

Abstract
Neural circuits that determine the perception and modulation of pain remain poorly understood. The prefrontal cortex (PFC) provides top-down control of sensory and affective processes. While animal and human imaging studies have shown that the PFC is involved in pain regulation, its exact role in pain states remains incompletely understood. A key output target for the PFC is the nucleus accumbens (NAc), an important component of the reward circuitry. Interestingly, recent human imaging studies suggest that the projection from the PFC to the NAc is altered in chronic pain. The function of this corticostriatal projection in pain states, however, is not known. Here we show that optogenetic activation of the PFC produces strong antinociceptive effects in a rat model (spared nerve injury model) of persistent neuropathic pain. PFC activation also reduces the affective symptoms of pain. Furthermore, we show that this pain-relieving function of the PFC is likely mediated by projections to the NAc. Thus, our results support a novel role for corticostriatal circuitry in pain regulation.
Keywords: affective, nucleus accumbens, optogenetics, pain, prefrontal cortex
Introduction
Brain circuits that regulate pain are not well understood. The classic descending modulatory pathway involves neurons from the midbrain and brainstem. In this circuit, pain activates neurons in the periaqueductal gray (PAG), which project to the rostral ventromedial medulla (RVM), which in turn projects to the spinal cord to regulate nociceptive signals (Fields et al., 1983; Morgan et al., 1989, 1991; Ossipov et al., 2014). This is a dynamic and bidirectional pathway, and it can both inhibit and facilitate pain. The contribution and mechanism of the cortical and limbic circuits in pain modulation, in comparison, are poorly characterized.
The prefrontal cortex (PFC) provides top-down control of sensory and affective processes (Ressler and Mayberg, 2007; Fuster, 2009; Arnsten et al., 2012). Human fMRI studies show that chronic pain causes prefrontal gray matter loss and altered functional connectivity between the PFC and other regions (Apkarian et al., 2004; Baliki et al., 2006; Geha et al., 2008; Moayedi et al., 2011; Kucyi et al., 2014). Increased prefrontal activity is found in chronic pain patients after cognitive behavioral therapy (Jensen et al., 2012). Meanwhile, animal studies have demonstrated that synaptic changes within the PFC occur in both acute and chronic pain models (Apkarian et al., 2005; Metz et al., 2009; Ji et al., 2010; Li et al., 2010; Ji and Neugebauer, 2011; Obara et al., 2013; Hung et al., 2014). Animal studies have further shown that electrical stimulation or opioid receptor activation in the PFC can inhibit nociception (Cooper, 1975; Hardy and Haigler, 1985; Hardy, 1985).
A major prefrontal target is the nucleus accumbens (NAc; Beckstead and Norgren, 1979; Sesack et al., 1989; Brog et al., 1993; Ishikawa et al., 2008). The NAc is known to regulate reward and aversion-based behavior (Arai et al., 1994; Kanju et al., 2008; Reynolds and Berridge, 2008; Damgaard et al., 2010; Lammel et al., 2011; Navratilova and Porreca, 2014), and it has been shown to play a role in pain regulation (Gear et al., 1999; Becerra et al., 2001; Magnusson and Martin, 2002; Becerra and Borsook, 2008; Geha et al., 2008; Gear and Levine, 2009; Baliki et al., 2010; Goffer et al., 2013; Navratilova and Porreca, 2014). Imaging data show that chronic pain alters connections between the PFC and NAc (Baliki et al., 2010, 2012). However, the exact function of this corticostriatal circuit in pain regulation is not established.
In this study, we examined the role of the PFC–NAc projection in pain regulation in a rat chronic neuropathic pain [spared nerve injury (SNI)] model. We found that optogenetic activation of the PFC provided significant antinociceptive effects, and it also relieved the aversive quality and depressive symptoms of pain. These results suggest that the PFC can regulate both sensory and affective pain symptoms. More importantly, we found that this pain-inhibiting effect is mediated by the prefrontal projection to the NAc. Thus, our study directly demonstrates a novel and important role for corticostriatal circuitry in pain regulation.
Materials and Methods
Animals.
All procedures in this study were approved by the New York University School of Medicine Institutional Animal Care and Use Committee as consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (publication number 85-23) to ensure minimal animal use and discomfort. Male Sprague Dawley rats were purchased from Taconic Farms and kept at Mispro Biotech Services Facility in the Alexandria Center for Life Science, with controlled humidity, room temperature, and 12 h (6:30 A.M. to 6:30 P.M.) light/dark cycle. Food and water were available ad libitum. Animals arrived to the animal facility at 250–300 g and were given on average 7 d to adjust to the new environment before the onset of any experiments.
Virus construction and packaging.
Recombinant adeno-associated virus (AAV) vectors were serotyped with AAV1 coat proteins and packaged by the viral vector core at the University of Pennsylvania. Viral titers were 5 × 1012 particles/ml for AAV1.hSyn.ChR2-eYFP.WPRE.hGH and AAV1.hSyn.eYFP.WPRE.hGH (Fenno et al., 2011; Nelson et al., 2013; Schneider et al., 2014).
