

Abstract
We employed a minimalist approach for design of an allosterically controlled retroaldolase. Introduction of a single lysine residue into the nonenzymatic protein calmodulin led to a 15,000-fold increase in the second order rate constant for retroaldol reaction with methodol as a substrate. The resulting catalyst AlleyCatR is active enough for subsequent directed evolution in crude cell bacterial lysates. AlleyCatR's activity is allosterically regulated by Ca2+ ions. No catalysis is observed in the absence of the metal ion. The increase in catalytic activity originates from the hydrophobic interaction of the substrate (∼2000-fold) and the change in the apparent pKa of the active lysine residue.
Keywords: protein design, aldolase, enzyme catalysis, metalloproteins, calmodulin
Introduction
Nature has created a variety of effective enzymes that catalyze complex chemical reactions with remarkable stereo- and regioselectivity in the ultimately “green” solvent—water. After decades of concerted efforts to understand the principles of enzymatic activities we are starting to create new protein catalysts for (un)natural reactions. Apart from the inherent practical value of obtaining new catalysts with improved properties, protein design provides fundamental knowledge about enzymatic function from its successes and failures. De novo design of proteins has been very successful from a structural perspective, but the goal of designing function remains much more elusive. Indeed, current state-of-the-art methodologies can only provide a reasonable starting point for subsequent directed evolution.1 Currently, approaches to designing protein function can be broadly placed in three broad categories: (1) Theozyme-based,2 which relies on placing a finely tuned, theoretically created transition state—“theozyme” into a protein backbone; (2) Iterative,3 which combines the theozyme approach with rational tuning of the active site to achieve most impressive catalytic efficiency; (3) Minimalistic,4–6 which focuses on the general feasibility of catalysis without trying to match all possible interactions exactly. While catalytic efficiency of minimalistic designs is generally lower than that of more sophisticated methods, this approach is very computationally inexpensive and it allows for straightforward analysis of the contributions by various design components (e.g., substrate binding, active residue functional tuning, etc.). Turnover numbers of minimalist designs can be improved by directed evolution to be on par with the best examples of the proteins obtained by other approaches.7
Mechanistically, retroaldolases represent the most complex designed enzymes to date.8,9 The reaction they catalyze proceeds through a covalent iminium intermediate normally arranged via the side chain of a lysine residue followed by hydrolysis (Fig. 1). Retroaldolase assays commonly use substrates containing functional groups that provide distinct absorbance and/or fluorescent profiles. Methodol (4-hydroxy-4-(6-methoxy-2-naphthyl)-2-butanone) is one of the most commonly used model substrates for this reaction; it has been extensively benchmarked in a variety of different systems ranging from micelles10 and peptides11,12 to proteins8,9,13,14 and antibodies.15,16 Not unexpectedly for such a complex task, computational protein design produces retroaldolases of modest efficiency with kcat/KM values ranging from 0.0041 to 0.74 M−1 s−1 and rate acceleration (kcat/kuncat) of up to 2.4 × 104. Nonetheless, even designs with relatively low activity do serve as suitable starting points for successful directed evolution to yield impressive catalysts with catalytic efficiency reaching 850 M−1s−1.8 Here we present our results on using the minimalist approach to create allosterically regulated retroaldolases.
Figure 1.
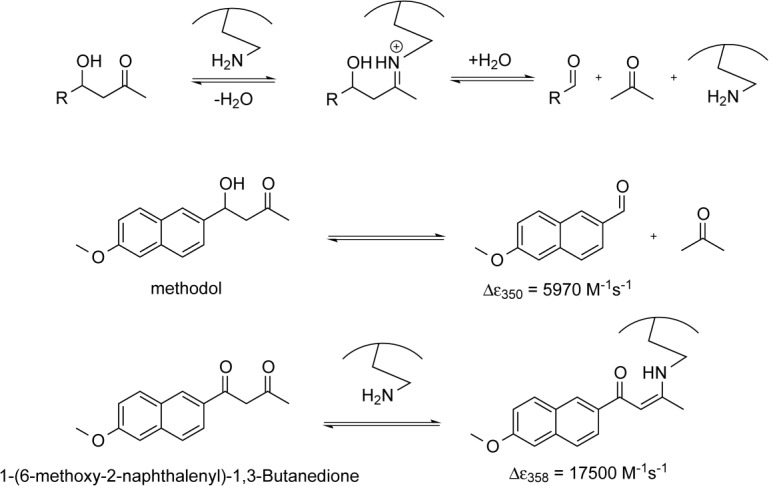
General scheme of the lysine-catalyzed retroaldol reaction pathway (top). Cleavage of 4-hydroxy-4-(6-methoxy-2-naphthyl)−2-butanone (methodol) to 6-methoxy-2-naphthaldehyde and acetone (middle). Formation of the enamine between the catalyst and diketone 1-(6-methoxy-2-naphthalenyl)−1,3-butanedione (bottom).
Results
The bare minimum requirements for a successful retroaldolase include a substrate binding pocket and a lysine residue. Importantly, the pKa of the ε-amino group of the lysine residue needs to be properly modulated to form the iminium intermediate at neutral or near neutral pH values. As most of the substrates for the retroaldol reaction are hydrophobic, introducing a lysine residue in (or near) the hydrophobic binding pocket will likely decrease the side chain pKa, effectively killing two birds with one stone. This requirement poses stringent constraints on the protein scaffold; however, the protein has to have enough stability to withstand a highly disruptive introduction of a charged residue in the hydrophobic core.
