

Abstract
GLT-1 (EAAT2; slc1a2) is the major glutamate transporter in the brain, and is predominantly expressed in astrocytes, but at lower levels also in excitatory terminals. We generated a conditional GLT-1 knock-out mouse to uncover cell-type-specific functional roles of GLT-1. Inactivation of the GLT-1 gene was achieved in either neurons or astrocytes by expression of synapsin-Cre or inducible human GFAP-CreERT2. Elimination of GLT-1 from astrocytes resulted in loss of ∼80% of GLT-1 protein and of glutamate uptake activity that could be solubilized and reconstituted in liposomes. This loss was accompanied by excess mortality, lower body weight, and seizures suggesting that astrocytic GLT-1 is of major importance. However, there was only a small (15%) reduction that did not reach significance of glutamate uptake into crude forebrain synaptosomes. In contrast, when GLT-1 was deleted in neurons, both the GLT-1 protein and glutamate uptake activity that could be solubilized and reconstituted in liposomes were virtually unaffected. These mice showed normal survival, weight gain, and no seizures. However, the synaptosomal glutamate uptake capacity (Vmax) was reduced significantly (40%). In conclusion, astrocytic GLT-1 performs critical functions required for normal weight gain, resistance to epilepsy, and survival. However, the contribution of astrocytic GLT-1 to glutamate uptake into synaptosomes is less than expected, and the contribution of neuronal GLT-1 to synaptosomal glutamate uptake is greater than expected based on their relative protein expression. These results have important implications for the interpretation of the many previous studies assessing glutamate uptake capacity by measuring synaptosomal uptake.
Keywords: glia, ischemia, plasticity, seizures, spillover, transport
Introduction
The precise regulation by glutamate transporters of extracellular glutamate concentration is critical for the normal function of excitatory synapses and for protection of the brain against excitotoxicity. There are five subtypes of Na+-dependent glutamate transporters, EAAT1 (GLAST), EAAT2 (GLT-1), EAAT3 (EAAC1), EAAT4, and EAAT5 (Danbolt, 2001). The use of synaptosomes was introduced 40 years ago to study glutamate uptake in the brain (Levi and Raiteri, 1973a,b; McLennan and Haldeman, 1973; Roach et al., 1973), and early studies provided evidence that glutamate is accumulated by a high affinity uptake system in synaptic terminals and synaptosomes in the pigeon (Beart, 1976) and in the rat (Iversen and Storm-Mathisen, 1976; Divac et al., 1977; Gundersen et al., 1993). Since then, glutamate transport has been shown to be important in a wide variety of neurobiological phenomena, and synaptosomal uptake of glutamate is widely used to assay changes in glutamate transport related to experimental manipulations.
Based on immunoadsorption of transport activity GLT-1 was shown to account for ∼95% of the total glutamate uptake activity in young adult forebrain tissue (Danbolt et al., 1992; Haugeto et al., 1996). This conclusion was confirmed by deletion of the GLT-1 gene in mice (Tanaka et al., 1997; Holmseth et al., 2012a), as well as by electrophysiological recordings of glutamate transporter currents (Otis and Kavanaugh, 2000). Deletion of the GLT-1 gene eliminates >95% of the glutamate uptake activity in forebrain homogenates and leads to premature death due to intractable seizures (Tanaka et al., 1997). GLT-1 is highly expressed in glial membranes (Danbolt et al., 1992; Lehre et al., 1995). Early evidence up to the present indicated that GLT-1 mRNA was expressed in neurons (Torp et al., 1994; Trotti et al., 1995; Schmitt et al., 1996; Berger and Hediger, 1998; Berger et al., 2005), but the presynaptic glutamate transporter evaded detection for a long time (Danbolt, 2001). GLT-1 was first shown to be expressed in axon terminals in the hippocampus (Chen et al., 2004; Furness et al., 2008), and subsequently also in axon terminals of the somatosensory cortex (Melone et al., 2009) and the striatum (Petr et al., 2013). Approximately 5–10% of hippocampal GLT-1 protein is neuronal (Furness et al., 2008). Here we have generated a conditional knock-out of GLT-1 to dissect the contributions of astrocytic and neuronal GLT-1 to glutamate homeostasis in the intact brain. We show that selective deletion of GLT-1 in astrocytes is sufficient to reproduce the lethal phenotype of the global GLT-1 knock-out. In contrast, the consequences of selective GLT-1 deletion in neurons were undetectable with basic phenotyping approaches (e.g., measurement of weight gain, survival, electroencephalographic and spontaneous seizures, SHIRPA protocol for basic behavioral assessment). However, in agreement with d-aspartate immunocytochemistry (Furness et al., 2008), we show here by a genetic approach that uptake activity measured in crude synaptosomes correlates better with neuronal GLT-1 than with astrocytic GLT-1. These results imply that the many studies that have relied on synaptosomal uptake to assess glutamate uptake activity need reinterpretation.
Materials and Methods
Generation of GLT-1 conditional knock-out mice
The conditional targeting construct was generated using the method of recombination-mediated genetic engineering (recombineering) developed by Liu et al. (2003). The specific locations of elements in the final construct are defined by their positions in the Genome Reference Consortium Mouse Build 38 (GRCm38). Briefly, a 12.2 kb fragment (2:102731514–102743743) of the mouse slc1a2 gene, containing exons 2, 3, and 4, was subcloned by recombineering from the bacterial artificial chromosome, RP23-361H22 (CHORI BACPAC Resource Center), into a plasmid containing a thymidine kinase (TK) selection cassette. A loxP site was inserted 217 bp (2:102739471) upstream of exon 4 (2:102739689–102739939). A neomycin (neo) selection cassette flanked by FRT sites and a single downstream loxP site was inserted 419 bp (2:102740358–102740394) downstream of exon 4 to generate the final targeting construct (Fig. 1A).
Figure 1.

Generation of GLT-1 conditional knock-out mice. A, Targeted locus; a schematic of the targeted locus showing the relevant elements of the targeting construct. Cre recombinase recognition sites are represented as closed triangles. Flp recombinase recognition sites are represented as open triangles. The arrows identify the positions of screening primers. The target sequence for primer PLF resides on genomic DNA upstream of the targeting construct. The target sequence for primer LoxPR resides within the region introduced with the isolated loxP site upstream of exon 4. Presence of the 7.9 kb amplicon verifies correct recombination of the left arm and verifies retention of the isolated loxP site. The target sequence for NeoF resides within the introduced neo selection cassette. The target sequence for PRR resides on genomic DNA downstream of the targeting construct. Presence of the 3.3 kb amplicon verifies correct insertion of the right arm. B, Conditional allele; the structure of the targeted locus after Flp recombinase-mediated excision of the neo cassette. The resultant allele can be conditionally inactivated by the action of Cre recombinase, which will excise exon 4. C, Homologous recombination verification. Initial DNA samples from resistant colonies were pooled in groups of two and screened for homologous recombination by long range PCR for correct insertion of the right arm (amplicon 3.3 kb). Positive pools were rescreened as individual samples for correct insertion of the left arm and retention of the upstream loxP site (amplicon 7.9 kb). Positive clones were expanded and rescreened. Results for three correctly targeted clones (D33–54, 67, 90) and one nontargeted control clone are shown in C. D, Demonstration of in vivo excision. We mated GLT-1flox/flox mice with a mouse line that expresses Cre in all cell types under the control of a ubiquitous promoter, EIIa, which expresses Cre-recombinase in the whole embryo from early development. The protein expression in mice with conditional inactivation of one (GLT-1+/Δ; EIIaCre) and both (GLT-1Δ/Δ; EIIaCre) GLT-1 alleles was compared with protein expression in wild-type mice (GLT-1+/+) and mice heterozygous (GLT-1+/−) for a null allele. Inactivation of one conditional allele produced a decrease in expression comparable to that in mice heterozygous for the null allele; inactivation of both alleles (GLT-1Δ/Δ; EIIaCre) results in complete loss of detectable GLT-1. GLT-1 labeling shows monomer (∼60 kDa), dimer (∼120 kDa), and multimer (∼250 kDa).
The construct was linearized with SalI and electroporated into J1 ES cells (Li et al., 1992) from 129SvJ mice and subsequently selected with appropriate drugs. Genomic DNA from resistant colonies was screened by long range PCR (KOD Extreme Hot Start DNA Polymerase, EMD Millipore) for homologous recombination. Left arm primers (PLF: GACTAGCATTCTCTGGGGTGCAGCTTGGGAGTTGC, LoxPR: TTAAGGGTTATTGAATATGATCGGAATTGGGCTGCAGGAATT) amplify a 7.9 kb fragment after homologous recombination (Fig. 1A,C). Right arm primers (NeoF: TGGGGCAGGACAGCAAGGGGGAGGAT; PRR: GTGGTATTGGCCTCCTCAGATGGAGGTGCCACCA) amplify a 3.3 kb fragment after homologous recombination (Fig. 1A,C). Clones indicating appropriate homologous recombination were subjected to karyotype analysis and clones with a high number of cells with 40 chromosomes were injected into C57BL/6 blastocysts (Robertson, 1987). Chimeric animals were mated and offspring were assessed for germline transmission of the targeted allele. In some cases, clones were subjected to a second round of transfection with the pOG231 plasmid (courtesy of Stephen O'Gorman, Case Western Reserve University, Cleveland, Ohio) that contains a gene that encodes a Cre-NLS protein and that is driven by a CMV promoter. Subsequent subclones were analyzed by genotyping for those that underwent the appropriate recombination of loxP sites.
Even though the neomycin resistance cassette resides in an intron, it can have unpredictable effects on the expression of the gene of interest and other nearby genes. Therefore, we removed the neomycin cassette in vivo by mating mice that were homozygous for the conditional GLT-1 knock-out allele with mice that expressed the FLP1 recombinase gene driven by the Gt(ROSA)26Sor promoter (JAX Stock no. 003946; Fig. 1B).
We mated GLT-1flox/flox mice (with a 129SvJ × C57BL/6 background) with a mouse line that expresses Cre in all cell types under the control of a ubiquitous promoter, EIIa, that expresses Cre-recombinase in the whole embryo from early development. Immunoblot analysis for GLT-1 demonstrated Cre excision of the floxed GLT-1 allele in vivo (Fig. 1D). A rabbit anti-GLT-1 antibody directed against the N-terminus (Chen et al., 2002) stained a monomer band at ∼60 kDa, a dimer band at ∼120 kDa, and a multimer ∼250 kDa (Chen et al., 2002, 2004). Brain lysate from a mouse heterozygous for the constitutive null allele (GLT-1+/−) was run for comparison.
To generate the astrocytic knock-out of GLT-1 (GLT-1Δ/Δ;GFAP-Cre ERT2), we bred male or female mice that carried the conditional GLT-1 knock-out allele with female or male mice that carried the inducible human glial fibrillary acidic protein hGFAP-CreERT2 (GFAP-CreERT2, with C57BL/6 background (Casper et al., 2007).
The breeding scheme to generate astrocytic GLT-1 knock-outs was as follows:
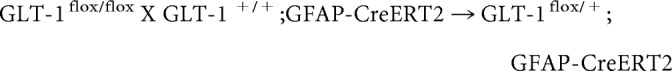 |
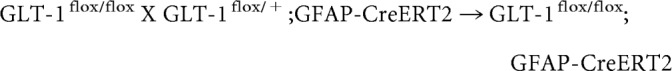 |
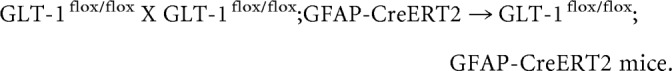 |
P5 pups were treated daily with tamoxifen (T5648, Sigma-Aldrich; 33 mg/kg, i.p. or oral gavage in sunflower oil) for 4–5 consecutive days (Ganat et al., 2006). Tamoxifen solutions were made fresh for a given litter and not frozen.
