystal structure of a type II dihydrofolate reductase catalytic ternary complex. Biochemistry. 2007;46:14878–14888. doi: 10.1021/bi701532r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng J, Grubbs J, Dave A, Goswami S, Horner CG, Howell EE. Radical redesign of a tandem array of four R67 dihydrofolate reductase genes yields a functional, folded protein possessing 45 substitutions. Biochemistry. 2010;49:7384–7392. doi: 10.1021/bi1005943. [DOI] [PubMed] [Google Scholar]
- 19.Feng J, Goswami S, Howell EE. R67, the other dihydrofolate reductase: rational design of an alternate active site configuration. Biochemistry. 2008;47:555–565. doi: 10.1021/bi701455x. [DOI] [PubMed] [Google Scholar]
- 20.Martinez M, Pezo V, Marlière P, Wain-Hobson S. Exploring the functional robustness of an enzyme by in vitro evolution. EMBO J. 1996;15:1203–1210. [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuang P, Yin M, Holland J, Peterson CB, Howell EE. Artificial duplication of the R67 dihydrofolate reductase gene to create protein asymmetry: effects on protein activity and folding. J Biol Chem. 1993;268:22672–22679. [PubMed] [Google Scholar]
- 22.Dam J, Rose T, Goldberg ME, Blondel A. Complementation between dimeric mutants as a probe of dimer-dimer interactions in tetrameric dihydrofolate reductase encoded by R67 plasmid of E. coli. J Mol Biol. 2000;302:235–250. doi: 10.1006/jmbi.2000.4051. [DOI] [PubMed] [Google Scholar]
- 23.Miller GP, Benkovic SJ. Stretching exercises? Flexibility in dihydrofolate reductase catalysis. Chem Biol. 1998;5:R105–R113. doi: 10.1016/s1074-5521(98)90616-0. [DOI] [PubMed] [Google Scholar]
- 24.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 25.Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci USA. 2006;103:15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodrigues JV, Prosinecki V, Marrucho I, Rebelo LPN, Gomes CM. Protein stability in an ionic liquid milieu: on the use of differential scanning fluorimetry. Phys Chem Chem Phys. 2011;13:13614–13616. doi: 10.1039/c1cp21187k. [DOI] [PubMed] [Google Scholar]
- 27.Bastien D, Ebert MCCJC, Forge D, Toulouse J, Kadnikova N, Perron F, Mayence A, Huang TL, Vanden Eynde JJ, Pelletier JN. Fragment-based design of symmetrical bis-benzimidazoles as selective inhibitors of the trimethoprim-resistant, type II R67 dihydrofolate reductase. J Med Chem. 2012;55:3182–3192. doi: 10.1021/jm201645r. [DOI] [PubMed] [Google Scholar]
- 28.Blakley R. Crystalline dihydropteroylglutamic acid. Nature. 1960;188:231–232. [Google Scholar]
- 29.Schägger H. Tricine-SDS-PAGE. Nat Protoc. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 30.Savitzky A, Golay MJE. Smoothing and differentiation of data by simplified least squares procedures. Anal Chem. 1964;36:1627–1639. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information