Calmodulin (CaM) is one of the most abundant proteins in eukaryotic organisms. This nonenzymatic calcium-binding messenger protein is capable of binding to a variety of different substrates through its hydrophobic pocket, making it a suitable scaffold to accommodate substrates for various chemical reactions. Moreover, CaM is allosterically regulated—binding of calcium ions to EF-hands of this protein results in a large conformational change that exposes the protein's hydrophobic region to solvent. CaM is a very stable protein in the metal bound state, therefore introduction of a number of charged groups in the hydrophobic pocket can be tolerated without noticeable unfolding.7 Additionally, calmodulin can be reprogrammed to allosterically respond to metal ions other than calcium.17 Previously, we have successfully used calmodulin to create allosterically regulated catalysts of Kemp elimination. We have shown that even a single Phe to Glu mutation conferred catalytic activity onto this nonenzymatic protein.5 The resulting 74-residue long protein catalyst, nicknamed AlleyCat, was further improved using seven rounds of directed evolution to achieve turnover numbers on par with those of the enzymes designed by the best computational algorithms and catalytic antibodies.7
We set to explore whether calmodulin could adopt the ability to catalyze retroaldol reaction using methodol as a substrate. Using our minimalist approach, we first performed a docking experiment with AutoDock Vina of the C-terminal domain of calmodulin to ensure that its relatively featureless hydrophobic cavity could indeed accommodate the Michaelis complex [Fig. 2(A)]. The docking experiment indeed showed that the substrate associates with the protein and seven residues (F92, L105, V108, M109, M124, A128, F141) that are in direct van der Waals contact with the docked substrate were identified as possible locations for a catalytic lysine residue [Fig. 2(B)]. Next we have computationally varied residues in the positions identified above to lysine and calculated the Rosetta score for the mutants, lysine rotamers were allowed to vary. All of the seven mutations were well tolerated by the scaffold [Fig. 2(C)]. We then attempted to predict whether these mutants may produce an effective transition state for imine formation. To establish feasibility of catalysis we assumed that the terminal amine of the lysine residue must be no more than 3 Å away from the carbonyl carbon and the Namine–Ccarbonyl-Ocarbonyl angle must be within the range between 90° and 150°. Transition state possibility was clear for some mutants [Fig. 2(D)], but due to a large number of possible substrate poses and lysine rotamers we were not able to reliably rank the mutants in order of expected activity. Therefore, we prepared all above mentioned mutants and screened them for ability to catalyze the retroaldol reaction. The screening results are shown in Figure 3.
Figure 2.
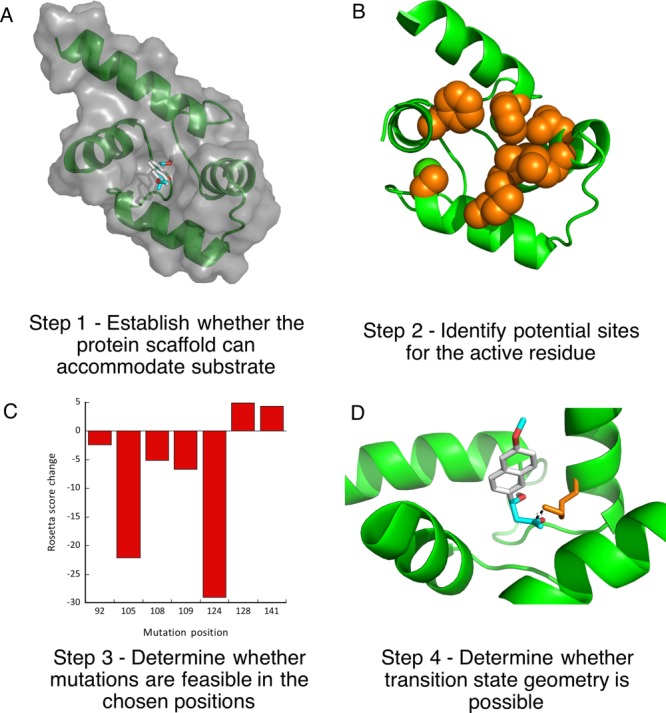
Overview of the design.
Figure 3.
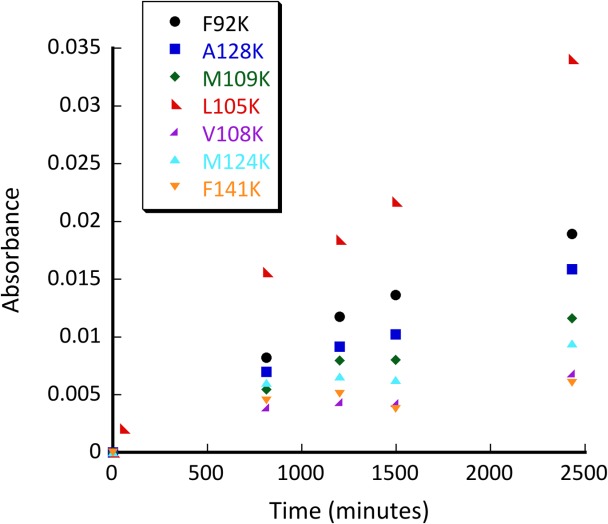
Initial screen for retroaldolase activity exerted by various calmodulin mutants: F92K (black circle), A128K (blue square), M109K (green diamond), L105K (red left-facing triangle), V108K (purple right facing triangle), M124K (M124K bright blue triangle), F141K (orange downward facing triangle). Conditions: pH 8.0; 50 µM methodol; 50 μM protein.