To generate a neuronal knock-out of GLT-1 (GLT-1Δ/Δ;Syn-Cre), we bred male mice that carried the conditional GLT-1 knock-out allele with female mice (Rempe et al., 2006) that carried the synapsin I promoter-driven Cre recombinase allele (with C57BL/6 background, JAX Stock no. 003966). We used a similar breeding approach to generate the neuronal GLT-1 knock-outs as for the astrocytic GLT-1 knock-outs, substituting synapsin-Cre for GFAP-CreERT2, and introducing synapsin-Cre only through the females.
All animal experiments were performed in accordance with NIH guidelines, and were approved by the Children's Hospital Boston Institutional Animal Care and Use Committee. Both male and female adult mice were used in the experiments. For each particular experiment, animal ages are given in the Materials and Methods section for that experiment.
Light and electron microscopic immunocytochemistry
Materials for electron microscopic detection of GLT-1.
(1) Immunoreagents and other chemicals. For the detection of GLT-1a, a monoclonal antibody harvested from hybridoma culture medium and directed against the C terminus of GLT-1a (anti-GLT-1a antibody) was used. This antibody was a gift from Dr Jeffrey Rothstein (Johns Hopkins University, Baltimore, MD). Biotinylated anti-mouse IgG and the avidin-biotin peroxidase complex (ABC Elite kit) were purchased from Vector Laboratories. Electron microscopic (EM) grade paraformaldehyde, glutaraldehyde, EM grids, and the resin EPON812 were purchased from EM Sciences. All other chemicals were purchased from Sigma Chemicals. (2) Animals. Brains were collected from the mice at 5–9 weeks of age. Hippocampi of the following genetically modified mice were analyzed by EM-immunocytochemistry (ICC) for the ultrastructural pattern of GLT-1 distribution: mice with genetic deletion of GLT-1 within neurons (GLT-1Δ/Δ;Syn-Cre; referred to as syn/cre+, n = 3); GLT-1flox/flox littermates without Syn-Cre (referred to as syn/cre−, n = 2); GLT-1Δ/Δ;GFAP-Cre/ERT2 mice (GFAP/cre+, n = 3); GLT-1flox/flox (GFAP/cre−, n = 2). In addition, specificity of antibody labeling was verified by analyzing brains of mice with global knock-out of GLT-1.
Procedures for EM detection of GLT-1.
Mice were deeply anesthetized using urethane (i.p., 0.34 g/g body weight), then killed by transcardial perfusion with 200 ml of 0.1 m phosphate buffer (PB), pH 7.4, containing 4% formaldehyde and 0.1% glutaraldehyde, delivered using a peristaltic pump set at a flow rate of 20 ml/min. On the day after transcardial perfusion, brains were sectioned using a vibratome at a thickness setting of 50 μm, then treated for 30 min with 1% sodium borohydride in 0.1 m PB within 4 h after vibratome sectioning, so as to terminate the aldehyde fixation. After immersion for 30 min, sections were rinsed repeatedly in 0.1 m PB, so as to remove unreacted sodium borohydride. Vibratome sections containing the hippocampus of different genotypes of mice were cut on the same day, collected in 0.01 m PB containing 0.9% sodium chloride (PBS), set at a pH 7.4. These sections were stored free-floating in a cold room (4°C) in PBS containing 0.05% sodium azide as preservative.
To visualize GLT-1-immunoreactive processes, vibratome sections were incubated overnight at room temperature under constant agitation in a buffer consisting of PBS-azide with 1% bovine serum albumin (BSA), together with the anti-GLT-1a antibody (original concentration is 6.7 mg/ml) at the following dilutions: 1:100, 1:500, 1:1000, 1:3000, 1:10,000, 1:30,000, and 1:60,000. Subsequently, the unbound antibodies were removed by rinsing sections in PBS, then incubated for 1 h at room temperature in the PBS-azide/BSA buffer containing biotinylated goat anti-mouse IgG at a dilution of 1:200. Sections were rinsed again, and then incubated in the A+B solution from the Vector Laboratories ABC Elite kit. Following rinses in PBS to remove unbound secondary antibodies, sections were immersed in PBS containing 3,3′-diaminobenzidine HCl (DAB; 10 mg tablets from Sigma Chem, dissolved in 44 ml of PBS) The peroxidase reaction product was begun by adding hydrogen peroxide (4 μl of 30% hydrogen peroxide per 44 ml of DAB solution) and terminated at the end of 12 min by rinsing sections repeatedly in PBS. Vibratome sections were postfixed using 2% glutaraldehyde in PBS.
Following this immunocytochemical procedure, the vibratome sections were processed by a conventional EM procedure, consisting of postfixation by immersion in 1% osmium tetroxide/0.1 m phosphate buffer for 1 h, then dehydrated using graded concentrations of alcohol, up to 70%, then postfixed overnight using 1% uranyl acetate, dissolved in 70% ethanol. On the following day, dehydration continued up to 100%, then was rinsed in acetone, and infiltrated in EPON 812 (EM Sciences), which was cured by heating the tissue at 60°C, while sandwiched between two sheets of Aclar plastic, with lead weights placed on top of the Aclar sheets, so as to ensure flatness of the EPON-embedded sections. These flat-embedded vibratome sections were re-embedded in Beem capsules filled with EPON 812, and then ultrathin-sectioned at a plane tangential to the vibratome sections. The number of ultrathin sections collected from any one animal's brain section depended on the natural curvature that the section took on, even while cured under lead weights. Usually, a minimum of 10 ultrathin sections needed to be collected from the surface-most portion of each vibratome section, where immunoreactivity was expected to be maximal. Tissues were sampled for each anatomical layer of the CA1 of hippocampus (stratum oriens, stratum pyramidale, stratum radiatum, and stratum lacunosum-moleculare). The ultrathin sections were collected onto formvar-coated, 400 mesh thin-bar nickel grids. A subset of these ultrathin sections was counter-stained with Reynold's lead citrate.
Quantification of the expression level of GLT-1 at presynaptic axon terminals and postsynaptic dendritic spines and by astrocytes.
Digital EM images were captured from ultrathin sections by an experimenter that was kept blind about the genotype of the animal from which brain tissue was collected. Images were captured using a 1.2 megapixel Hamamatsu CCD camera from AMT from the JEOL 1200XL EM. Images were captured strictly from regions of the ultrathin section where the vibratome section surface could be verified by the transition of neural tissue to purely EPON matrix. Asymmetric axo-spinous synapses were identified based on the cluster of round clear vesicles on the presynaptic side and the presence of prominently electron-dense postsynaptic densities (PSDs) along the intracellular surface of the membrane facing the vesicle-containing axon terminals. Once these asymmetric axo-spinous synapses were identified, the synapse was scored as containing the DAB reaction product within the axon terminal or within the spine or both. Whenever present, the fine astrocytic processes that immediately abutted the asymmetric synapses were scored as exhibiting immunoreactivity or not. Other astrocytic processes in the same micrograph (5 μm × 5 μm in area, captured at a magnification of 40,000×) were evaluated as containing or not containing DAB reaction product along the membrane or intracellularly. The brain region analyzed was consistently the CA1 field stratum radiatum of the dorsal hippocampus.
All axon terminals and dendritic spines forming asymmetric synapses were analyzed, strictly in the order that they were encountered, so as to ensure randomness of sampling. For every group of 10 synapses, the proportion of labeled axon terminals and spines was determined. This procedure was repeated 10 and 15 times (i.e., for 100 and 150 synapses) for the syn/cre− and syn/cre+ tissues, respectively and 16 and 17 times for the GFAP/cre− and GFAP/cre+ tissues, respectively. Astrocytes labeled within 35 micrographs containing the 160 synapses of the GFAP/cre− tissue and within 23 micrographs containing the 170 synapses of the GFAP/cre+ tissue were quantified as the proportion of astrocytes labeled, per micrograph.
Primary SHIRPA screen
We used a modified SHIRPA protocol (Rogers et al., 1997), which is composed of 29 separate parameters that provide a wide range of measurements of muscle, cerebellar, sensory, neuropsychiatric, and autonomic functions. Briefly, each mouse (the astrocytic GLT-1 knock-outs and their littermate mice were 28–67 weeks old; the neuronal GLT-1 knock-outs and their control mice were 12–52 weeks old) was placed into a clear viewing jar (L = 18 cm, W = 10 cm, H = 19 cm) and its behavior and morphology were observed, such as body position, tremor, coat appearance, and whiskers. Subsequently, the mouse was transferred to an arena (L = 28 cm, W = 28 cm, H = 21 cm) that consisted of nine evenly sized squares for testing the transfer arousal and other motor behaviors, such as locomotor activity, gait, tail elevation, and touch escape. Afterward, a sequence of manipulations with tail suspension was performed, such as positional passivity, skin color, trunk curl, and limb grasping. Visual placing and reflexes were also recorded. Throughout this procedure, vocalization was recorded. All behaviors were scored to provide a quantitative assessment of each parameter.
Scalp EEG and tethered EEG
EEG recordings.
Scalp EEG signals were acquired for 20–40 min with two thin silver/silver-chloride Teflon-coated EEG subdermal wire electrodes (Ives EEG Solutions), with a reference contact positioned over the dorsal snout at midline, and one active contact over the parietal region. Mice tolerated the electrodes without signs of local pain or discomfort after initial subcutaneous placement. The mice were gently restrained on a wooden platform with two broad Velcro straps positioned over the torso behind the forelimbs and in front of the hindlimbs, under brief isoflurane anesthesia. The mice were closely observed during this period for occurrence of clinical seizures and video-recorded for post hoc visualization of the correct timing of onset and duration of seizures. The tethered EEG technique consisted of 48–72 h long recordings in freely moving animals. Two-channel subdural electrodes were implanted bilaterally in all animals. The EEG cables were connected to the tether with a low torque commutator and connector assembly (Plastics One).
All the EEG measurements were recorded using a Cadwell EEG system (Cadwell Laboratories) at a sampling rate of 400 Hz. The signals were filtered in the bandpass cutoff range of 1–70 Hz along with a 60 Hz Notch filter. The animals used in EEG studies were at 13–68 weeks of age.
Digital spike count analysis.
After acquiring data, the power in frequency bands was analyzed for seizures. Automated seizure detection relied on metrics that use the Fourier transform of the signal and also an array of nonlinear EEG features of the signal. A library of reference events was built by marking seizures and nonseizure artifacts by the visual inspection of a few hours of EEG signals from each animal. After building a large enough library, the detection software used the previously derived metrics to compare the raw signals with the library reference events. The EEG signal events within 1 s of separation from one another that were the most similar to reference seizure events were thus automatically classified as seizures (Chang et al., 2011; Goodrich et al., 2013). A spike was defined as one or more sharp deflections, of voltage exceeding background activity. A spike-train was defined as a continuous run of such spikes lasting at least 1 s.
Synaptosomal uptake of l-[3H]glutamate and d-[3H]aspartate
Sodium-dependent transport of l-[3H]glutamate and the nonmetabolizable analog, d-[3H]aspartate was measured as previously described (Petr et al., 2013). Mice (10–30 weeks of age) were anesthetized using ether, brains were removed, and the forebrain dissected, weighed and homogenized in 20× ice-cold 0.32 m sucrose. Samples were centrifuged at 800 × g for 10 min at 4°C. The supernatant was poured into a fresh tube and centrifuged at 20,000 × g for 20 min at 4°C. The pellet was then resuspended in 40× ice-cold 0.32 m sucrose and centrifuged at 20,000 × g for 20 min at 4°C. The washed pellet was then resuspended in 50× ice-cold 0.32 m sucrose by homogenization. The isolated crude synaptosomes were kept on ice and used immediately for uptake assay, or frozen for immunoblotting. Glass tubes containing 450 μl of buffer (in mm: NaCl 140 or choline chloride 140, KCl 2.5, CaCl2 1.2, MgCl2 1.2, K2HPO4 1.2, glucose 10, Tris base 5, HEPES 10) with 10 μm l-glutamate or d-aspartate [10 μm l-glutamate or d-aspartate] and 0.005 μm l-[3H]glutamate or d-[3H]aspartate (PerkinElmer) were preincubated for 5 min at 37°C in the absence or presence of inhibitors. Glutamate uptake into synaptosomes was initiated by adding 50 μl of crude synaptosomes to each tube and incubating at 37°C for 30 s. To stop uptake activity, 2 ml of ice-cold choline buffer was added to the tube, vortexed, and plunged into ice-water slurry. The samples were filtered through Whatman GF/C filter paper prewetted with 2 ml choline buffer, and then the filters were washed three times with 2 ml ice-cold choline buffer. Radioactivity on the filters was measured by liquid scintillation counting (TRI-CARB2200CA, PACKARD; Long Island Scientific). The radioactivity taken up by the synaptosomes in the absence of sodium was subtracted from that taken up in the presence of sodium to isolate sodium-dependent transport. Inhibitors of glutamate transporters dihydrokainic acid (DHK) and dl-threo-β-benzyloxyaspartic acid (TBOA) were obtained from Tocris Bioscience. Nigericin (Sigma-Aldrich) was used to isolate the net uptake component in the synaptosomal assay. To test nigericin-sensitive uptake in synaptosomes, we preincubated the uptake system (uptake buffer and synaptosomes) with or without nigericin (3 μm) for 5 min at 37°C, and then added 10 μm glutamate (including 0.005 μm l-[3H]glutamate) into each reaction tube to start the uptake (37°C, 30 s).