Drugs.
Two,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX), purchased from Tocris Bioscience, was resuspended in 0.9% saline to a concentration of 0.55 nmol/μl and 0.5 μl was injected in each side. Infusions into guide cannula were given 15 min before behavioral assays. An equal volume of saline was injected as control.
Stereotaxic cannula implantation and intracranial viral injections.
As described previously (Goffer et al., 2013), rats were anesthetized with isoflurane (1.5–2%). In all experiments, virus was delivered to the prelimbic region of the prefrontal cortex (PL-PFC) only. For the PL-PFC, rats were bilaterally injected with 0.5 μl of AAV1.hSyn.ChR2-eYFP.WPRE.hGH or AAV1.hSyn.eYFP.WPRE.hGH at a rate of 0.1 μl/10 s using a 26 gauge 5 μl Hamilton syringe at anteroposterior (AP) +2.9 mm, mediolateral (ML) ±0.6 mm, and dorsoventral (DV) −3.6 mm. The microinjection needles were left in place for an additional 10 min, raised 1 mm, and left for another minute to allow for diffusion of virus particles away from injection site and to minimize spread of viral particles along the injection tract. Rats were then implanted with 26 gauge bilateral guide cannula (PlasticsOne) in the PL-PFC: AP +2.9 mm, ML ±0.6 mm, DV −2.6 mm. For the NAc core, rats were stereotaxically implanted with two 26 gauge guide cannulas bilaterally: AP +1.6 mm, ML ±2.9 mm, DV −5.6 mm, with tips angled 8° toward the midline. Cannulas were held in place by dental acrylic and patency was maintained with occlusion stylets. For behavioral experiments, 200 μm core optical fibers were threaded through guide cannulas.
For intracranial injections of NBQX, solutions were loaded into two 30 cm lengths of PE-50 tubing attached at one end to 10 μl Hamilton syringes filled with distilled water and at the other end to 33 gauge injector cannulas, which extended 1 or 2 mm beyond the implanted guides for the PL and NAc core, respectively. Over a period of 100 s, 0.5 μl was injected bilaterally. Injector cannulas were kept in place for another 60 s before removal from guides to allow diffusion of solution into the brain. Following the removal of injector cannulas from cannula guides, stylets were replaced, and animals were subject to behavior tests. Behavior tests were done 15 min after intracranial NBQX injections.
After animals were killed, cryogenic brain sections were collected with a thickness of 20 μm using a Microm HM525 cryostat and analyzed for cannula localization with histological staining; animals with improper cannula placements, low viral expression, or viral expression outside the PL-PFC were excluded from the study.
SNI surgery.
SNI surgery was previously described in detail (Decosterd and Woolf, 2000; Wang et al., 2011). Briefly, under isoflurane anesthesia (1.5–2%), the skin on the lateral surface of the right thigh of rat was incised and a section made through the biceps femoris muscle to expose three branches of the sciatic nerve: sural, common peroneal, and tibial nerves. The common peroneal and tibial nerves were tied with nonabsorbent 5-0 silk sutures at the point of trifurcation. The nerves were then cut distal to each knot, and ∼5 mm of the distal ends were removed. In sham surgeries (control), above nerves were dissected but not cut. Muscle layer was then sutured close, while the skin was stapled. Staples were removed before behavioral testing.
Immunohistochemistry.
Rats were deeply anesthetized with isoflurane and transcardially perfused with ice-cold PBS followed by 4% paraformaldehyde (PFA) in PBS. Brains were fixed in PFA overnight and then transferred to 30% sucrose in PBS to equilibrate for 3 d. Then, 20 μm coronal sections were cut on a cryostat and washed with PBS for 10 min. Sections were washed with PBS with 0.3% Triton X-100 (PBST) for 10 min, then incubated in PBST and 3% normal donkey serum (NDS) for 1 h at room temperature. Sections were incubated with primary antibody, anti-NeuN (Millipore; 1:500) overnight in PBST plus 3% NDS at 4°C. Sections were then washed in PBS and incubated with secondary antibody conjugated to Cy3 for 2 h at room temperature in PBST plus 3% NDS. Sections were washed in PBS and coverslipped with Vectashield mounting medium with DAPI. Images were acquired using a Nikon eclipse 80i microscope with a DS-U2 camera head.
In vivo electrophysiology.