Several mutants have shown substantial retroaldol activity, with L105K clearly being the best. We have chosen the two most active proteins (CaM L105K and CaM F92K) for in depth characterization. The mutations have not substantially disrupted the overall protein fold as evidenced by the circular dichroism data (Fig. 4). The mutants maintained well-defined alpha helical confirmation essentially identical to that of the CaM scaffold. Michaelis-Menten graphs (Fig. 5) have shown no saturation at the substrate concentrations studied, thus individual kcat and KM values cannot be established. The enzymatic efficiency (kcat/KM) values are given in Table I. Activity of CaM L105K reached 0.038 M−1 s−1, on par with some of the more sophisticated designs. Deprotonation of the side chain amino group of the active residue made it more nucleophilic and improved its ability to form an iminium intermediate with methodol. The pH profiles of the reaction were consistent with a large decrease in effective pKa of the lysine residue (from 10.6 in free amino acid to 7.6 for CaM L105K) resulting from its location in the hydrophobic pocket of calmodulin (Fig. 6). In all cases we have confirmed product formation and kinetic parameters using HPLC to exclude the possibility of side reactions (see Supporting Information for more detail).
Figure 4.

CD spectra of CaM (A), CaMWN (B), CaM L105K (C) and CaM F92K (D). Buffer conditions: 2 mM HEPES (pH 7.5); 2 mM CaCl2; 30 mM NaCl; 30 µM protein.
Figure 5.
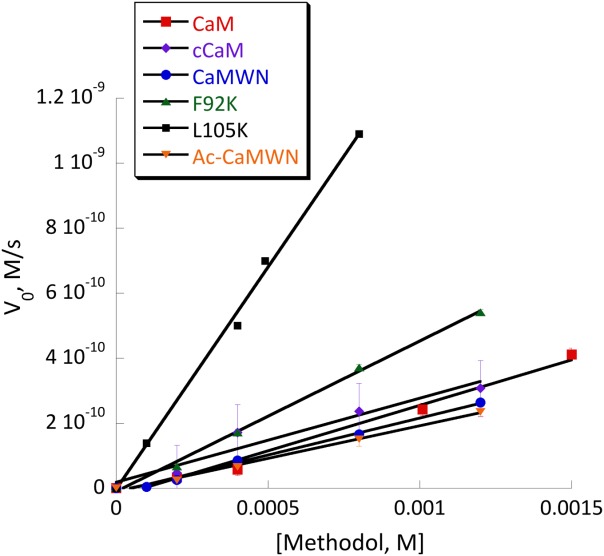
Michaelis-Menten plot of retro-aldol activity catalyzed by CaM (red squares), cCaM (purple diamonds), cCaMWN (blue circles), CaM F92K (green triangles), CaM L105K (black squares) and Ac-cCaMWN (orange triangles). Conditions: 20 mM HEPES (pH 7); 100 mM NaCl; 10 mM CaCl2; substrate final concentration: 0.1–1.5 mM; 40 μM protein.
Table I.
Catalytic Efficiencies of Various Calmodulin Derivatives in Retroaldol Reaction Using Methodol as a Substrate at pH 7.5
| Protein | (kcat/KM), M−1 s−1 |
|---|---|
| CaM | 0.006 ± 0.001 |
| cCaM | 0.007 ± 0.002 |
| CaMWN | 0.005 ± 0.001 |
| Ac-CaMWN | 0.004 ± 0.002 |
| CaM F92K | 0.012 ± 0.001 |
| CaM L105K | 0.038 ± 0.002 |
Figure 6.
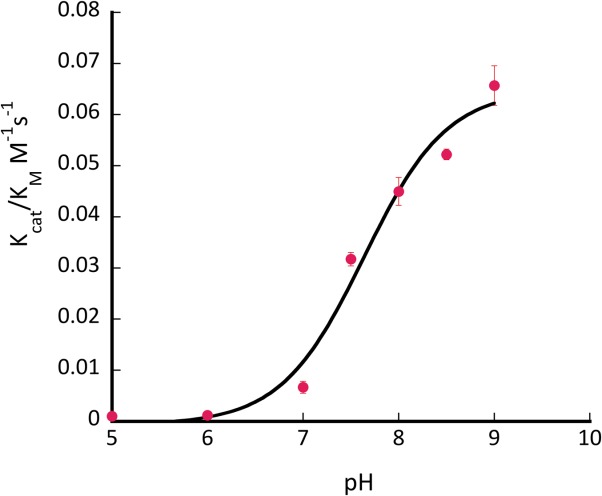
pH profile of activity of CaM L105K. Conditions: pH 6−9.5; 0.75 mM methodol; 40 μM protein.
To test allosteric regulation of the resulting catalysts, we have conducted kinetic assays both in the presence and in the absence of calcium ions (Fig. 7). F92K and L105K were active only in the presence of Ca2+ - removing the metal ion resulted in at least a 100-fold drop in catalytic efficiency. This result confirmed that the original allosteric nature of the protein was still preserved in the catalysts, and it provided an important reference point to ensure that the catalytic activity could be only supported by the open conformation of calmodulin consistent with the design.
Figure 7.
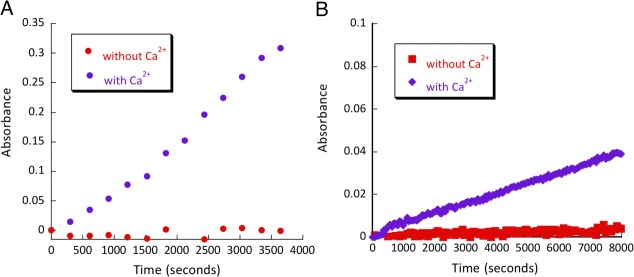
Calcium dependence assay of (A) CaM L105K calcium (purple diamonds), CaM L105K no calcium (red squares) and (B) L105K C-terminal calcium (purple diamonds), L105K C-terminal no calcium (red squares), 80 µM of each protein.