In some experiments, we compared the glutamate uptake in uncentrifuged homogenate, P1 and P2 fractions derived from neuronal GLT-1 knock-outs and littermate controls. After homogenizing the forebrain in 20× ice-cold 0.32 m sucrose, we diluted a portion of the homogenate to 50× with 0.32 m sucrose and kept it on ice until use. A P1 pellet was collected on another portion of the same homogenate after a 800 × g spin at 4°C and was resuspended in 50× 0.32 m sucrose and kept on ice until use. The supernatant from the P1 spin was spun at 20,000 × g for 20 min at 4°C, resuspended to 40× in 0.32 m sucrose, respun, and resuspended to 50×. Glutamate uptake was then compared in these three fractions.
Western blot
Following a previously published procedure (Petr et al., 2013), the crude synaptosomes isolated from mouse forebrain were centrifuged at 20,000 × g for 20 min at 4°C, then resuspended in lysis buffer [1% SDS, 0.05 m PB and 1× Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific, catalog #78440), pH 7.4]. Protein concentration was quantified using the Bio-Rad DC protein assay. Five micrograms (∼7 μl) of protein from each sample was mixed with 5 μl 4× Laemmli buffer (250 mm Tris-HCl, pH 6.8, 8% SDS, 40% glycerol, 8% β-mercaptoethanol, 0.02% bromophenol blue; Boston BioProducts catalog #BP-110R) and lysis buffer (∼8 μl) to a final volume of 20 μl. The mixed samples were subjected to SDS-PAGE using 4–20% acrylamide gels and transferred to Immun-BlotPVDF membrane (Bio-Rad Laboratories). Membranes were stained with Ponceau S solution (Boston Bioproducts) to visualize protein. Membranes were incubated for 1 h at room temperature in 5% milk in TBST (Tris-buffered saline, pH 7.4, with 0.1% Triton X-100). Membranes were then incubated overnight at 4°C with antibody against the N-terminus of GLT-1 (anti-nGLT-1 antibody, 1.16 μg/ml; Chen et al., 2002) diluted in 5% milk in TBST. Membranes were washed three times, each for 5 min, and then incubated for 1 h at room temperature with horseradish peroxidase-coupled secondary antibody (goat anti-rabbit at 1:10000; Bio-Rad Laboratories) diluted with 5% milk in TBST. Membranes were washed three times, each for 5 min, and then incubated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) for 5 min. The membranes were exposed to Hyblot CL Autoradiography Film (Denville Scientific), and films were developed with a Kodak 2000 processor. Membranes were then reprobed with chicken anti-β-tubulin antibody (Millipore) diluted at 1:10,000 to assess for equivalent protein loading. Immunoblots were scanned and digitized using ImageJ software to obtain the optical density of the bands. The anti-nGLT1 antibody often stained a monomer band at ∼60 kDa, a dimer band at ∼120 kDa, and sometimes a multimer at ∼250 kDa (Chen et al., 2002, 2004). All monomer, dimer (if present), and multimer bands (if present) were used for GLT-1 quantification.
Uptake of l-[3H]glutamate in proteoliposomes
Reconstitution of glutamate transporters into proteoliposomes.
This was done as previously described (Beckstrøm et al., 1999). Briefly, crude mouse (14–21 weeks of age) forebrains were homogenized in 30 volumes 50 mm Tris-acetate, pH 7.4. The homogenate was divided into aliquots of 0.5 ml and centrifuged (14,500 rpm, 20 min, 4°C; Beckman, 18.1 rotor). The crude membrane pellets were collected and stored at −80°C until used. The frozen pellets were mixed with 25 volumes solubilization buffer (12.5 g/L cholic acid, 500 mm NaCl, 5 mm EDTA, 100 mm NaPi, pH 7.4), incubated (10 min) on ice, and then centrifuged (39,000 × g, 20 min, 4°C). The supernatant was mixed with a phospholipid cholate salt mixture, incubated on ice and gel filtered (Trotti et al., 1995) to remove detergent and sodium ions on Sephadex G50 spincolumns equilibrated with the internal medium containing 0.12 m KPi, pH 7.4, and 1% glycerol. For electrophoresis, the pellets were dissolved in 1% SDS containing 10 mm NaPi, pH 7.4, and 1 mm PMSF. Immunoblotting was performed as described before (Holmseth et al., 2009) using chemiluminescence (Supersignal West Dura, Pierce) in combination with antibodies to GLT-1 (anti-B12 Ab#360, 0.2 μg/ml; Holmseth et al., 2012b) or antibodies to GLAST (anti-A522 Ab#314, 0.2 μg/ml; Holmseth et al., 2009).
l-[3H]glutamate or [3H]GABA uptake in proteoliposomes.
The glutamate transport activity was determined as described previously (Danbolt et al., 1990). Briefly, the uptake reaction was started by adding 20 μl of the above proteoliposomes suspension into 500 μl medium containing 150 mm NaCl, 1% glycerol, 1.5 μm valinomycin (a selective potassium ionophore to ensure a negative membrane potential), 1 μCi l-[3H]glutamate or [3H]GABA (GE Healthcare) at room temperature. After incubation (10 s for glutamate uptake, 5 min for GABA uptake), the reaction was terminated by dilution in 2 ml ice-cold 150 mm NaCl, 1% glycerol and filtration through Millipore cellulose nitrate acetate filters (0.45 μm pores). The filters were washed and dissolved for liquid scintillation counting.
Antibodies
To assay the protein expression of GLT-1 in astrocytic and neuronal GLT-1 knock-out mice by immunoblotting, rabbit anti-nGLT-1 antibody was used at 1.16 μg/ml, which was raised against the N-terminal of GLT-1 protein (Chen et al., 2002). To detect the presence of GLT-1 by light microscopic (LM) and EM-ICC, a monoclonal antibody directed against the C-terminus of GLT-1a (anti-GLT-1a) was used that was generously provided by Dr Jeffrey Rothstein (Johns Hopkins University). The original concentration of anti-GLT-1a was 6.7 mg/ml; 1:30,000 dilution (0.22 μg/ml) was used for GFAP/cre− and GFAP/cre+ tissues; 1:10,000 dilution (0.67 μg/ml) was used for syn/cre− and syn/cre+ tissues. Antibodies used in the proteoliposome experiments were antibodies against GLT-1 (anti-B12 Ab#360, 0.2 μg/ml; Holmseth et al., 2012b) or antibodies against GLAST (anti-A522 Ab#314, 0.2 μg/ml; Holmseth et al., 2009).
Statistics
GraphPad Prism v5.01 and Statistica v10 software were used for statistical analyses. Group data were calculated as mean ± SEM. Nonparametric analysis was used to analyze the frequency of GLT-1 immunoreactivity occurring within astrocytic processes or presynaptic axon terminals across two genotype groups (conditional GLT-1 knock-out group vs their littermate controls). Probability of survival was estimated by Kaplan–Meier method and statistical significance calculated by the log-rank (Mantel–Cox) analysis. Two-way ANOVA and Bonferroni's post-test were used to analyze the body weight between genotype and age of mice. Unpaired t test was performed to compare the means of two groups of data in all other experiments. A factor was considered statistically significant if it had p < 0.05. Saturation analysis data were analyzed using Prism 5 (GraphPad Software) to determine Vmax and Km values.
Results
Verification of the cell-type-specific conditional deletion of GLT-1
Previous studies established that GLT-1 expression is predominantly but not exclusively astrocytic. Within the hippocampus, GLT-1 is also expressed within axon terminals forming asymmetric, and thus excitatory, synapses (Chen et al., 2004). To investigate the roles played by astrocytic versus neuronal GLT-1, we created two lines of conditional knock-outs; one aimed to delete GLT-1 within astrocytes through Cre expression driven by a tamoxifen inducible GFAP-promoter (Ganat et al., 2006) and another aimed to delete GLT-1 within neurons through Cre expression driven by the synapsin I promoter (JAX Stock no. 003966). To produce animals deficient in GLT-1 in astrocytes, we administered tamoxifen postnatally (P5–P9), following a previously published study (Guo et al., 2013).
Deletion of GLT-1 was detected by immunoblot using anti-nGLT-1 antibody in the crude synaptosomes isolated from astrocytic and neuronal GLT-1 knock-out mice and their controls (Fig. 2). Among the four GFAP/cre+ mice that we tested, two of them had almost complete loss of GLT-1 expression (one such example is illustrated in Fig. 2A, left blot), whereas the other two GFAP/cre+ mice also showed reduction of GLT-1 protein compared with GFAP/cre− mice (Fig. 2A, right blot, one such example illustrated). On average, the GLT-1 band density in the crude synaptosomal preparations of GFAP/cre+ mice was greatly reduced to 18.0 ± 10.3% (*p < 0.05, n = 4) of their littermate controls (GFAP/cre−, n = 4; Fig. 2B). The variability of GLT-1 knockdown between mutant mice may be due to variability in tamoxifen absorption after intraperitoneal or oral gavage, or other factors. Immunoblot analysis using anti-nGLT-1 antibody demonstrated no change in the expression of GLT-1 protein in crude synaptosomes from syn/cre+ mice (97.6 ± 4.5% of control, n = 3, p > 0.05) compared with the syn/cre− mice (n = 3; Fig. 2C,D). This is expected as only ∼6% of total GLT-1 protein is expressed in neurons (Furness et al., 2008). Immunoblots of GLT-1 expression in whole forebrain homogenates isolated from astrocytic or neuronal GLT-1 knock-outs and their wild-type (WT) controls showed similar results (Fig. 8A,C, insets). In the same immunoblots (Fig. 8) we assayed for GLAST expression and found no significant change in expression in the conditional knock-outs (Fig. 8A,C, insets). In addition, we found no change in EAAC1 expression (data not shown).
Figure 2.

The Cre-driver GFAP produces great loss of GLT-1 expression in conditional GLT-1 knock-out mice, whereas synapsin-Cre produces no detectable loss of GLT-1 protein. A, Protein immunoblot analysis using anti-nGLT-1 antibody in crude synaptosomes isolated from GLT-1Δ/Δ;GFAP-Cre ERT2 mice (referred to as GFAP/cre+) and their littermate controls (GFAP/cre−): in two GFAP/cre+ mice, the GLT-1 protein was almost completely knocked out (left blot); in another two GFAP/cre+ mice, the GLT-1 protein was greatly but not completely (∼35% remaining) deleted (right blot). B, Pooling densitometry results from the four experiments that were performed, there was an ∼80% reduction of GLT-1 expression (*p < 0.05) in GFAP/cre+ (n = 4) mice compared with controls (n = 4). C, Immunoblot assay of GLT-1 expression in GLT-1Δ/Δ;Syn-Cre mice (referred to as syn/cre+) compared with control mice (syn/cre−). D, Densitometry analysis of GLT-1 expression in syn/cre+ and syn/cre− mice revealed no change (p > 0.05, n = 3). GLT-1 monomer is at ∼60 kDa (dimer at ∼120 kDa and multimer at ∼250 kDa). All GLT-1 forms (if present) were included in the densitometry analysis.