Approximately 2 weeks after viral injection, animals were anesthetized with 2% isoflurane and secured to a stereotaxic apparatus. After exposing the skull, a 2.5-mm-diameter hole was drilled in the skull above the target region and a durotomy was carefully performed. A microwire array containing an optical fiber positioned 1 mm above the tips of six surrounding electrodes was then slowly lowered into place with the stereotaxic apparatus, using the coordinates mentioned above. The optical fiber was connected to a 473 nm laser, which was connected to a transistor-to-transistor logic (TTL) pulse-generating box (Tucker-Davis Technologies). An extra output on the TTL box and the headstage were then connected to the acquisition system to simultaneously record laser pulses and brain activity (Blackrock Microsystems). The signal was high-pass filtered online with a 250 Hz threshold to eliminate low-frequency events. Filtered 30 kS/s traces of all channels were imported into Matlab and plotted.
Slice preparation.
Acute slices of the PFC and the NAc were prepared from 8-week-old male Sprague Dawley rats. Animals were deeply anesthetized with a 1:1 ketamine/xylazine mixture and decapitated. The brain was rapidly removed and placed in ice-cold dissection buffer containing the following (in mm): 87 NaCl, 75 sucrose, 2 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 25 NaHCO3, 1.3 ascorbic acid, and 10 dextrose, bubbled with 95/5% O2/CO2, pH 7.4. Slices (300–400 μm thick) were prepared with a vibratome (Leica), then placed in warm dissection buffer (33–35°C) for <30 min, then transferred to a holding chamber containing artificial CSF (ACSF) at room temperature (ACSF in mm: 124 NaCl, 2.5 KCl, 1.5 MgSO4, 1.25 NaH2PO4, 2.5 CaCl2, and 26 NaHCO3). Slices were kept at room temperature (22–24°C) for ≥30 min before use. For experiments, slices were transferred to the recording chamber and perfused (2–2.5 ml min−1) with oxygenated ACSF at 33°C. PFC recordings were done 2 weeks after viral injections into the PFC, and NAc recordings were done 4 weeks after viral injections.
Whole-cell recordings.
Somatic whole-cell recordings were made from pyramidal cells in the prelimbic cortex and medium spiny neurons in the NAc, in either current-clamp or voltage-clamp mode with a Multiclamp 700B amplifier (Molecular Devices) using infrared differential interference contrast video microscopy (Olympus). Patch pipettes (3–8 MΩ) were filled with intracellular solution (in mm: 135 K-gluconate, 5 NaCl, 10 HEPES, 5 MgATP, 10 phosphocreatine, and 0.3 GTP). Data were filtered at 2 kHz, digitized at 10 kHz, and analyzed with Clampfit 10 (Molecular Devices). Optical stimulation (470 nm) was applied with a custom fit LED and driver (Mightex Systems). The stimulation pattern was similar to that used in vivo (20 Hz pulse trains; 10 ms on, 40 ms off cycle) at 200–600 mA. In both NAc and PFC, light was applied through a 40× objective over the field of view of the patched cell. NBQX (25 μm) was used to block excitatory synaptic transmission from the prefrontal projecting axons to the NAc.
Animal behavioral tests.
Animals used for behavior received either AAV1.hSyn.ChR2-eYFP.WPRE.hGH or AAV1.hSyn.eYFP.WPRE.hGH (control group) in the PL-PFC. Behavioral tests with optical stimulation in the PFC were done ≥2 weeks after viral injection, and tests with stimulation in the NAc core were done 6–8 weeks after injection to ensure optimal expression.
Before behavioral tests, stylets were removed, and cannula guides were inspected for obstructions. Multimode optical fibers (numerical aperture, 0.37; 200 μm core; Thorlabs) were precisely cut to the length of the guide cannulas to restrict the light locally. The fiber was connected to a 473 nm laser diode through an FC/PC adapter (Shanghai Dream Lasers). Laser intensity was measured with a power meter (Thorlabs) before being placed into cannulas and secured into place. Laser was delivered using a TTL pulse-generating box (Tucker-Davis Technologies). The output of the laser was split evenly to two optical fibers (for bilateral stimulation) using a splitter. Before behavior tests, laser output in each of the two fibers was verified with a power meter to ensure that equal power was provided. A laser protocol that included alternating light-on and light-off epochs for 30 s each was provided for the duration of the mechanical and cold allodynia tests, Hargreaves test, conditioned place preference (CPP) test, sucrose preference test (SPT), and the forced swim test (FST). Within the light-on epoch, the laser was pulsed at 20 Hz with 10 ms pulse length. After behavioral tests, fibers were carefully removed from the animals' cannula guides and stylets were replaced. Animals with occluded cannula guides were excluded from analysis.
Mechanical allodynia test.
A traditional Dixon up-down method with von Frey filaments was used to measure mechanical hypersensitivity as described previously (Chaplan et al., 1994; Bourquin et al., 2006; Wang et al., 2011). In brief, rats were individually placed into Plexiglas chambers over a mesh table and acclimated for 20 min before the onset of examination. Beginning with 2.55 g, von Frey filaments in a set with logarithmically incremental stiffness (0.45, 0.75, 1.20, 2.55, 4.40, 6.10, 10.50, 15.10 g) were applied to the lateral one-third of right paws (in the distribution of the sural nerve) of rats. A 50% withdrawal threshold was calculated as described previously (Wang et al., 2011).