The results of this control experiment were a bit puzzling—while no activity over the background rate was observed in the absence of calcium, every single mutant produced in our study had some activity in the presence of the metal ion. This prompted us to investigate the ability of calmodulin itself to catalyze retroaldol reaction. Indeed, we were surprised to discover that CaM in the presence of calcium is capable of facilitating retroaldol reaction in methodol. In the absence of calcium, no activity above background was observed. The catalytic efficiency of this reaction was small, but it is similar to that of some of the successful computational designs. To confirm this observation, we decided to mutate all lysine and histidine residues in CaM to exclude adventitious reactivity. To simplify cloning and analysis we focused on the C-terminal domain of CaM (cCaM) only. cCaM also catalyzed retroaldol reaction with a catalytic efficiency similar to that of full CaM (Table I). The pH profile of cCaM's activity (Supporting Information Figure S1) showed no well-defined maxima or minima, which suggested no protonatable residues are involved in this reaction. cCaM has four lysine residues located mostly in the metal binding loops. We had also decided to mutate the only histidine in this protein to exclude any nucleophilicity resulting from the side chain residues. From aligning calmodulins from various organisms we had identified five naturally occurring mutations known to maintain the protein's function: K77A, K94R, H107I, K115N, K148L. The resulting protein nicknamed CaMWN (Calmodulin Without Nucleophiles) was recombinantly expressed and characterized. As expected, its CD signature was indicative of the typical calmodulin fold (Fig. 4). CaMWN also has shown above background activity (kcat/KM = 0.005 ± 0.001 M−1 s−1) in retroaldol reaction consistent with the results obtained for native CaM. Additionally, to exclude the possibility that the N-terminal amine is involved in retro-aldol activity we acetylated CaMWN's N-terminus using sulfo-NHS-acetate. The resulting protein, Ac-CaMWN, has also shown above background activity (kcat/KM = 0.004 ± 0.002 M−1 s−1, Table I). This suggests that CaM's activity was due to the hydrophobic pocket alone and not due to naturally occurring lysines, histidine, or the N-terminal amine.
To confirm that the retroaldol reaction proceeds via the mechanism proposed in Figure 1, we have tested the reactivity of CaM L105K and CaM toward 1-(6-methoxy-2-naphthalenyl)−1,3-butanedione, the diketone analog of methodol. This molecule is capable of forming the imine intermediate with a distinct spectroscopic feature but cannot undergo the subsequent retroaldol reaction (Fig. 1). The results of this experiment are shown in Figure 8. CaM L105K readily formed the iminium intermediate, whereas CaM did not react with the diketone. These results have shown that the observed increase in the background rate of methodol by CaM could be attributed solely to its hydrophobic association with the protein.
Figure 8.
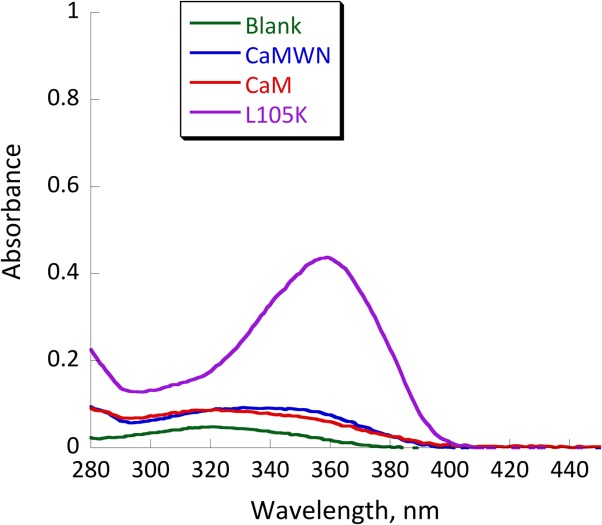
UV-vis spectra of 25 µM 1-(6-methoxy-2-naphthalenyl)−1,3-butanedione (the diketone analog of methodol) in buffer (green), with 25 µM CaM (red trace) and 25 µM CaM L105K (purple trace). Conditions: 20 mM HEPES (pH 7); 100 mM NaCl; 10 mM CaCl2; equilibration time, 24 h.
As discussed above, catalytic efficiency of computationally designed enzymes was almost universally improved by subsequent directed evolution. Successful directed evolution experiments require careful selection of methods to generate and screen mutant libraries. Diversity of libraries can be generated through saturation mutagenesis, error-prone PCR, gene shuffling, as well as using more rational predictive methods. Strategic combination of various methods allows for creation of focused libraries with higher probability of success.18 High throughput screening in crude cell lysate can be utilized to screen thousands of mutants in bacterial lysates to identify variants with improved activity. We have tested whether CaM F92K (the less efficient of the two best mutants) had enough retroaldolase activity to be screened in E. coli crude cell lysate—the necessary condition for directed evolution. Fluorescence of the crude cell lysates containing the CaM F92K variant was substantially higher than that of the lysates of the cells with no calmodulin-expressing plasmid or the lysates producing CaM (Fig. 9). These results confirm that the retroaldolases designed using the minimalist approach could, in principle, be further improved by screening the fluorescence of crude cell lysates from a mutant library.
Figure 9.