Figure 8.

L-[3H]glutamate uptake into proteoliposomes prepared from astrocytic or neuronal GLT-1 knock-out and their controls correlated with GLT-1 protein expression. Deletion of GLT-1 in astrocytes leads to a major reduction in glutamate uptake (A), whereas deletion in neurons hardly affects the glutamate uptake activity (C). A, l-[3H]glutamate uptake into proteoliposomes prepared from the forebrains of wild-type (GFAP/cre−, n = 3) and GLT-1Δ/Δ;GFAP-Cre/ERT2 mice (GFAP/cre+, n = 3; ***p < 0.001). C, l-[3H]glutamate uptake in proteoliposomes prepared from the forebrains of wild-type (syn/cre−, n = 3) and GLT-1Δ/Δ;Syn-Cre mice (syn/cre+, n = 3; p = 0.065). Application of DHK (0.5 mm) inhibited ∼95% glutamate uptake activity and application of PMB-TBOA (1 μm) inhibited ∼98% uptake. Also, addition of nigericin (Nig., 3 μm) abolished all the uptake activity (A, C). The blots in A and C were developed with antibodies anti-A522 (Ab#314; 0.2 μg/ml) for GLAST and anti-B12 (Ab#360; 0.2 μg/ml) for GLT-1 (cKO refers to GFAP/cre+ in A, and refers to syn/cre+ mice in C). The blots from global GLT-1 and GLAST KO mice were used as negative controls for antibody specificity. B, D, GABA uptake activity in the same preparations of proteoliposomes as A and C, respectively. Note that liposome preparations that had a major reduction in taking up l-[3H]glutamate, took up [3H]-GABA as well as those prepared from wild-type tissue, implying that the reconstitution process has been successful.
LM and EM were performed upon the hippocampal tissue of the GFAP/cre+ and GFAP/cre− littermates so as to quantify the extent of reduction of GLT-1 within astrocytic profiles in the GFAP/cre+ line and to verify that GLT-1 expression within axon terminals forming asymmetric synapses was retained. Application of an anti-GLT-1a monoclonal antibody of dilutions ranging from 1:200 to 1:60,000, including a dilution of 1:30,000 (shown) yielded robust immunolabeling throughout the synaptic layers of the hippocampus of GFAP/cre− mice, leaving the pyramidal cell layer relatively unlabeled (Fig. 3A, asterisks). Inspection of these tissues at a higher-magnification indicated that many of the immunoreactive processes were of astrocytes (Fig. 3B, small arrow). Additionally, immunoreactive puncta, too small to be identifiable, were evident throughout the stratum radiatum and stratum lacunosum moleculare (slm). EM can readily reveal profiles that are astrocytic, based on the irregular, acutely angled contour of the plasma membrane that surrounds the ovoid profiles of axons and dendrites (Chen et al., 2004). EM revealed that the small puncta observed in the synaptic layers of the hippocampus of GFAP/cre− brains consisted of a mixture of astrocytic processes (Fig. 3C, asterisks), as well as axon terminals forming asymmetric synaptic junctions (Fig. 3C, LT), the latter characteristic of excitatory synapses. Approximately one-half of the astrocytic processes were GLT-1 immunoreactive (Fig. 3H), whereas ∼12.5% of the presynaptic axon terminals at asymmetric synapses were GLT-1-immunoreactive (Fig. 3G).
Figure 3.

Genetic deletion of GLT-1 within GFAP-expressing astrocytes exhibit reduced expression of GLT-1 in astrocytic processes but no reduction in axons forming asymmetric synapses. LM-ICC using a monoclonal anti-GLT-1a antibody at a dilution of 1:30,000. There was reduced immunolabeling within the hippocampus of GLT-1Δ/Δ;GFAP-Cre/ERT2 mice (GFAP/cre+; D, E), relative to the labeling exhibited by the hippocampus (HPC) of GLT-1flox/flox mice (GFAP/cre−; A, B). For both genotypes, the stratum with the least labeling was the pyramidal cell layer (A, B, D, E, asterisks). At a higher-magnification (B, E), it was evident that astrocytic labeling was reduced (B, E, arrows point to labeled astrocytes). A few labeled astrocytes persist within the GFAP/cre+ tissue (E, double arrow). Electron micrographs revealed that both genotypes exhibit labeling of a subpopulation of axons of passage (a in F) and axon terminals forming asymmetric synaptic junctions onto spines, which, presumably, are excitatory (LT = labeled presynaptic terminal; UT = unlabeled presynaptic terminal; arrowheads point to PSDs). Although the wild-type tissue showed prominent GLT-1-immunolabeling within astrocytic processes (C, white asterisks), more of the astrocyte processes within the GFAP/cre+ tissue were devoid of GLT-1-labeling (F, black asterisks). G, H, The outcome from quantitative analyses of tissue from three GFAP/cre+ and two GFAP/cre− animals, conducted while blind to genotype. This procedure revealed no statistically significant difference in the number of presynaptically labeled asymmetric synapses across the two genotypes (p = 0.15 by the nonparametric K–S test; G). The frequency of encounter with GLT-1-immunolabeled astrocytic profiles (H) was determined for the same set of the micrographs used to analyze synapse-labeling frequency. Nonparametric statistical analysis revealed a highly significant reduction in the encounter with immunolabeled astrocytic processes within the GFAP/cre+ tissue (H; ***p < 0.0001). bv, Blood vessels; LGN, lateral geniculate nucleus; slm, stratum lacunosum moleculare; LT, labeled terminals; UT, unlabeled terminals. Calibration bars: A, D, 500 μm; B, E, 50 μm; C, F, 500 nm.
Sections obtained from brains of the GFAP/cre+ mice yielded much lighter immunolabeling in the synaptic neuropil (Fig. 3D). Inspection of these tissues at a higher-magnification revealed nearly complete elimination of immunolabeling of astrocytic processes and of the unidentifiable puncta, thereby revealing only sparse immunolabeling of profiles that appeared astrocytic (Fig. 3E, black double arrows). Quantitative EM-ICC was performed to compare the GLT-1 immunolabeling pattern of tissue from three GFAP/cre+ and two GFAP/cre− brains. The rate of encounter with GLT-1-immunoreactive astrocytic and presynaptic processes within the stratum radiatum of CA1 was tallied, while keeping the experimenter blind of the genotype. The frequency of immunolabeling of the presynaptic axon terminal forming asymmetric axo-spinous junctions was quantified by counting the number of axon terminals immunolabeled per group of 10 randomly encountered synapses and repeating this procedure 16 times for the WT tissue from two animals and 17 times for the knock-out (KO) tissue from two animals. Tissue immunolabeled with the anti-GLT-1a antibody at the dilution of 1:30,000 revealed immunolabeling within axon terminals forming asymmetric synapses that was not significantly different across the genotypes (12.5 ± 3% for the GFAP/cre− and 20 ± 3% for the GFAP/cre+ brains; p = 0.15 by the K–S nonparametric test; Fig. 3G). In contrast, when frequency of encounter with GLT-1-immunolabeled astrocytic profiles (Fig. 3H) was determined for the same set of the micrographs used to analyze presynaptic labeling frequency (35 micrographs for the WT and 23 micrographs for the KO), the frequency of astrocytic immunolabeling per unit area of synaptic neuropil was found to be reduced from 55 ± 5% in the GFAP/cre− brains down to 9 ± 6% in the GFAP/cre+ (p < 0.001 by the K–S nonparametric test; Fig. 3H).
A similar light and electron microscopic evaluation of the syn/cre+ and syn/cre− brains was undertaken to determine the extent of reduction of GLT-1 within axon terminals and to verify that GLT-1 within astrocytes was retained. Light microscopic evaluation of syn/cre+ brains (Fig. 4D,E) revealed a GLT-1-immunolabeling pattern that was similar to that of the syn/cre− littermates (Fig. 4A,B). Both tissues showed immunolabeling throughout the synaptic neuropil using the anti-GLT-1a antibody at a dilution of 1:10,000 (shown) up to 1:60,000. A difference in neuropil labeling became evident by EM. The frequency of immunolabeling within axon terminals forming asymmetric synapses was determined for every group of 10 asymmetric synapses, encountered through random sampling (ensured by analyzing strictly in the order of encounter along the surface portion of vibratome sections) of the synaptic neuropil in stratum radiatum of the hippocampal CA1, while keeping the observer blind to the genotype. This procedure was repeated 10 times across brains of two syn/cre− animals, thereby sampling 100 synapses altogether and was repeated 15 times across the brains of three syn/cre+ animals, thereby sampling 150 synapses altogether. Both genotypes of tissue exhibited intense immunolabeling within astrocytes (Fig. 4C,F). In contrast, the frequency of immunolabeling within axon terminals forming asymmetric synapses was reduced from 16 ± 4% (1.6 per 10 synapses encountered) down to 2 ± 2% (0.2 per 10 synapses encountered; p < 0.01 by the K–S nonparametric test; Fig. 4I).
Figure 4.

Hippocampi with genetic deletion of GLT-1 in neurons exhibit reduced expression of GLT-1 in axons forming asymmetric synapses onto dendritic spines. D, E, LM-ICC labeling of GLT-1 in the hippocampi of animals with genetic deletion of GLT-1 within neurons (GLT-1Δ/Δ;Syn-Cre; referred to as syn/cre+), whereas A, B, show LM-ICC from GLT-1flox/flox littermates without Syn-Cre (referred to as syn/cre−). LM-ICC used the same monoclonal antibody as in previous figure, at a dilution of 1:10,000, and revealed dense labeling in all layers of the hippocampus (HPC), except for the pyramidal cell layer (A, B, D, E, G, H, black asterisks). At a higher-magnification, intense labeling of cells with morphological features of astrocytes (arrowheads) are evident throughout stratum radiatum (B) and stratum lacunosum-moleculare (slm; E). EM of the same brains (C, F) revealed the most intense labeling to be within astrocytic processes (white asterisks), many of which encircled asymmetric synapses with dark PSDs (C, F, arrowheads), indicating that these synapses were excitatory. Some of axons and presynaptic axon terminals forming asymmetric synaptic junctions within the syn/cre− tissue also exhibited GLT-1-immunoreactivity (C, LT). Most of the presynaptic axon terminals forming asymmetric synaptic junctions within syn/cre+ brains were unlabeled for GLT-1 (C, F, UT). Using the same antibody at the same dilution and processed in parallel with the syn/cre+ and syn/cre− tissue, LM-ICC of tissue with global knock-out of GLT-1 (Tanaka et al., 1997) exhibited no labeling throughout these layers of the hippocampus (H), whereas tissue collected from littermates without genetic deletion of GLT-1 revealed strong labeling (G). Quantitative analysis (I), conducted blind to genotype of tissue, using three syn/cre+ and two syn/cre− animals, confirmed reduction in the frequency of GLT-1-immunoreactivity within presynaptic axon terminals forming excitatory synaptic junctions onto spines in stratum radiatum (SR) of the hippocampus of syn/cre+ tissue (**p < 0.005 by the nonparametric K–S analysis). The values represent mean ± SEM. Calibration bars: A, D, 500 μm; B, E, G, H, 50 μm; C, F, 500 nm. bv, Blood vessels; slm, stratum lacunosum moleculare; LGN, lateral geniculate nucleus; UG, unlabeled glia.