Cold allodynia test.
Animals were individually placed into Plexiglas chambers as mentioned above and acclimated for 20 min. A drop of acetone was applied to the lateral plantar surface of the paws (in the distribution of the sural nerve). As previously described (Hao et al., 1999; Jørum et al., 2003; Wang et al., 2011), the following scoring system was applied: 0, no visible response or startle response lasting <0.5 s; 1, paw withdrawal lasting <5 s; 2, withdrawal lasting 5–10 s, with or without licking of the paws; 3, prolonged repetitive withdrawal lasting >10 s. Acetone was applied five times to each paw, and an average score was calculated. Cold hypersensitivity tests were typically done after mechanical allodynia tests on the same day, and observers were blinded to the test conditions.
Hargreaves test (Plantar test).
The Hargreaves test was performed to evaluate thermal hyperalgesia (Hargreaves et al., 1988). We used a radiant heat-emitting device (37370-Plantar Test, Ugo Basile) to produce acute noxious thermal stimuli and measured the latency to paw withdrawal. Rats were placed individually in a clear plastic chamber and left to acclimatize before the test. A mobile infrared heat generator with an aperture of 10 mm in diameter was aimed at the plantar surface of rat hindpaws, and an infrared intensity of 40 was used. The latency to paw withdrawal was recorded automatically. Minimum and maximum cutoffs were assigned at 1 and 30 s, respectively. Paw withdrawals due to locomotion or weight shifting were not counted, and the trials were repeated. The measurements were repeated five times at 5 min intervals on each paw, and the averages of the five measurements were taken and analyzed.
CPP.
CPP experiments were conducted in a standard three-compartment apparatus (Stoelting) consisting of two large compartments of equal size (45 × 40 × 35 cm) joined by a tunnel (40 × 9 × 35 cm). The movements of rats in each chamber were automatically recorded by a camera and analyzed with the ANY-maze software (Stoelting). The CPP protocol was modified from King et al. (2009), and it included preconditioning, conditioning, and testing phases. Preconditioning was performed across 3 d for SNI-treated or sham-treated rats. During preconditioning, all animals were exposed to the environment with full access to all chambers for 30 min each day. On day 3, the movement of each rat was recorded for 15 min and analyzed to verify the absence of any preconditioning chamber preference. Animals spending >80% or <20% (time spent) of the total time in any chamber were eliminated from further testing or analysis (∼20% of total animals). Following the preconditioning phase, the rats underwent conditioning for 4 d with alternating treatment-chamber pairings in the morning and afternoon. Half of the rats received light (laser) treatment-chamber pairing in the morning and no light treatment-chamber pairing in the afternoon. The other half of the rats received no light treatment-chamber pairing in the morning and light treatment-chamber pairing in the afternoon. Both light versus no light treatments and chamber pairings were counterbalanced, and ≥4 h separated morning and afternoon sessions. During conditioning, rats were placed in the paired chamber without access to the other compartments in the presence or absence of light treatment for 30 min. On the test day, the animals were placed into the neutral chamber and had access to all chambers for a total of 15 min. There were no light treatments on test days. During these 15 min, animal movements in each of the chambers were recorded, and the time spent in either of the treatment chambers was analyzed by the ANY-maze software. Increased time spent in a chamber associated with either light or no light treatment indicates preference for that chamber.
SPT.
As described previously (Wang et al., 2011; Goffer et al., 2013), before testing, animals were trained for 2–7 d to drink from two bottles (1% sucrose solution vs water). On the test day, these rats were acclimated to the test room for ≥20 min. Two bottles (1% sucrose solution vs water) were presented to each animal for 1 h. At the end of each test, sucrose preference was calculated as volume of sucrose consumed divided by total liquid consumption for each rat. Rats were not deprived of water or food. They were allowed to eat and drink ad libitum before the test.
FST.
As described previously (Wang et al., 2011; Goffer et al., 2013), animals were acclimated to the test room. On the first session of the test, each animal was placed for 15 min into a standard clear Porsolt chamber with water at 25°C filled to 25 cm. Afterward, the animal was taken out of the chamber, dried, and put back in its home cage. Twenty-four hours later, the animal was placed into the Porsolt chamber again under the same conditions for 5 min. The second session was recorded and analyzed. For experiments using optogenetics, light treatment occurred during the second session. Immobility was defined as a lack of movement of the hindpaws lasting >1 s. Two independent observers, blinded to the test conditions, examined and graded the total time of immobility for each rat using the ANY-maze software for video playback.
Locomotor activities.
Rats were placed in a 0.5 × 0.5 m chamber with an overhead camera. Using video analysis software (ANY-maze), animal movements were tracked during a 30 min test. Within the software, the total distance traveled was computed for 10 bins of 3 min. Later, videos were correlated with automatic results to verify tracking accuracy.