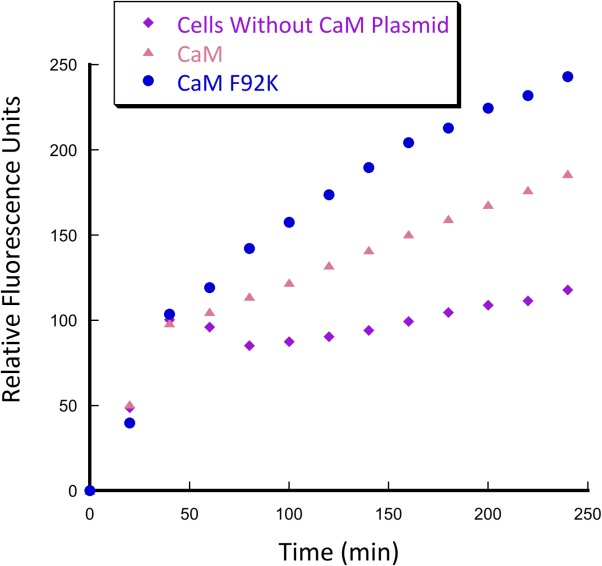
Crude cell lysate screening for retroaldol activity of BL21 (DE3) pLysS E. coli cells containing plasmids encoding CaM and CaM F92K genes. Conditions: 20 mM HEPES (pH 7.5); 100 mM NaCl; 10 mM CaCl2; 0.2% Triton X-100; 700 µM substrate.
Discussion
Critical evaluation of recent successes in computational protein design has shown that while impressive results have been achieved in certain cases, there is substantial room for improvement.1 It is painfully clear that computational design alone has a hard time getting it right from the first try. Nowhere is it more obvious than in the case of retroaldol reaction—despite much effort, enzymatic efficiencies of the designed proteins are many orders of magnitude below that of their natural counterparts. Still there are many reasons to celebrate achievements in the field. Many successful and failed attempts of designing proteins greatly improved our fundamental understanding of catalysis and provided insight into the evolution of enzymatic function.19 Moreover, essentially any computationally designed enzyme could be further improved by subsequent directed evolution. With an advent of new methodology for creation of focused libraries20,21 and advanced robotic methods for high-throughput screening, chances of substantial improvement of the starting designs are getting higher.
It is also becoming clear that relying on the transition state alone for designing function is not sufficient. All parts of the enzymatic cycle, from the Michaelis product formation all the way to product release, need to be considered.22 We need to account for multiple factors that effect proteins: the pKa of surrounding residues, thermodynamic stability of the fold, and protein dynamics. Explicit consideration of all these parameters using sophisticated calculations quickly becomes prohibitively computationally expensive, thus simplifications are necessary.
Minimalist approach is considering only a handful of various parameters at once to allow for direct evaluation of contribution of the basic physico-chemical parameters such as pKa of the active residue and hydrophobic interactions of the substrate to catalysis. Using a nonenzymatic protein as a scaffold for design can be particularly instructive. Calmodulin possesses no known enzymatic activity, thus its hydrophobic cavity is truly featureless in terms of catalyzing chemical reactions. Moreover, CaM is allosterically regulated providing not only a useful practical feature but an important reference point to ensure that the designed activity indeed comes from the designed interactions.
Placing a single lysine residue into calmodulin confers retroaldolase activity on this nonenzymatic protein. The enzymatic efficiency of the best mutant, CaM L105K nicknamed AlleyCatR, is 0.038 M−1 s−1, and is on par with that of other designed retroaldolases.9,13,23 This value represents a 104 improvement in second order rate constant compared to lysine in solution and the estimated ratio of kcat/kuncat is also >104. Interestingly, most of the rate acceleration can be attributed to hydrophobic association of the substrate with protein based on the activity of native CaM.
Our results are in good agreement with the analysis of Lasilla et al.24 who conducted a detailed study on the factors contributing to catalytic activity of a designed retroaldolase RA61. By varying substrate hydrophobicity Lasilla et al. were able to establish that the naphthalene ring association with the protein accounts for 500-fold improvement of the rate, however contribution of the desolvation and full substrate binding were not established. Allosteric regulation of calmodulin allows for a more direct measurement of rate acceleration provided by substrate binding in the absence of any designed interactions.
The observed second order rate constant of ∼ 0.005 M−1 s−1 for unmutated calmodulin suggests that increase in the rate of the retroaldol reaction due to non-specific interactions can reach about 2000-fold. This value provides an important reference point to evaluate performance of the existing and future retroaldolases. On the one hand it is humbling that some of the successful designs show activity on the order of 0.005 M−1 s−1. On the other hand it is fascinating that designs with modest activity can be improved by several orders of magnitude after just a few rounds of directed evolution. We expect that computationally inexpensive minimalist approach will help expand the repertoire of useful protein catalysts.
It is also interesting to compare our results to other examples of minimalist approach that employ peptides for retroaldol reaction11 and mechanistically similar oxaloacetate decarboxylation.25,26 In both cases introduction of multiple lysine residues into helical bundles formed by either genetically encoded or β-peptides led to an increase in catalytic activity of 500- to 3000-fold above the background rate. Substantially higher relative improvement observed in AlleyCatR can be attributed to its ability to bind substrate and position the catalytic residue in a more favorable geometry; properties that are more difficult to achieve using relatively featureless peptidic catalysts.
Finally, our results may shed some light on how proteins evolve to assume new functionality. Catalytic promiscuity of proteins has long been implicated as one of the major paths for the creation of enzymes.19 We demonstrated that even a small nonenzymatic protein can adopt various catalytic activities (Kemp eliminase, retroaldolase, esterase) solely due to one amino acid mutation, which is very likely to occur in nature. This may indicate existence of “hidden” catalytic promiscuity, where catalytic activity not present in the original scaffold can be substantially increased as a result of a point mutation and subsequently evolutionary improved.