Finally, to validate the specificity of the anti-GLT-1a monoclonal antibody, the same antibody at a dilution of 1:10,000 was applied to the hippocampus of the brain with global knock-out of GLT-1 (Tanaka et al., 1997) and to WT littermates. The GLT-1 labeling within WT brain was as observed earlier for the GFAP/cre− and syn/cre− tissue, with robust labeling throughout the synaptic neuropil, including the stratum lacunosum-moleculare and relatively less immunolabeling in the pyramidal cell layer (Fig. 4G, asterisk). In contrast, the hippocampal tissue of the global knock-out brain was devoid of astrocytic labeling as well as the unidentifiable small puncta. EM confirmed that the hippocampal tissue of the global knock-out was devoid of immunoreactivity in astrocytes, as well as in neurons, as was shown earlier using other GLT-1 antibodies (Chen et al., 2004). These data in aggregate confirmed the cell-type-specific deletion of GLT-1 in neurons and in astrocytes.
Contribution of astrocytic and neuronal GLT-1 to l-[3H]glutamate uptake in crude synaptosomes
To further validate the inactivation of the GLT-1 gene in the conditional knock-out mice, and compare the contributions of GLT-1-mediated uptake in neurons and in astrocytes, we assayed glutamate uptake in crude forebrain synaptosomes. l-[3H]glutamate uptake into crude synaptosomes isolated from the forebrains of astrocytic or neuronal GLT-1 KO mice and their littermate controls (14–19 weeks of age) was assayed. Interestingly, there was only a minor, insignificant, decrease in the uptake of l-[3H]glutamate into synaptosomes from astrocytic GLT-1 KO mice (GFAP/cre+, n = 4), compared with the controls (GFAP/cre−, n = 4; Fig. 5A; uptake reduced to 85.5 ± 4.7% of control, p = 0.58). Application of DHK (300 μm), a selective inhibitor of GLT-1 (Bridges et al., 1999), resulted in a further decrease in glutamate uptake (reduced to 21.0 ± 0.7% of control, n = 4) in GFAP/cre+ mice, revealing the contribution of neuronal GLT-1 plus the astrocytic GLT-1 residue to the glutamate uptake in crude synaptosomes. The uptake in GFAP/cre− mice (n = 4) was decreased to 25.6 ± 2.2% of control by application of 300 μm DHK. In contrast, neuronal deletion of GLT-1 produced a major loss in synaptosomal uptake of glutamate, which was reduced to 44.3 ± 3.4% (p < 0.05, n = 5) of the controls in these experiments using a single concentration (10 μm) of glutamate (Fig. 5B). Pharmacological inhibition by DHK (300 μm) resulted in decrease in syn/cre− (reduced to 26.9 ± 3.3% of controls, n = 5) and syn/cre+ (reduced to 17.2 ± 2.8% of controls, n = 5) mice. The uptake activities in all genotypes were greatly inhibited, to 15.0 ± 2.8% (GFAP/cre−), 9.0 ± 1.1% (GFAP/cre+), 14.7 ± 3.4% (syn/cre−), and 11.7 ± 1.8% (syn/cre+) of control, respectively, by 30 μm TBOA, which is a broad-spectrum competitive high affinity glutamate transporter antagonist (Fig. 5A,B). Increasing the concentration of TBOA to 300 μm almost completely abolished the glutamate uptake in all the genotypes (reduced to 4.5 ± 1.3%, 3.5 ± 1.2%, 2.8 ± 1.3% and 2.7 ± 0.8% of control in GFAP/cre−, GFAP/cre+, syn/cre−, and syn/cre+ mice, respectively; Fig. 5A,B). Given prior genetic evidence that synaptosomal glutamate uptake is almost exclusively mediated by GLT-1 (Tanaka et al., 1997), the discrepancy between the effectiveness of DHK and TBOA in inhibiting glutamate uptake in synaptosomes from the conditional knock-outs and control animals seemed likely to be due to the relatively low affinity of DHK (Robinson et al., 1993; Chen et al., 2002; Kabakov and Rosenberg, 2009), and the fact that in these assays we used 10 μm glutamate. Indeed, we found that, at a glutamate concentration of 0.5 μm, 300 μm DHK abolished ∼85% of the glutamate uptake in crude synaptosomes isolated from syn/cre+ mice and their controls (data not shown).
Figure 5.

Synaptosomal uptake of l-[3H]glutamate was significantly decreased in neuronal GLT-1 knock-out but not in astrocytic GLT-1 knock-out. A, Astrocytic knock-out of GLT-1 (GLT-1Δ/Δ;GFAP-Cre ERT2 referred to as GFAP/cre+; n = 4) produced no significant decrease (p = 0.58) of l-[3H]glutamate uptake into crude synaptosomes compared with the littermate controls (GFAP/cre−, pooled data from 4 experiments that were performed). B, Neuronal knock-out of GLT-1 (GLT-1Δ/Δ;Syn-Cre referred to as syn/cre+; pooled data from five experiments that were performed) resulted in a significant loss (*p < 0.05) of l-[3H]glutamate uptake into crude synaptosomes compared with syn/cre− controls (n = 5). Results of pharmacological inhibition by 300 μm DHK, 30 μm TBOA and 300 μm TBOA are also shown (A, B). C, D, Saturation analysis of glutamate uptake into synaptosomes isolated from astrocytic and neuronal GLT-1 knock-out mice and their littermate controls. No apparent change was observed in the Vmax value obtained for GFAP/cre+ mice compared with GFAP/cre− mice (a representative experiment of 2 that were performed in C). However, the Vmax value obtained for syn/cre+ mice was decreased (∼40% reduction) compared with that of syn/cre− mice (n = 4, p < 0.05). Pooled results of four experiments are shown in D. No change of Km values was found in GFAP/cre+ (n = 2) or syn/cre+ mice (n = 4) compared with their controls. E, F, Results using d-[3H]aspartate uptake to measure glutamate transport activity in astrocytic (n = 3, p > 0.05) and neuronal GLT-1 knock-out mice (n = 3, *p < 0.05) were similar to those using l-[3H]glutamate. Results are pooled from three separate experiments for each genotype.
We also performed saturation assay of glutamate uptake into crude synaptosomes from the mutant mice and their littermate controls to determine maximal velocity (Vmax) and glutamate binding affinity (Km) values for glutamate transport (Fig. 5C,D). Vmax values determined using GFAP/cre+ brain synaptosomes were, in two experiments, 11.1 (Fig. 5C) and 19.2 pmol/μg/min, similar to values obtained using GFAP/cre− brain synaptosomes [11.7 (Fig. 5C) and 18.3 pmol/μg/min]. Km values were 9.7 (Fig. 5C) and 10.2 μm for the GFAP/cre− mice and 12.4 (Fig. 5C) and 13.0 μm for the GFAP/cre+ mice. The mean Vmax value of syn/cre+ synaptosomes in four experiments was 7.2 ± 0.9 pmol/μg/min (n = 4), significantly lower (40.5%) than that of syn/cre− synaptosomes (12.1 ± 1.8 pmol/μg/min, n = 4, p < 0.05). There was no difference in Km values between the syn/cre− (9.7 ± 0.6 μm, n = 4) and syn/cre+ mice (11.8 ± 0.9 μm, n = 4, p > 0.05). Pooled data from the four experiments that were performed are shown in Figure 5D.
Similar results were observed when d-[3H]aspartate was used instead of l-[3H]glutamate for the measurement of synaptosomal uptake in astrocytic or neuronal GLT-1 knock-out mice (Fig. 5E,F). No significant reduction was found in the d-aspartate uptake in the astrocytic GLT-1 knock-out mice (reduced to 87.5 ± 14.5% of control, n = 3, p = 0.49; Fig. 5E). Neuronal deletion of GLT-1 caused a significant decrease of d-aspartate uptake (reduced to 51.3 ± 5.4% of control, n = 3, p < 0.05; Fig. 5F).
Phenotypic characterization
We examined lifespan and weight gain, and performed a basic behavioral screen (SHIRPA) in the astrocytic and neuronal GLT-1 knock-out mice. The Kaplan–Meier survival curves showed that GFAP/cre+ mice (n = 38) died significantly (p < 0.01) earlier than the control mice (GFAP/cre−, n = 24), with median survival at 23 weeks of age (Fig. 6A). We found four GFAP/cre− mice died within the time period of observation. The death of control GFAP/cre− mice may have been caused by the side effects of tamoxifen administration (Andersson et al., 2010). The weight gain of GFAP/cre+ mice was significantly less than GFAP/cre− mice starting from 10 weeks of age (21.1 ± 0.9 g, n = 24 compared with 24.9 ± 0.7 g, n = 20, p < 0.01; Fig. 6B). However, both the mortality and diminished weight gain are less severe than reported for the global GLT-1 knock-outs, which had a 50% mortality at 6 weeks on the original genetic background and gained weight more slowly from early life compared with control animals (Tanaka et al., 1997). We performed the same evaluation on the neuronal GLT-1 knock-outs. The survival curves of syn/cre+ mice (n = 10) and their littermate controls (syn/cre−, n = 10) were indistinguishable (p = 1.000) up to 70 weeks of age (Fig. 6C). The weight gain of syn/cre+ (n = 19) was the same as controls (syn/cre−, n = 17) up to 24 weeks of age (Fig. 6D).
Figure 6.

Mortality and weight deficit were found in astrocytic GLT-1 knock-out, but not in neuronal GLT-1 knock-out. A, Kaplan–Meier survival curves of GLT-1Δ/Δ;GFAP-Cre ERT2 mice (GFAP/cre+, n = 38) compared with control animals (GFAP/cre−, n = 24). The curves were statistically different (p < 0.01). B, Body weight (mean ± SE) of GFAP/cre+ mice (n = 24) and their littermate controls (GFAP/cre−, n = 20; *p < 0.05, **p < 0.01, ***p < 0.001). C, Kaplan–Meier survival curves of GLT-1Δ/Δ;Syn-Cre mice (syn/cre+; n = 10) compared with control animals (syn/cre−, n = 10). No difference was detected between these two curves (p = 1.000). D, Body weight (mean ± SD) of syn/cre+ mice (n = 19) and their littermate controls (syn/cre−, n = 17). No difference was detected in the body weight of syn/cre+ mice compared with syn/cre− mice. E, F, No significant effects on total SHIRPA scores were observed by conditional deletion of GLT-1 in astrocytes (GFAP/cre+, n = 9) or in neurons (syn/cre+, n = 18) compared with controls (GFAP/cre, n = 12; syn/cre−, n = 19).
The primary SHIRPA screen provides a comprehensive assessment of general health and behavior of genetically altered mice (Brooks and Dunnett, 2009). Overall, the performance of the astrocytic GLT-1 knock-out mice (GFAP/cre+) and the controls (GFAP/cre−; excluding body weight) were similar (p > 0.05) in SHIRPA testing: total SHIRPA scores of GFAP/cre+ (n = 9) and GFAP/cre− mice (n = 12) were 16.6 ± 2.7 and 15.8 ± 1.3, respectively (Fig. 6E). No significant difference (p > 0.05) was found between neuronal GLT-1 knock-out (syn/cre+, n = 18) and the control mice (syn/cre−, n = 19) in the SHIRPA screen: total SHIRPA scores of syn/cre+ and syn/cre− were 19.8 ± 1.0 and 20.4 ± 1.0, respectively (Fig. 6F).
Electroencephalographic assessment by scalp and tethered recording
It has been previously reported that global GLT-1 knock-out mice die due to intractable seizures (Tanaka et al., 1997). However, we did not observe any spontaneous clinical seizures in either astrocytic or neuronal GLT-1 knock-out mice. To test for electrographic seizures, we performed 20 min scalp EEG recording on these mice. Representative baseline EEG (Fig. 7Ai) and spike-trains (seizures; Fig. 7Aii) are shown in Figure 7A. We found more electrical seizure events in astrocytic GLT-1 knock-out mice (GFAP/cre+, n = 6) compared with the controls (GFAP/cre−, n = 7). The difference in the number of automatically detected spike-trains per 20 min of EEG was statistically significant (18.7 ± 7.2 for GFAP/cre+ vs 1.9 ± 0.5 for GFAP/cre−; unpaired t test, p = 0.027; Fig. 7B, first two bars). No significant increase in the seizure events was detected in the neuronal GLT-1 knock-out mice (syn/cre+; 0.47 ± 0.24, n = 3) relative to the control group (syn/cre−; 0.2 ± 0.2, n = 4; p = 0.429, unpaired t test; Fig. 7B, second two bars). We then performed tethered EEG in freely moving animals for 48–72 h to increase the probability of detecting seizure events with long-term recordings. The number of digitally counted spike-trains per 24 h of tethered EEG recording was identical between the neuronal GLT-1 knock-out (syn/cre+; 8.9 ± 6.2, n = 6) mice and the WT controls (syn/cre−; 9.4 ± 4.9, n = 6, unpaired t test, p = 0.95; Fig. 7B, last two bars).