Statistics.
The results of behavioral experiments were given as mean ± SEM. For mechanical and cold allodynia, a two-way ANOVA with repeated measures and post hoc multiple-pairwise comparison Bonferroni's tests were used to compare the 50% withdrawal threshold or cold score for SNI-treated and sham-treated rats. Two-way ANOVA was also used to compare withdrawal threshold or cold score in SNI-treated and sham-treated rats with either channelrhodopsin-2 (ChR2)-enhanced yellow fluorescent protein (eYFP) or eYFP-only treatment. A two-tailed Student's t test was used to compare the effects of NBQX versus saline. A paired Student's t test was used to analyze the results from the Hargreaves test. For CPP tests, differences in time spent in each chamber before conditioning (preconditioning) and after conditioning (test) were analyzed using a two-way ANOVA with repeated measures followed by post hoc Bonferroni's tests. For the SPT, a two-tailed Student's t test was used to compare SNI-treated and sham-treated rats as well as to compare the effects of NBQX versus saline, and a two-way ANOVA with post hoc Bonferroni's tests was used to compare ChR2-eYFP versus eYFP-only treatment in sham and SNI groups. A two-tailed Student's t test was used for the analysis of total fluid consumption, sucrose consumption, and water consumption. A two-tailed Student's t test was used for the analysis of FSTs. A two-way ANOVA with repeated measures was used to compare locomotor activities for rats receiving light treatment versus no light treatment over 30 min. For all tests, a p value <0.05 was considered statistically significant. All data were analyzed using GraphPad Prism Version 6 software (GraphPad).
Results
SNI causes persistent pain
We studied the SNI model, a well known model for chronic neuropathic pain (Decosterd and Woolf, 2000; Wang et al., 2011; Goffer et al., 2013). We surgically resected the tibial and common peroneal branches of the sciatic nerve, causing permanent nerve injury and neuropathic pain (Decosterd and Woolf, 2000). Three days after SNI, rats experienced mechanical and cold allodynia (Fig. 1A,B; p < 0.0001). Sensory allodynia after SNI persisted for ≥14 d, as has been described earlier (Decosterd and Woolf, 2000; Wang et al., 2011; Goffer et al., 2013). In contrast, control rats that underwent sham operation did not display sensory allodynia (Fig. 1A,B).
Figure 1.
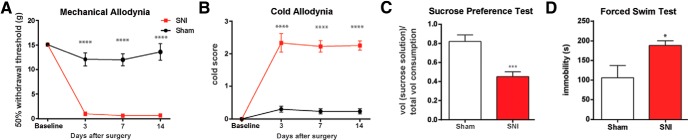
SNI evokes sensory and depressive symptoms of chronic pain. A, B, SNI-treated rats developed mechanical and cold allodynia, compared with sham-treated rats. Two-way ANOVA with repeated measures and Bonferroni's post-test, n = 6, p < 0.0001. C, SNI-treated rats developed decreased preference for sucrose in the SPT, a test of anhedonia. Student's t test, n = 8, p < 0.001. D, SNI-treated rats developed decreased motivation or behavioral despair, as indicated by prolonged immobility on the FST. Student's t test, n = 4, p < 0.05. Error bars show mean and SEM.
A key affective feature of pain is anhedonia, as shown by patients who lose interest in daily pleasurable activities or work (Dworkin and Gitlin, 1991; Miller and Cano, 2009). Anhedonia in rodents can be tested by the SPT (Nestler and Hyman, 2010). Recently, we and others have reported depression-like behaviors including anhedonia in rats with chronic pain (Gonçalves et al., 2008; Hu et al., 2009; Wang et al., 2011; Kim et al., 2012; Goffer et al., 2013). When we applied the SPT to rats 14 d after the SNI, we found that these rats, compared with control, showed decreased preference for sucrose (Fig. 1C; p < 0.001). These results confirmed previous findings and indicate that, in addition to developing sensory pain symptoms, SNI-treated rats also developed anhedonia.
Another important affective feature of pain, particularly in patients who suffer from persistent pain, is decreased motivation (Romano and Turner, 1985; Dworkin and Gitlin, 1991; Miller and Cano, 2009). Decreased motivation can be assessed by the FST (Porsolt et al., 1978). Increased immobility on the FST has been used as a standard measure for decreased motivation or behavioral despair in rodents (Porsolt et al., 1978), and numerous studies have demonstrated clinically relevant pharmacological validity of this measure (Nestler and Hyman, 2010). We and others have previously shown increased immobility on the FST in various rodent models of pain as well (Suzuki et al., 2007; Gonçalves et al., 2008; Hu et al., 2009; Wang et al., 2011; Kim et al., 2012; Goffer et al., 2013). As expected, we found that SNI, compared with sham operation, caused an increase in immobility (Fig. 1D; p < 0.05), indicating decreased motivation that accompanies neuropathic pain. Anhedonia and behavioral despair are hallmark features of depression (Nestler and Hyman, 2010). Thus, together, results on the SPT and FST confirm previous findings and demonstrate the presence of depressive symptoms of persistent neuropathic pain.