Materials and Methods
Chemicals and reagents
Reagents and buffers were purchased from BioBasic, ChemIpex, VWR and Santa Cruz Biotechnology. DNA oligonucleotides were purchased from Integrated DNA Technologies. Enzymes for cloning were purchased from Promega and New England Biolabs. TEV protease was expressed and purified according to literature procedure.27 4-hydroxy-4-(6-methoxy-2-naphthyl)−2-butanone (methodol) and 1-(6-methoxy-2-naphthalenyl)−1,3-butanedione were synthesized according to literature procedures.28,29
Computational studies
A high resolution crystal structure of calmodulin (PDB entry 1CLL)30 served as a starting point of the design. The side chain placement was computationally optimized with fixed backbone (fixbb protocol) using Rosetta31 prior to subsequent computational work. The structures of the R- and S-isomers of methodol were drawn with MarvinSketch, their geometry was optimized with UCSF Chimera package.32 The final preparation for docking was done with ADT 1.5.6. AutoDock Vina33 was used to dock methodol in the C-terminal domain of the obtained CaM model. The docking parameters were systematically varied in independent runs. Forty poses with the best score were selected determination of the possible mutation sites. Residues in direct contact with the docked substrate (F92, L105, V108, M109, M124, A128, F141) were selected for computational mutagenesis. Mutations were computationally introduced by changing the amino acid identity in positions 92, 105, 108, 109, 124, 128, 141 to lysine and side chain placement was again computationally optimized with fixed backbone (fixbb protocol) using Rosetta, rotamers were allowed to vary. The Rosetta score for the mutants were compared to that of the original structure and reported in Figure 2. To test for the possibility of transition state geometry, the subtsrate was docked again into the computationally generated models of lysine mutants as described above. To establish feasibility of catalysis we assumed that the terminal amine of the lysine residue must be no more than 3 Å away from the carbonyl carbon and the Namine–Ccarbonyl-Ocarbonyl angle must be within the range between 90° and 150° in the docked structures.
Cloning and mutagenesis
All genes were cloned into pEXP5-NT expression vector. The vector contains an N-terminal His6-tag and a Tobacco etch virus (TEV) protease recognition site (ENLYFQ/S). Site directed mutagenesis was done with Pfu Turbo polymerase using manufacturer's protocols. The sequences for all genes are given in Table S1, Supporting Information.
Protein expression and purification
The vectors containing the genes of interest were transformed into E. coli BL21(DE3) pLysS cells and expressed using isopropyl β-D-1-thiogalactopyranoside (IPTG) induction in Luria Bertani (LB) medium. The cells were sonicated and centrifuged in a buffer containing 25 mM Tris, 20 mM imidazole, 10 mM CaCl2, and 300 mM NaCl (pH 8.0) on ice with protease inhibitor PMSF (phenylmethylsulfonyl fluoride) added. Lysed contents were centrifuged at 20000g at 4°C for 1 h and the supernatant was applied onto a Ni-NTA column (Clontech). The proteins were eluted with buffer containing 25 mM TRIS, 500 mM imidazole, 10 mM CaCl2, and 300 mM NaCl (pH 8.0) and then subjected to TEV digestion to remove the purification tags. After nickel column chromatography, proteins were exchanged into a buffer containing 75 mM NaCl and 50 mM TRIS (pH 8) with a BioRad 10 DG desalting column. TEV protease was added to 6 mL of protein using 1:10 OD280(TEV): OD280(protein) ratio in a buffer containing 1 mM of dithiothreitol (DTT) and 0.5 mM of ethylenediaminetetraacetic acid (EDTA). The solution was mixed, sterilized using a 0.22-μm filter, and incubated overnight at 34°C. Following the digest, DTT, EDTA, and low molecular weight compounds were removed with a BioRad 10 DG desalting column. The obtained solution was loaded on Ni-NTA column to remove cleaved affinity tags and then exchanged into 25 mM HEPES (pH 7.5), 10 mM CaCl2, 100 mM NaCl buffer. SDS-PAGE gels of the purified proteins are given in Supporting Information Figures S2 and S3. The N-terminus of CaMWN was aceylated to produce Ac-CaMWN using Sulfo-NHS-Acetate (ProteoChem). 20-fold molar excess Sulfo-NHS-Acetate was added to CaMWN in buffer containing 100 mM sodium phosphate (pH 7.4), 100 mM NaCl and the reaction was allowed to proceed for 2 h at room temperature. The NHS ester excess was removed using a BioRad 10 DG desalting column. Acetylation of CaMWN was confirmed by MALDI-TOF.
Kinetic assays
Absorbance measurements were done on an Agilent UV/VIS model 8453 spectrophotometer. The absorbance of the product was measured at 350 nm, each measurement was done in triplicate. For Michaelis-Menten experiments methodol stock solution in acetonitrile was added to protein solution (40 µM) in buffer containing 25 mM HEPES (pH 7.5), 10 mM CaCl2, 100 mM NaCl. The resulting reaction mixture contained 3.5% acetonitrile and the substrate concentration ranged from 50 μM to 1.5 mM. Kinetic parameters for proteins (kcat/KM) were obtained by fitting the data to linear portion the Michaelis-Menten equation v0 = (kcat/KM)[E]0[S]0.
For pH profile studies the following buffers were used at the 25 mM concentration: citrate (pH 5.0), MES (pH 6.0 and 6.5), HEPES (pH 7.0 and 7.5), TRIS (pH 8.0 and 8.5) and TAPS (pH 9.0). The protein concentration was 40 μM, the substrate concentration was 700 µM, the reaction mixture contained 1.4% acetonitrile. The apparent pKa value of the lysine was obtained from fitting the pH dependence data to following equation: kcat/KM = (kcat/KM)protonated+(kcat/KM)deprotonated ×10−pKa/(10−pH + 10−pKa).