Figure 7.

Electroencephalographic seizures were observed in astrocytic GLT-1 knock-out mice, but not in neuronal GLT-1 knock-out mice. A, Representative EEG traces (from astrocytic GLT-1 knock-out) showing: (Ai) sample baseline EEG without spikes; (Aii) sample EEG shows two characteristic automatically detected spike-trains, indicated by a black bar, with spiking activity lasting >1 s in each case. B, The number of automatically detected spike-trains per 20 min of scalp EEG recording in GLT-1Δ/Δ;GFAP-Cre ERT2 mice (GFAP/cre+, n = 6) was significantly higher than in the GFAP/cre− controls (n = 7, p = 0.027; B, first two bars); the number of automatically detected spike-trains per 20 min of scalp EEG recording in GLT-1Δ/Δ;Syn-Cre mice (syn/cre+, n = 3) was not different from syn/cre− controls (n = 4, p = 0.429; B, middle two bars); the number of digitally counted spike-trains per 24 h of tethered EEG recording in syn/cre+ mice (n = 6) were not significantly different from syn/cre− controls (n = 6, p = 0.95; B, last two bars).
The l-[3H]glutamate uptake into proteoliposomes
In the glutamate uptake experiments reported above, we found a discrepancy between the low protein level of neuronal GLT-1 (∼6% of total GLT-1 protein; Furness et al., 2008) and the high uptake activity (∼40% of total; Fig. 5D) mediated by it in the crude synaptosomes. This discrepancy between GLT-1 protein distribution and uptake activity may be due to less efficient resealing of astroglial membranes than of axon-terminals after homogenization of the tissue. Another possibility is post-translational modifications making neuronal GLT-1 more active than astroglial GLT-1. To test these hypotheses, forebrains from astrocytic and neuronal GLT-1 knock-out mice were solubilized and reconstituted in artificial membranes (liposomes). The uptake activity in these liposomes correlated with the labeling of immunoblots: the glutamate uptake in proteoliposomes prepared from astrocytic GLT-1 knock-out mice (GFAP/cre+, n = 3) was 24.2 ± 4.1% of the uptake into proteoliposomes from control animals (GFAP/cre−, n = 3, p < 0.001; Fig. 8A). No significant decrease in uptake was produced by deleting GLT-1 in neurons (n = 3, p = 0.065; Fig. 8C). It should be noted that the glutamate uptake in proteoliposomes is predominantly due to GLT-1 (Holmseth et al., 2012a). As expected, therefore, addition of DHK (0.5 mm), a selective inhibitor of GLT-1 at this concentration (Bridges et al., 1999), inhibited ∼95% glutamate uptake activity. Addition of PMB-TBOA (1 μm; Shimamoto, 2008), a nonselective glutamate transporter inhibitor, inhibited ∼98% of the uptake. Also, as expected, the uptake depended on the electrochemical gradients, as is illustrated by the fact that addition of nigericin (3 μm), an ionophore that performs electroneutral exchange of alkali metal cations with H+ (Pressman and Fahim, 1982) and thereby runs down the sodium and potassium gradients, abolished all uptake activity (Fig. 8A,C). The immunoblots in Figure 8A,C show the expression of GLT-1 and GLAST in the animals that were used in this assay (GFAP/cre+ and syn/cre+ are referred as “cKO”; GFAP/cre− and syn/cre− are referred as “WT”; Fig. 8A,C, insets). The SDS extracts from global GLT-1 (Tanaka et al., 1997) and GLAST (Watase et al., 1998) knock-out mice (referred to as “KO”; Fig. 8C, inset) were used as negative controls for antibody specificity. The conditional deletion of GLT-1 did not affect the [3H]-GABA uptake in proteoliposomes compared with those prepared from WT tissue, indicating that the reconstitution process was successful (Fig. 8B,D). These data do not give any indication that neuronal GLT-1 molecules differ from astroglial GLT-1 molecules (e.g., due to different post-translational modifications) as the reconstitutable uptake activity correlates with the immunoblots, but support the hypothesis of inefficient sealing of astrocyte membranes in the crude synaptosome preparation.
Comparison of uptake of l-[3H]glutamate in different fractions of homogenized brain
Because of the possibility that the astrocyte contribution to glutamate uptake into crude synaptosomes in these experiments was underestimated by using the P2 pellet (crude synaptosomes) for these studies, as is conventional, we compared glutamate uptake in uncentrifuged homogenate, the P1 pellet that is normally discarded, and the P2 pellet (Fig. 9A). We found a similar loss of glutamate uptake in these three different preparations (Fig. 9A), all produced from the same neuronal GLT-1 knock-out mice (n = 3) and their littermate controls (n = 3) in three separate experiments. In Figure 9A, glutamate uptake was reduced to 67.4 ± 6.2% (p < 0.01), 65.7 ± 3.5% (p < 0.001), and 65.9 ± 5.6% (p < 0.01) of their controls in uncentrifuged homogenate, P1 and P2 pellet, respectively. These results suggest that the significant decrease of glutamate uptake into crude synaptosomes prepared from the P2 pellet is not due to excluding astrocyte membranes from this preparation.
Figure 9.

Investigation of the basis for disproportionate glutamate uptake by neuronal GLT-1 in synaptosomes. A, Similar reduction of glutamate uptake was detected in the uncentrifuged homogenate (**p < 0.01), P1 (***p < 0.001), and P2 (crude synaptosomes; **p < 0.01) fractions isolated from neuronal GLT-1 KO mice (syn/cre+; n = 3) compared with their controls (syn/cre−; results pooled from three experiments that were performed). B, C, Approximately 50% inhibition of net uptake of l-[3H]glutamate by 3 μm nigericin (nig) was found in crude synaptosomes isolated from GFAP/cre+ (**p < 0.01, n = 3), syn/cre+ (**p < 0.01, n = 3), and their control mice (GFAP/cre−: ***p < 0.001, n = 3; syn/cre−: *p < 0.05, n = 3), respectively. Results are pooled from three experiments. D, Nigericin-sensitive component (net uptake fraction) of glutamate uptake in GFAP/cre+, syn/cre+ and littermate control mice was calculated from data shown in B and C. No significant differences were observed between the net uptake fractions of the different genotypes (n = 3 for each genotype).
Synaptosomes perform net uptake of l-[3H]glutamate
Glutamate transporters, like other transporters, participate in exchange reactions (Danbolt and Storm-Mathisen, 1986; Pines and Kanner, 1990). Externally added radioactively labeled substrate can accumulate inside synaptosomes either by exchange with internal K+ (net uptake) or by exchange with internal substrate (heteroexchange). These two processes occur at the same time (Volterra et al., 1996; Bergles and Jahr, 1998) and importantly, with similar rates (Zhou et al., 2014). These considerations raise the question of how much of the apparent uptake of radioactive substrates into terminals is due to net uptake or to heteroexchange. Nerve terminals have high levels of internal glutamate (Storm-Mathisen and Ottersen, 1990; Gundersen et al., 1996). The cytosolic concentration is unknown, but can potentially be high if glutamate leaks out from the synaptic vesicles. This implies that terminals have a greater potential for heteroexchange than astrocytes because glutamate is quickly metabolized in the latter resulting in lower internal levels. Here we have attempted to estimate how much is net uptake and how much is exchange in the synaptosome preparation. The ionophore nigericin (Pressman and Fahim, 1982) was used to compromise the transmembrane gradients. In all genotypes, application of 3 μm nigericin resulted in reduction of glutamate uptake into crude synaptosomes to ∼50% of uptake in the absence of nigericin (Fig. 9B,C). The nigericin-sensitive fraction of uptake (net uptake) in each genotype is shown in Figure 9D (GFAP/cre−: 53.4 ± 2.3%, GFAP/cre+: 44.3 ± 5.0%, syn/cre−: 57.6 ± 13.9%, syn/cre+: 55.6 ± 6.3% of the total uptake in each genotype; n = 3 for each). These data indicate that net uptake mediated by astrocytic GLT-1 (remainder in syn/cre+; 55.6 ± 6.3%) is not significantly greater than net uptake mediated by neuronal GLT-1 (remainder in GFAP/cre+; 44.3 ± 5.0%; p = 0.23). These data further demonstrate a significant net uptake component even in axon terminals that have been subjected to the harsh treatment of tissue homogenization, indicating that the “excess” glutamate uptake measured by radioactive substrates in axon terminals is not due to excess heteroexchange into these structures.
Discussion
The elusive presynaptic glutamate transporter
Nerve terminal uptake of glutamate has been a puzzle for more than 40 years (Danbolt, 2001, section 4.2). Specifically, whether glutamatergic axon terminals express glutamate transporters has been very controversial over this period of time. GLT-1 was detected in axon terminals in the hippocampus (Chen et al., 2004; Furness et al., 2008), and it was shown to be the only glutamate transporter in terminals (Furness et al., 2008; Holmseth et al., 2012a). However, whether glutamate clearance by glutamatergic axon terminals has any functional significance, given the very high levels of expression of GLT-1 and GLAST in astrocyte processes near excitatory synapses (Lehre and Danbolt, 1998) and the relatively low levels of expression of GLT-1 in axon terminals (Furness et al., 2008) remains uncertain (Tzingounis and Wadiche, 2007).
We report the creation of a conditional GLT-1 knock-out mouse line allowing deletion of GLT-1 in neurons without significantly affecting astrocytes and vice versa. We found that deletion of GLT-1 in astrocytes had the most serious consequences (excess mortality, lower body weight, and spontaneous seizures) while deletion in neurons did not give rise to any apparent neurological abnormalities. This is in agreement with the notion that the clearance of extracellular glutamate is mostly the responsibility of glial glutamate transporters. However, we found that neuronal GLT-1 but not astrocytic GLT-1 contributed significantly to glutamate uptake in crude synaptosomes isolated from forebrain.
The physiological relevance of expression of GLT-1 in axon terminals
The functional significance of the expression of glutamate transporters in presynaptic terminals needs to be determined. Although the primary SHIRPA behavioral screen has a broad coverage, it should be kept in mind it is not particularly sensitive (Brooks and Dunnett, 2009). Thus, more subtle changes might have gone undetected. In this context, it is worthwhile to recall that it took a long time to discover neurological abnormalities in mice and humans lacking the EAAC1 glutamate transporter. EAAC1 deficiency turns out to be associated with dicarboxylic aminoaciduria and obsessive-compulsive disorders in human (Bailey et al., 2011; Brandl et al., 2012; Walitza et al., 2010), and neurodegeneration (as well as dicarboxylic aminoaciduria) in mice (Aoyama et al., 2006; Zhou and Danbolt, 2013).
The GLT-1 in terminals may be unlikely to have any noticeable impact on diffusion of glutamate out of the synaptic cleft or on ambient glutamate in the presence of astrocyte processes. The reason for this is that the amount of GLT-1 protein is only ∼10% of that expressed in astroglia, and it is distributed in most of the axon-terminal membranes, thereby not being concentrated in the synapses (Furness et al., 2008). This implies that the density is not expected to be sufficient to capture any major proportion of released glutamate molecules (Holmseth et al., 2012a, see their Discussion concerning the densities of EAAC1). Exceptions to this prediction may be excitatory synapses which do not have adjacent astrocyte processes (∼40% of the synapses in the CA1 region of the hippocampus) and diffusion pathways between excitatory synapses that are not bounded by glial membranes (∼2/3 of the nearest neighbor pathways in the CA1 region; Ventura and Harris, 1999). Axon-terminal GLT-1 will catalyze direct recycling of released glutamate thereby short-circuiting the so-called glutamate-glutamine cycle (Conti and Melone, 2006; McKenna, 2007; Coulter and Eid, 2012; Hertz, 2013). In fact, there is some support in the literature for the notion that this may be physiologically relevant (Bjørnsen et al., 2014), but more studies are required.