Activation of neurons in the PL-PFC inhibits neuropathic pain
We used an in vivo optogenetic approach to study the role of PFC in pain modulation. We injected AAV encoding light-sensitive ChR2 fused to e-YFP (ChR2-eYFP; Fenno et al., 2011; Witten et al., 2011; Nelson et al., 2013; Schneider et al., 2014) into neurons of the PL-PFC. Two weeks after viral injections, we found stable histologic expression of ChR2-eYFP that is restricted to the PL-PFC (Fig. 2A). Costaining with NeuN, a neuronal marker, confirms that ChR2s are expressed in neurons. To ensure that these ChR2s are functional, we performed electrophysiological recordings (Fig. 2B). First, we conducted whole-cell patch-clamp recordings in brain slices containing the PL-PFC. Here we found that light activation of the ChR2s in acute brain slices reliably elicited action potential spikes in neurons (Fig. 2C). To ensure that ChR2s are functional in the brain, we then conducted in vivo optrode recordings in the PL-PFC of anesthetized animals. We found that laser pulses elicited prefrontal spikes in vivo as well (Fig. 2D). This combination of immunohistological and electrophysiological data demonstrates that the optogenetic method can indeed activate neurons in the PL-PFC.
Figure 2.
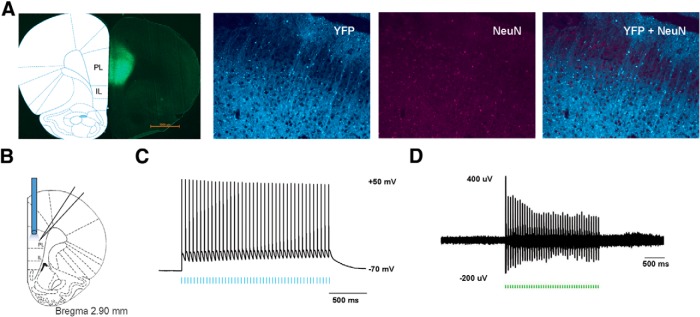
Functional expression of ChR2 in PL-PFC neurons. A, Histologic expression of ChR2-eYFP in the PL-PFC 14 d after viral injection. ChR2-eYFP was found to be restricted to neurons in the PL-PFC region. NeuN, Neuronal marker. B, Schematic showing electrophysiological recordings were conducted at the site of light delivery. Data were collected from prefrontal neurons 14 d after viral injections in the PL-PFC. C, Whole-cell patch-clamp recording showed that direct illumination with an LED in brain slices containing the PL-PFC elicited spikes with high fidelity in prefrontal neurons. LED pulses are represented below in blue. D, In vivo recording at the virus injection site with simultaneous photoactivation demonstrated that laser pulses reliably triggered spikes in PL-PFC neurons. Laser pulses are represented below in green.
We then examined the role of the PL-PFC in pain regulation. We first assessed the sensory component of pain by measurements of allodynia during PL-PFC photostimulation 14 d after the SNI or sham procedure (Fig. 3A,D). We compared rats injected with ChR2 to (control) rats injected with YFP only. We found that photoactivation of the PL-PFC completely reversed mechanical and cold allodynia in SNI-treated rats, as shown by an increase in mechanical threshold to pain and a decrease in cold sensitivity in the ChR2 group compared with the eYFP group (Fig. 3B,C; p < 0.0001). As expected, animals treated with sham surgery displayed no allodynic symptoms (Fig. 3B,C). Together these data indicate that activation of the PL-PFC can increase sensory thresholds for neuropathic pain.
Figure 3.