The initial screen of mutants as well as the calcium dependence studies were done on a Thermo Labsystems Multiskan Spectrum platereader using 96-well plates monitoring absorbance at 350 nm, in 200-μL reaction volumes. The protein concentration was 80 μM and the methodol concentration was 700 μM in the buffer containing 20 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.5. In the calcium dependence experiment the “no-calcium” buffer contained no added calcium chloride and was treated with EDTA to a final concentration of 50 μM to ensure complete removal of metal ions.
Circular dichroism spectroscopy
CD data were acquired on a Jasco J-715 CD spectrometer using a step scan mode averaging over three runs. A quartz cuvette with a 1 mm path length was used for all expriments. The protein concentration was kept at 25 μM in a buffer containing 2 mM HEPES (pH 7.5), 2 mM CaCl2, and 30 mM NaCl. Care was taken that the sample absorbance never exceeded 1.5 at all wavelengths to produce reliable MRE values.
High performance liquid chromatography
Chromatographic separation was performed on a Shimadzu Prominence UFLC system (model 27625) using an Agilent reverse phase C18 (5 µM; 4.6 × 150 mm) HPLC column monitoring absorbance at 254 nm and 280 nm. The gradient run started with 100% Solvent A and was as follows; 0% Solvent B at 1.00 min, 100% Solvent B at 11.5 min, 0% Solvent B at 12 min and was performed at 1.5 mL/min flow rate. Solvent A was water containing 0.1% trifluoroacetic acid and Solvent B was 90%/9.9%/0.1% acetonitrile/water/trifluoroacetic acid. A representative trace is given in the Supporting Information (Fig. S4). Integrated absorbance of the substrate at 280 nm and the product was corrected for different extinction coefficients.
Crude cell lysate screening
To test the ability of CaM F92K and CaM to catalyze retroaldol reaction, plasmids containing the corresponding genes were transformed into E. coli BL21 (DE3) pLysS cells. Additionally, BL21 (DE3) pLysS cells grown under the chloramphenicol selection originating from the pLysS plasmid were used as a background reference. Individual colonies were cultured into 20 mL of Luria Bertani (LB) containing 100 μg/mL of ampicillin (or 37 μg/mL chloramphenicol) for 6–7 h at 37°C. Then, 200 mL of LB with the appropriate antibiotic were inoculated by 2 mL of the seed culture and grown to an OD600 of ∼0.5 at 37°C; protein expression was induced by addition of 1 mM IPTG (Isopropyl β-d−1-thiogalactopyranoside) and the cultures were grown for 2 h at 30°C. The cells were harvested by centrifugation and lysed with a buffer containing 20 mM HEPES, 10 mM CaCl2, 100 mM NaCl, 0.2% triton-X, pH 7.5. Methodol (to the final concentration of 700 μM) was added to the lysate and the reaction was followed by monitoring fluorescence on a Molecular Devices Gemini fluorescence plate-reader; excitation wavelength, 330 nm, and emission wavelength, 458 nm.
Acknowledgments
The authors thank Prof. Robert P. Doyle for the use of his CD instrument.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Korendovych IV, DeGrado WF. Catalytic efficiency of designed catalytic proteins. Curr Opin Struct Biol. 2014;27:113–121. doi: 10.1016/j.sbi.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tantillo DJ, Chen J, Houk KN. Theozymes and compuzymes: theoretical models for biological catalysis. Curr Opin Chem Biol. 1998;2:743–750. doi: 10.1016/s1367-5931(98)80112-9. [DOI] [PubMed] [Google Scholar]
- 3.Privett HK, Kiss G, Lee TM, Blomberg R, Chica RA, Thomas LM, Hilvert D, Houk KN, Mayo SL. Iterative approach to computational enzyme design. Proc Natl Acad Sci USA. 2012;109:3790–3795. doi: 10.1073/pnas.1118082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeGrado WF, Wasserman ZR, Lear JD. Protein design, a minimalist approach. Science. 1989;243:622–628. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 5.Korendovych IV, Kulp DW, Wu Y, Cheng H, Roder H, DeGrado WF. Design of a switchable eliminase. Proc Natl Acad Sci USA. 2011;108:6823–6827. doi: 10.1073/pnas.1018191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rufo CM, Moroz YS, Moroz OV, Stohr J, Smith TA, Hu X, DeGrado WF, Korendovych IV. Short peptides self-assemble to produce catalytic amyloids. Nat Chem. 2014;6:303–309. doi: 10.1038/nchem.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moroz OV, Moroz YS, Wu Y, Olsen AB, Cheng H, Mack KL, McLaughlin JM, Raymond EA, Zhezherya K, Roder H, Korendovych IV. A single mutation in a regulatory protein produces evolvable allosterically regulated catalyst of nonnatural reaction. Angew Chem Int Ed Engl. 2013;52:6246–6249. doi: 10.1002/anie.201302339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giger L, Caner S, Obexer R, Kast P, Baker D, Ban N, Hilvert D. Evolution of a designed retro-aldolase leads to complete active site remodeling. Nat Chem Biol. 2013;9:494–498. doi: 10.1038/nchembio.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, III, Hilvert D, Houk KN, Stoddard BL, Baker D. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt J, Ehasz C, Epperson M, Klas K, Wyatt J, Hennig M, Forconi M. The effect of the hydrophobic environment on the retro-aldol reaction: comparison to a computationally-designed enzyme. Org Biomol Chem. 2013;11:8419–8425. doi: 10.1039/c3ob41898g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller MM, Windsor MA, Pomerantz WC, Gellman SH, Hilvert D. A rationally designed aldolase foldamer. Angew Chem Int Ed Engl. 2009;48:922–925. doi: 10.1002/anie.200804996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka F, Fuller R, Barbas CF. Development of small designer aldolase enzymes: catalytic activity, folding, and substrate specificity. Biochemistry. 2005;44:7583–7592. doi: 10.1021/bi050216j. [DOI] [PubMed] [Google Scholar]