The discrepancy between low expression of GLT-1 protein in neurons and significant contribution to synaptosomal glutamate uptake
A previous study reported the unexpected observation that GLT-1 in terminals accounted for approximately half of the 3H-d-aspartate uptake observed in hippocampal slices despite GLT-1 in axon terminals representing <10% of total GLT-1 protein (Furness et al., 2008). Heteroexchange is a confound of any transport assay using radioactive substrate (Kanner and Schuldiner, 1987), and an exchange cycle mediated by glutamate transporters has been characterized (Kanner and Bendahan, 1982; Kavanaugh et al., 1997). If (a) heteroexchange had been 10 times faster than net uptake and (b) terminals mostly performed exchange, then this could explain the apparent similar rates of uptake in glia and terminals despite 10 times differences in GLT-1 levels. However, the rates of uptake and heteroexchange are similar (Zhou et al., 2014). Further, as shown in the present study, terminals appear to perform net uptake. Thus, the relatively high capacity of uptake by GLT-1 into terminals relative to GLT-1 in astrocytes is not an in vitro artifact simply due to differential rates of net uptake and heteroexchange.
The simplest explanation why the synaptosome assay preferentially measures neuronal GLT-1 may be differences in mechanical properties of the cells. It is easy to imagine that terminals only connected via thin axons to soma can reseal to form tight compartments during homogenization of the tissue. In contrast, only small membrane fragments from astrocytes are likely to do the same (Dodd et al., 1981; Nakamura et al., 1993). If so, most of the GLT-1 in astrocytes may be in leaky compartments and thereby go undetected in the assay. In contrast, when all membranes, whether leaky or not, are solubilized and reconstituted in liposomes (artificial membranes), then there should not be any preference for neuronal GLT-1 provided neuronal and astroglial GLT-1 molecules behave the same way. This is in agreement with what we observed. The predominance of terminal labeling in hippocampal slices is harder to explain solely by different mechanical properties. Therefore, additional mechanisms may be at play. For instance, it should be considered whether the substrate leaks out from astrocytes via various forms of anion channels (Malarkey and Parpura, 2008).
Another possibility is that a substantial fraction of GLT-1 protein expressed in astrocytes is not functional, either because of compartmentalization, or because of a functional impairment related to cellular context. GLT-1 distributes between the plasma membrane and intracellular compartments (Zelenaia et al., 2000; Kalandadze et al., 2004; Susarla and Robinson, 2008; Foran et al., 2014), and transporter protein localized in intracellular compartments, in cells or crude synaptosomes, would not be expected to participate in the uptake of extracellular glutamate. Regarding the second possibility, it is well recognized that the cycling rate of glutamate transporters is too slow for them to remove glutamate in the time frame of a fast synaptic event, and that the control they exert on the concentration profile of synaptically released glutamate is due to their rapid binding and expression at high density (Tzingounis and Wadiche, 2007). Interestingly, fine astrocytic processes adjacent to synapses do not contain mitochondria (Peters et al., 1991): most perisynaptic glial processes are <100 nm in diameter whereas mitochondria are typically upward of 200 nm in diameter. These fine processes may therefore be metabolically handicapped with regard to being able to provide adequate ATP to keep up with the energy requirements of active transport. The essential function of GLT-1 (and GLAST) in these fine processes may be accomplished by binding, with net transport possibly disfavored by high packing density, membrane lipid environment, lack of energy producing metabolic machinery or other factors extrinsic to the transporters.
Changes in synaptosomal uptake of glutamate are likely due, in part, to neuronal GLT-1
The assay of synaptosomal uptake of glutamate is widely used in different brain regions to study glutamate transport activity in a variety of physiological and pathological conditions, response to drug treatment (Levenson et al., 2002; Pita-Almenar et al., 2006; Li et al., 2009; Inquimbert et al., 2012) and neurodegeneration (Danbolt, 2001; Maragakis and Rothstein, 2004; Sheldon and Robinson, 2007).
Reduced synaptosomal glutamate uptake has been reported in the postmortem Alzheimer's disease (AD) brains (Hardy et al., 1987; Procter et al., 1988). Soluble amyloid-β (Aβ) protein, which is correlated strongly to the pathology of AD, significantly reduced the uptake of radiolabeled glutamate to ∼70% of the control in crude synaptosomes isolated from hippocampus (Li et al., 2009), suggesting a possible pathogenetic mechanism in AD pathology involving interaction between soluble Aβ and GLT-1. However, work done in liposomes with reconstituted glutamate transporters failed to detect any differences in uptake activity between AD brains and controls (Beckstrøm et al., 1999). Given our findings in the present study, the discrepancy in these findings may be due to the different preparations used.
Previously, we found a lack of correlation between immunoblotting and synaptosomal uptake results in a study of a mouse model of Huntington's disease (Petr et al., 2013). This lack of correlation may now be explained by the limitation of the synaptosomal preparation itself: astrocytic GLT-1 is not proportionately represented by the synaptosomal uptake assay. Therefore, in assessing glutamate transport activity, it appears important to compare the uptake activity in synaptosomes with that in liposomes to detect the contributions from neuronal and astrocytic GLT-1, respectively. Importantly, changes in synaptosomal glutamate uptake that were observed in past studies to be induced by a variety of manipulations may reflect, at least in part, changes in uptake mediated by GLT-1 in neurons.
Footnotes
This work was supported by William Randolph Hearst Foundation, Deutsche Forschungsgemeinschaft, Hereditary Disease Foundation, Tommy Fuss Fund, Children's Hospital Intellectual and Developmental Disabilities Research Center Core Grant HD 18655, NINDS RO1NS066019, NEI Core Grant EY13079, NIMH R21-MH091445, Novo Nordisk Fonden Grant 10959 and the University of Oslo. We thank Drs David Conner (Department of Genetics, Harvard Medical School) and Margaret Thompson (Mouse Gene Manipulation Facility, Boston Children's Hospital) for their invaluable assistance in the production of the mutant mouse used in this study; Nicole Sabaliauskas, Gauri Wable, and Tara Chowdhury for their assistance with electron microscopic tissue processing; and Drs Chinfei Chen and Josh Zimmerberg for helpful comments about the paper.
The authors declare no competing financial interests.
References
- Andersson KB, Winer LH, Mørk HK, Molkentin JD, Jaisser F. Tamoxifen administration routes and dosage for inducible Cre-mediated gene disruption in mouse hearts. Transgenic Res. 2010;19:715–725. doi: 10.1007/s11248-009-9342-4. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Bailey CG, Ryan RM, Thoeng AD, Ng C, King K, Vanslambrouck JM, Auray-Blais C, Vandenberg RJ, Bröer S, Rasko JE. Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J Clin Invest. 2011;121:446–453. doi: 10.1172/JCI44474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beart PM. An evaluation of l-glutamate as the transmitter released from optic nerve terminals of the pigeon. Brain Res. 1976;110:99–114. doi: 10.1016/0006-8993(76)90211-0. [DOI] [PubMed] [Google Scholar]
- Beckstrøm H, Julsrud L, Haugeto O, Dewar D, Graham DI, Lehre KP, Storm-Mathisen J, Danbolt NC. Interindividual differences in the levels of the glutamate transporters GLAST and GLT, but no clear correlation with Alzheimer's disease. J Neurosci Res. 1999;55:218–229. doi: 10.1002/(SICI)1097-4547(19990115)55:2<218::AID-JNR9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Berger UV, Hediger MA. Comparative analysis of glutamate transporter expression in rat brain using differential double in situ hybridization. Anat Embryol. 1998;198:13–30. doi: 10.1007/s004290050161. [DOI] [PubMed] [Google Scholar]
- Berger UV, DeSilva TM, Chen W, Rosenberg PA. Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. J Comp Neurol. 2005;492:78–89. doi: 10.1002/cne.20737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørnsen LP, Hadera MG, Zhou Y, Danbolt NC, Sonnewald U. The GLT-1 (EAAT2; slc1a2) glutamate transporter is essential for glutamate homeostasis in the neocortex of the mouse. J Neurochem. 2014;128:641–649. doi: 10.1111/jnc.12509. [DOI] [PubMed] [Google Scholar]
- Brandl EJ, Müller DJ, Richter MA. Pharmacogenetics of obsessive-compulsive disorders. Pharmacogenomics. 2012;13:71–81. doi: 10.2217/pgs.11.133. [DOI] [PubMed] [Google Scholar]
- Bridges RJ, Kavanaugh MP, Chamberlin AR. A pharmacological review of competitive inhibitors and substrates of high-affinity, sodium-dependent glutamate transport in the central nervous system. Curr Pharm Des. 1999;5:363–379. [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB. Tests to assess motor phenotype in mice: a user's guide. Nat Rev Neurosci. 2009;10:519–529. doi: 10.1038/nrn2652. [DOI] [PubMed] [Google Scholar]
- Casper KB, Jones K, McCarthy KD. Characterization of astrocyte-specific conditional knock-outs. Genesis. 2007;45:292–299. doi: 10.1002/dvg.20287. [DOI] [PubMed] [Google Scholar]
- Chang P, Hashemi KS, Walker MC. A novel telemetry system for recording EEG in small animals. J Neurosci Methods. 2011;201:106–115. doi: 10.1016/j.jneumeth.2011.07.018. [DOI] [PubMed] [Google Scholar]
- Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci. 2002;22:2142–2152. doi: 10.1523/JNEUROSCI.22-06-02142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci. 2004;24:1136–1148. doi: 10.1523/JNEUROSCI.1586-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti F, Melone M. The glutamine commute: lost in the tube? Neurochem Int. 2006;48:459–464. doi: 10.1016/j.neuint.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60:1215–1226. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Storm-Mathisen J. Na+-dependent “binding” of d-aspartate in brain membranes is largely due to uptake into membrane-bounded saccules. J Neurochem. 1986;47:819–824. doi: 10.1111/j.1471-4159.1986.tb00684.x. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Pines G, Kanner BI. Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glycoprotein from rat brain. Biochemistry. 1990;29:6734–6740. doi: 10.1021/bi00480a025. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Storm-Mathisen J, Kanner BI. An [Na+ + K+] coupled l-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience. 1992;51:295–310. doi: 10.1016/0306-4522(92)90316-T. [DOI] [PubMed] [Google Scholar]
- Divac I, Fonnum F, Storm-Mathisen J. High affinity uptake of glutamate in terminals of corticostriatal axons. Nature. 1977;266:377–378. doi: 10.1038/266377a0. [DOI] [PubMed] [Google Scholar]
- Dodd PR, Hardy JA, Oakley AE, Edwardson JA, Perry EK, Delaunoy JP. A rapid method for preparing synaptosomes: comparison, with alternative procedures. Brain Res. 1981;226:107–118. doi: 10.1016/0006-8993(81)91086-6. [DOI] [PubMed] [Google Scholar]