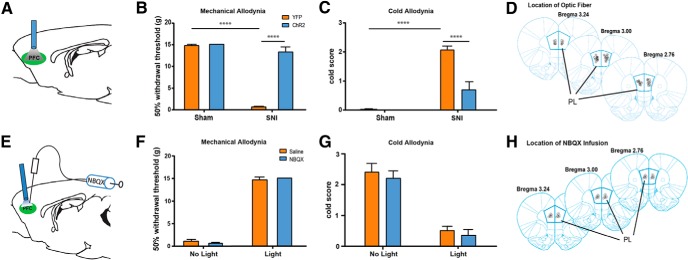
Activation of PL-PFC neurons relieves sensory allodynia in a rat chronic neuropathic pain (SNI) model. A, Optical fibers were positioned directly above viral injection sites in the PL-PFC. Photoactivation occurred during behavior tests. B, Illumination of ChR2-expressing neurons in the PL-PFC decreased mechanical allodynia in SNI-treated rats, but not in sham-treated rats. Light treatment did not have any effect on animals that received vectors containing eYFP only. Two-way ANOVA with Bonferroni's post-test, n = 8–9, p < 0.0001. Light treatment was performed ≥2 weeks after SNI/sham procedures. C, Illumination of ChR2-expressing neurons in the PL-PFC decreased cold allodynia in SNI-treated rats. Two-way ANOVA with Bonferroni's post-test, n = 8–9, p < 0.0001. Light treatment was performed ≥2 weeks after SNI/sham procedures. D, Schematic showing the locations of optic fibers. E, To test the contribution of local circuits within the PL-PFC to the antiallodynic effects of prefrontal activation, NBQX was injected into the PL-PFC before photoactivation and behavior assays. Tests were performed on SNI-treated rats that expressed ChR2. F, Compared with saline, NBQX infusion in the PFC did not alter mechanical allodynia in SNI-treated rats, with or without light activation of the PFC. Student's t test, n = 5 (light), n = 6–7 (no light), p > 0.05. G, NBQX infusion in the PFC did not alter cold allodynia in SNI-treated rats, with or without light activation of the PFC. Student's t test, n = 5 (light), n = 6–7 (no light), p > 0.05. H, Schematic showing the locations of NBQX infusion in the PL-PFC. Error bars show mean and SEM.
Prefrontal pyramidal neurons are known to form connections with local interneurons in the PFC. Thus, light activation of these pyramidal neurons could potentially activate local circuits within the PFC. To investigate the contribution of this local circuitry to pain regulation, we blocked local transmission within the PL-PFC with NBQX, an AMPA receptor antagonist (Fig. 3E,H). NBQX did not alter the allodynic thresholds in the absence of light activation of the PFC (Fig. 3F,G). We then applied light stimulation in the presence of NBQX so as to selectively activate prefrontal cells of projection pathways. We found that the application of NBQX in the PL-PFC did not alter the antinociceptive effect of PL-PFC activation (Fig. 3F,G). These experiments indicate that local circuits within the PL-PFC do not contribute significantly to the antinociceptive effects of prefrontal activation. Thus, the activation of distal projections from the PFC most likely provides these antinociceptive effects in the neuropathic pain model.
The effect of PFC activation on nociception can be due to an alteration of acute nociceptive threshold, or it can be due to specific changes to the central plasticity that occurs with chronic pain. To ensure that PFC activation continues to exert antinociceptive effects even after pain has become chronic, we measured the effect of PL-PFC activation on mechanical and cold allodynia 4 weeks after SNI. We found that activation of the PL-PFC was still able to relieve allodynia 4 weeks after the onset of pain (Fig. 4A,B; A, p < 0.001; B, p < 0.01). Next, to assess the possibility that PFC activation can alter acute nociception, we applied the Hargreaves test, a test that measures behavioral responses to acute noxious thermal stimuli (Hargreaves et al., 1988), to SNI-treated rats. We first measured the response to thermal stimuli in uninjured paws, and we found that PFC activation resulted in a significant increase in the latency to paw withdrawal (Fig. 4C; p < 0.05). These data indicate that PFC activation can indeed elevate the acute nociceptive threshold. Next, we measured the response to thermal stimuli in the injured paws, and we found that PFC activation also significantly increased the latency to withdrawal in these injured paws (Fig. 4D; p < 0.01). Thus, prefrontal stimulation appears to be able to reduce both acute nociception and hypersensitivity to evoked pain in the chronic neuropathic pain state.
Figure 4.
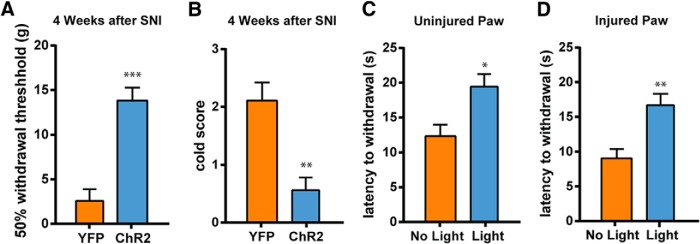
Activation of PL-PFC neurons elevates acute nociceptive threshold and relieves chronic neuropathic pain. A, Photoactivation of the PL-PFC relieved mechanical allodynia of the injured paws 4 weeks after SNI. Student's t test, n = 5, p < 0.001. B, Activation of the PL-PFC relieved cold allodynia of the injured paws 4 weeks after SNI. Student's t test, n = 4, p < 0.01. C, Activation of the PL-PFC increased the latency to withdrawal of the uninjured (contralateral) paws of SNI-treated rats on the Hargreaves test. Student's t test, n = 5, p < 0.05. D, Activation of the PL-PFC increased the latency to withdrawal of the injured paws of SNI-treated rats. Student's t test, n = 5, p < 0.01. Error bars show mean and SEM.