- 13.Althoff EA, Wang L, Jiang L, Giger L, Lassila JK, Wang Z, Smith M, Hari S, Kast P, Herschlag D, Hilvert D, Baker D. Robust design and optimization of retroaldol enzymes. Protein Science. 2012;21:717–726. doi: 10.1002/pro.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bjelic S, Nivon LG, Celebi-Olcum N, Kiss G, Rosewall CF, Lovick HM, Ingalls EL, Gallaher JL, Seetharaman J, Lew S, Montelione GT, Hunt JF, Michael FE, Houk KN, Baker D. Computational design of enone-binding proteins with catalytic activity for the Morita-Baylis-Hillman reaction. ACS Chem Biol. 2013;8:749–757. doi: 10.1021/cb3006227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilvert D. Critical analysis of antibody catalysis. Annual Rev Biochem. 2000;69:751–793. doi: 10.1146/annurev.biochem.69.1.751. [DOI] [PubMed] [Google Scholar]
- 16.Barbas CF, Heine A, 3rd, Zhong G, Hoffmann T, Gramatikova S, Björnestedt R, List B, Anderson J, Stura EA, Wilson IA, Lerner RA. Immune versus natural selection: antibody aldolases with enzymic rates but broader scope. Science. 1997;278:2085–2092. doi: 10.1126/science.278.5346.2085. [DOI] [PubMed] [Google Scholar]
- 17.Mack KL, Moroz OV, Moroz YS, Olsen AB, McLaughlin JM, Korendovych IV. Reprogramming EF-hands for design of catalytically amplified lanthanide sensors. J Biol Inorg Chem. 2013;18:411–418. doi: 10.1007/s00775-013-0985-5. [DOI] [PubMed] [Google Scholar]
- 18.Kazlauskas RJ, Bornscheuer UT. Finding better protein engineering strategies. Nat Chem Biol. 2009;5:526–529. doi: 10.1038/nchembio0809-526. [DOI] [PubMed] [Google Scholar]
- 19.Khersonsky O, Tawfik DS. Enzyme promiscuity: a mechanism and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 20.Reetz MT. Biocatalysis in organic chemistry and biotechnology: past, present and future. J Am Chem Soc. 2013;135:12480–12496. doi: 10.1021/ja405051f. [DOI] [PubMed] [Google Scholar]
- 21.Kourist R, Jochens H, Bartsch S, Kuipers R, Padhi SK, Gall M, Böttcher D, Joosten H-J, Bornscheuer UT. The α/β-hydrolase fold 3DM database (ABHDB) as a tool for protein engineering. ChemBioChem. 2010;11:1635–1643. doi: 10.1002/cbic.201000213. [DOI] [PubMed] [Google Scholar]
- 22.Rajagopalan S, Wang C, Yu K, Kuzin AP, Richter F, Lew S, Miklos AE, Matthews ML, Seetharaman J, Su M, Hunt JF, Cravatt BF, Baker D. Design of activated serine-containing catalytic triads with atomic-level accuracy. Nature Chem Biol. 2014;10:386–391. doi: 10.1038/nchembio.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bjelic S, Kipnis Y, Wang L, Pianowski Z, Vorobiev S, Su M, Seetharaman J, Xiao R, Kornhaber G, Hunt JF, Tong L, Hilvert D, Baker D. Exploration of alternate catalytic mechanisms and optimization strategies for retroaldolase design. J Mol Biol. 2014;426:256–271. doi: 10.1016/j.jmb.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lassila JK, Baker D, Herschlag D. Origins of catalysis by computationally designed retroaldolase enzymes. Proc Nat Acad Sci USA. 2010;107:4937–4942. doi: 10.1073/pnas.0913638107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnsson K, Allemann RK, Widmer H, Benner SA. Synthesis, structure and activity of artificial, rationally designed catalytic polypeptides. Nature. 1993;365:530–532. doi: 10.1038/365530a0. [DOI] [PubMed] [Google Scholar]
- 26.Taylor SE, Rutherford TJ, Allemann RK. Design, synthesis and characterisation of a peptide with oxaloacetate decarboxylase activity. Bioorg Med Chem Lett. 2001;11:2631–2635. doi: 10.1016/s0960-894x(01)00519-4. [DOI] [PubMed] [Google Scholar]
- 27.Blommel PG, Fox BG. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expres Purif. 2007;55:53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.List B, Barbas CF, Lerner RA. Aldol sensors for the rapid generation of tunable fluorescence by antibody catalysis. Proc Nat Acad Sci USA. 1998;95:15351–15355. doi: 10.1073/pnas.95.26.15351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dess DB, Martin JC. Readily accessible 12-I-5 oxidant for the coversion of primary and seconadry alcohols to aldehydes and ketones. J Org Chem. 1983;48:4155–4156. [Google Scholar]
- 30.Chattopadhyaya R, Meador WE, Means AR, Quiocho FA. Calmodulin structure refined at 1.7 A resolution. J Mol Biol. 1992;228:1177–1192. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- 31.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popović Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Meth Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information