- Foran E, Rosenblum L, Bogush A, Pasinelli P, Trotti D. Sumoylation of the astroglial glutamate transporter EAAT2 governs its intracellular compartmentalization. Glia. 2014;62:1241–1253. doi: 10.1002/glia.22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, Gundersen V, Holmseth S, Lehre KP, Ullensvang K, Wojewodzic M, Zhou Y, Attwell D, Danbolt NC. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2) Neuroscience. 2008;157:80–94. doi: 10.1016/j.neuroscience.2008.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganat YM, Silbereis J, Cave C, Ngu H, Anderson GM, Ohkubo Y, Ment LR, Vaccarino FM. Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. J Neurosci. 2006;26:8609–8621. doi: 10.1523/JNEUROSCI.2532-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma. 2013;30:1434–1441. doi: 10.1089/neu.2012.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen V, Danbolt NC, Ottersen OP, Storm-Mathisen J. Demonstration of glutamate/aspartate uptake activity in nerve endings by use of antibodies recognizing exogenous d-aspartate. Neuroscience. 1993;57:97–111. doi: 10.1016/0306-4522(93)90114-U. [DOI] [PubMed] [Google Scholar]
- Gundersen V, Ottersen OP, Storm-Mathisen J. Selective excitatory amino acid uptake in glutamatergic nerve terminals and in glia in the rat striatum: quantitative electron microscopic immunocytochemistry of exogenous (D)-aspartate and endogenous glutamate and GABA. Eur J Neurosci. 1996;8:758–765. doi: 10.1111/j.1460-9568.1996.tb01261.x. [DOI] [PubMed] [Google Scholar]
- Guo Z, Wang X, Xiao J, Wang Y, Lu H, Teng J, Wang W. Early postnatal GFAP-expressing cells produce multilineage progeny in cerebrum and astrocytes in cerebellum of adult mice. Brain Res. 2013;1532:14–20. doi: 10.1016/j.brainres.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cowburn R, Barton A, Reynolds G, Lofdahl E, O'Carroll AM, Wester P, Winblad B. Region-specific loss of glutamate innervation in Alzheimer's disease. Neurosci Lett. 1987;73:77–80. doi: 10.1016/0304-3940(87)90034-6. [DOI] [PubMed] [Google Scholar]
- Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honoré T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins formhomomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- Hertz L. The glutamate–glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front Endocrinol (Lausanne) 2013;4:59. doi: 10.3389/fendo.2013.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmseth S, Scott HA, Real K, Lehre KP, Leergaard TB, Bjaalie JG, Danbolt NC. The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience. 2009;162:1055–1071. doi: 10.1016/j.neuroscience.2009.03.048. [DOI] [PubMed] [Google Scholar]
- Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, Plachez C, Zhou Y, Furness DN, Bergles DE, Lehre KP, Danbolt NC. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci. 2012a;32:6000–6013. doi: 10.1523/JNEUROSCI.5347-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmseth S, Zhou Y, Follin-Arbelet VV, Lehre KP, Bergles DE, Danbolt NC. Specificity controls for immunocytochemistry: the antigen pre-adsorption test can lead to inaccurate assessment of antibody specificity. J Histochem Cytochem. 2012b;60:174–187. doi: 10.1369/0022155411434828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inquimbert P, Bartels K, Babaniyi OB, Barrett LB, Tegeder I, Scholz J. Peripheral nerve injury produces a sustained shift in the balance between glutamate release and uptake in the dorsal horn of the spinal cord. Pain. 2012;153:2422–2431. doi: 10.1016/j.pain.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen LL, Storm-Mathisen J. Uptake of (3H)glutamate in excitatory nerve endings in the hippocampal formation of the rat. Act Physiol Scand. 1976;96:22A–23A. [Google Scholar]
- Kabakov AY, Rosenberg PA. Evidence for change in current-flux coupling of GLT1 at high glutamate concentrations in rat primary forebrain neurons and GLT1a-expressing COS-7 cells. Eur J Neurosci. 2009;30:186–195. doi: 10.1111/j.1460-9568.2009.06809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalandadze A, Wu Y, Fournier K, Robinson MB. Identification of motifs involved in endoplasmic reticulum retention-forward trafficking of the GLT-1 subtype of glutamate transporter. J Neurosci. 2004;24:5183–5192. doi: 10.1523/JNEUROSCI.0839-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner BI, Bendahan A. Binding order of substrates to the sodium and potassium ion coupled l-glutamic acid transporter from rat brain. Biochemistry. 1982;21:6327–6330. doi: 10.1021/bi00267a044. [DOI] [PubMed] [Google Scholar]
- Kanner BI, Schuldiner S. Mechanism of transport and storage of neurotransmitters. CRC Crit Rev Biochem. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- Kavanaugh MP, Bendahan A, Zerangue N, Zhang Y, Kanner BI. Mutation of an amino acid residue influencing potassium coupling in the glutamate transporter GET-1 induces obligate exchange. J Biol Chem. 1997;272:1703–1708. doi: 10.1074/jbc.272.3.1703. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson J, Weeber E, Selcher JC, Kategaya LS, Sweatt JD, Eskin A. Long-term potentiation and contextual fear conditioning increase neuronal glutamate uptake. Nat Neurosci. 2002;5:155–161. doi: 10.1038/nn791. [DOI] [PubMed] [Google Scholar]
- Levi G, Raiteri M. GABA and glutamate uptake by subcellular fractions enriched in synaptosomes: critical evaluation of some methodological aspects. Brain Res. 1973a;57:165–185. doi: 10.1016/0006-8993(73)90574-X. [DOI] [PubMed] [Google Scholar]
- Levi G, Raiteri M. Detectability of high and low affinity uptake systems for GABA and glutamate in rat brain slices and synaptosomes. Life Sci I. 1973b;12:81–88. doi: 10.1016/0024-3205(73)90064-7. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-F. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2008;52:142–154. doi: 10.1016/j.neuint.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Glutamate transporters: animal models to neurologic disease. Neurobiol Dis. 2004;15:461–473. doi: 10.1016/j.nbd.2003.12.007. [DOI] [PubMed] [Google Scholar]
- McKenna MC. The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res. 2007;85:3347–3358. doi: 10.1002/jnr.21444. [DOI] [PubMed] [Google Scholar]
- McLennan H, Haldeman S. The actions of the dimethyl and diethyl esters of glutamic acid on glutamate uptake by brain tissue. J Neurochem. 1973;20:629–631. doi: 10.1111/j.1471-4159.1973.tb12164.x. [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, Conti F. Synaptic localization of GLT-1a in the rat somatic sensory cortex. Glia. 2009;57:108–117. doi: 10.1002/glia.20744. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Iga K, Shibata T, Shudo M, Kataoka K. Glial plasmalemmal vesicles: a subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia. 1993;9:48–56. doi: 10.1002/glia.440090107. [DOI] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP. Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J Neurosci. 2000;20:2749–2757. doi: 10.1523/JNEUROSCI.20-08-02749.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HD. The fine structure of the nervous system. New York: Oxford UP; 1991. [Google Scholar]
- Petr GT, Schultheis LA, Hussey KC, Sun Y, Dubinsky JM, Aoki C, Rosenberg PA. Decreased expression of GLT-1 in the R6/2 model of Huntington's disease does not worsen disease progression. Eur J Neurosci. 2013;38:2477–2490. doi: 10.1111/ejn.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines G, Kanner BI. Counterflow of l-glutamate in plasma membrane vesicles and reconstituted preparations from brain. Biochemistry. 1990;29:11209–11214. doi: 10.1021/bi00503a008. [DOI] [PubMed] [Google Scholar]
- Pita-Almenar JD, Collado MS, Colbert CM, Eskin A. Different mechanisms exist for the plasticity of glutamate reuptake during early long-term potentiation (LTP) and late LTP. J Neurosci. 2006;26:10461–10471. doi: 10.1523/JNEUROSCI.2579-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressman BC, Fahim M. Pharmacology and toxicology of the monovalent carboxylic ionophores. Annu Rev Pharmacol Toxicol. 1982;22:465–490. doi: 10.1146/annurev.pa.22.040182.002341. [DOI] [PubMed] [Google Scholar]
- Procter AW, Palmer AM, Francis PT, Lowe SL, Neary D, Murphy E, Doshi R, Bowen DM. Evidence of glutamatergic denervation and possible abnormal metabolism in Alzheimer's disease. J Neurochem. 1988;50:790–802. doi: 10.1111/j.1471-4159.1988.tb02983.x. [DOI] [PubMed] [Google Scholar]
- Rempe D, Vangeison G, Hamilton J, Li Y, Jepson M, Federoff HJ. Synapsin I Cre transgene expression in male mice produces germline recombination in progeny. Genesis. 2006;44:44–49. doi: 10.1002/gene.20183. [DOI] [PubMed] [Google Scholar]
- Roach MK, Davis DL, Pennington W, Nordyke E. Effect of ethanol on the uptake by rat brain synaptosomes of (3 H)DL-norepinephrine, (3 H)5-hydroxytryptamine, (3 H)GABA and (3 H)glutamate. Life Sci I. 1973;12:433–441. doi: 10.1016/0024-3205(73)90212-9. [DOI] [PubMed] [Google Scholar]
- Robertson EJ. Teratocarcinomas and embryonic stem cells: a practical approach. Washington DC: IRL; 1987. [Google Scholar]
- Robinson MB, Sinor JD, Dowd LA, Kerwin JF., Jr Subtypes of sodium-dependent high-affinity l-[3H]glutamate transport activity: pharmacologic specificity and regulation by sodium and potassium. J Neurochem. 1993;60:167–179. doi: 10.1111/j.1471-4159.1993.tb05835.x. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Püschel B, Jöns T, Kugler P. Expression of the glutamate transporter GLT1 in neural cells of the rat central nervous system: non-radioactive in situ hybridization and comparative immunocytochemistry. Neuroscience. 1996;71:989–1004. doi: 10.1016/0306-4522(95)00477-7. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K. Glutamate transporter blockers for elucidation of the function of excitatory neurotransmission systems. Chem Rec. 2008;8:182–199. doi: 10.1002/tcr.20145. [DOI] [PubMed] [Google Scholar]
- Storm-Mathisen J, Ottersen OP. Immunocytochemistry of glutamate at the synaptic level. J Histochem Cytochem. 1990;38:1733–1743. doi: 10.1177/38.12.1979340. [DOI] [PubMed] [Google Scholar]
- Susarla BT, Robinson MB. Internalization and degradation of the glutamate transporter GLT-1 in response to phorbol ester. Neurochem Int. 2008;52:709–722. doi: 10.1016/j.neuint.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Torp R, Danbolt NC, Babaie E, Bjørås M, Seeberg E, Storm-Mathisen J, Ottersen OP. Differential expression of two glial glutamate transporters in the rat brain: an in situ hybridization study. Eur J Neurosci. 1994;6:936–942. doi: 10.1111/j.1460-9568.1994.tb00587.x. [DOI] [PubMed] [Google Scholar]
- Trotti D, Volterra A, Lehre KP, Rossi D, Gjesdal O, Racagni G, Danbolt NC. Arachidonic acid inhibits a purified and reconstituted glutamate transporter directly from the water phase and not via the phospholipid membrane. J Biol Chem. 1995;270:9890–9895. doi: 10.1074/jbc.270.17.9890. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8:935–947. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–6906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Bezzi P, Rizzini BL, Trotti D, Ullensvang K, Danbolt NC, Racagni G. The competitive transport inhibitor l-trans-pyrrolidine-2,4-dicarboxylate triggers excitotoxicity in rat cortical neuron-astrocyte co-cultures via glutamate release rather than uptake inhibition. Eur J Neurosci. 1996;8:2019–2028. doi: 10.1111/j.1460-9568.1996.tb01345.x. [DOI] [PubMed] [Google Scholar]
- Walitza S, Wendland JR, Gruenblatt E, Warnke A, Sontag TA, Tucha O, Lange KW. Genetics of early-onset obsessive-compulsive disorder. Eur Child Adolesc Psychiatry. 2010;19:227–235. doi: 10.1007/s00787-010-0087-7. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Zelenaia O, Schlag BD, Gochenauer GE, Ganel R, Song W, Beesley JS, Grinspan JB, Rothstein JD, Robinson MB. Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Mol Pharmacol. 2000;57:667–678. doi: 10.1124/mol.57.4.667. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Danbolt NC. GABA and glutamate transporters in brain. Front Endocrinol (Lausanne) 2013;4:165. doi: 10.3389/fendo.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang X, Tzingounis AV, Danbolt NC, Larsson HP. EAAT2 (GLT-1; slc1a2) glutamate transporters reconstituted in liposomes argues against heteroexchange being substantially faster than net uptake. J Neurosci. 2014;34:13472–13485. doi: 10.1523/JNEUROSCI.2282-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]