The PFC is known to regulate both sensory perception and affective behaviors (Ressler and Mayberg, 2007; Arnsten et al., 2012). We next investigated whether activation of the PL-PFC can also relieve affective symptoms of pain. Two key affective features of pain are the aversive quality of pain and the depressed mood induced by pain (Dworkin and Gitlin, 1991; Ohayon and Schatzberg, 2003; Edwards et al., 2009; King et al., 2009; Miller and Cano, 2009; De Felice et al., 2013; Goffer et al., 2013). The CPP test has been established to capture the negative reinforcement associated with the seeking of pain relief, and hence this test can unmask the aversive quality of ongoing or tonic pain (King et al., 2009; De Felice et al., 2013). We used this test to assess the impact of PFC activation. During the conditioning phase of CPP, we applied light stimulation in one chamber, and no light in the opposite chamber (Fig. 5A,B). After a period of conditioning, we found that SNI-treated rats showed a significant preference for the chamber associated with light treatment (Fig. 5B; p < 0.05). In contrast, sham-treated rats showed no such preference (Fig. 5B; p > 0.05). These data complement results from allodynia and Hargreaves tests and demonstrate that PFC activation can relieve ongoing pain as well as acute or evoked pain. In addition, these data also indicate that PFC activation not only can modify nociception, but it can also reduce the aversive quality of pain.
Figure 5.

Prefrontal activation relieves the aversive quality and depressive symptoms associated with chronic pain. A, Schematic of the light treatment. B, Light activation in the PL-PFC induced chamber preference in SNI-treated, but not sham-treated, rats. Two-way ANOVA with repeated measures and Bonferroni's post-test was used to compare preconditioning with test values, n = 7, p < 0.05 for the SNI group, n = 5, p > 0.05 for the sham group. C, Photoactivation of the PL-PFC improved sucrose preference in animals with chronic pain. Two-way ANOVA with Bonferroni's post-test, n = 8–10, p < 0.0001. D, Photoactivation of the PL-PFC had no effect on total fluid consumption during the SPT in SNI-treated rats. Student's t test, p > 0.05. E, Activation of the PL-PFC caused a trend of increase in sucrose consumption during the SPT in SNI-treated rats. Student's t test, p > 0.05. F, Activation of the PL-PFC caused a trend of decrease in water consumption during the SPT in SNI-treated rats. Student's t test, p > 0.05. G, Photoactivation of the PL-PFC decreased immobility on the FST in SNI-treated rats. Student's t test, n = 5–9, p < 0.01. H, Locomotor activity was not altered by PL-PFC photoactivation. Two-way ANOVA with repeated measures, n = 4–8, p > 0.05. Error bars show mean and SEM.
To further test the impact of prefrontal activation on the affective component of pain, we also examined depression-like behaviors using the SPT and the FST (Nestler and Hyman, 2010). We performed SPT during photoactivation of the PFC (Fig. 5A). Here we found that, in addition to improving sensory allodynia, photoactivation of the PL-PFC also restored the sucrose preference of SNI-treated rats to the control level (Fig. 5C; p < 0.0001). To rule out the possibility that changes in sucrose preference are affected by an alteration in the drinking ability of SNI-treated rats after PFC activation, we analyzed sucrose, water, and total fluid consumption. We did not find any difference in the amount of total fluid consumption between rats that received PL-PFC activation and rats that did not (Fig. 5D). Instead, SNI-treated rats that received PL-PFC activation consumed a higher volume of sucrose solution (Fig. 5E) and a smaller volume of water during the SPT (Fig. 5F), although these differences were not statistically significant. These results indicate that photoactivation of the PL-PFC did not alter the ability of rats to drink fluid. Thus, according to the standard interpretation of the SPT, activation of the PL-PFC decreased symptoms of anhedonia, a salient affective feature of pain, in SNI-treated rats.
Next, we turned to the FST, another test for depression-like behaviors. When we photoactivated PL-PFC during the FST, we found that SNI-treated rats experienced a significant decrease in immobility (Fig. 5G, p < 0.01), suggesting that PL-PFC activation relieved behavioral despair associated with pain. To rule out the possibility that locomotor changes affected our behavior results, we evaluated locomotion during light treatment. We observed no locomotor changes during PL-PFC photoactivation (Fig. 5H), and thus decreased immobility on the FST is most likely the result of increased motivation or improvements in behavioral despair.
Overall, our data demonstrate that the activation of neurons in the PL-PFC can produce strong antinociceptive effects in the persistent neuropathic pain state, and this region also has an important role in modulating affective pain behaviors.
Activation of the corticostriatal circuit inhibits pain
We next investigated which output pathways might mediate the pain-inhibiting effect of PL-PFC. Previous pain studies have assessed the roles of PFC projections to the PAG, thalamus, and amygdala (Bushnell et al., 2013; Cardoso-Cruz et al., 2013; Ji and Neugebauer, 2014). There is recent evidence, however, that connections between the PFC and the NAc—an important projection within the reward circuitry (Koob and Volkow, 2010)—are altered by chronic pain (Baliki et al., 2012). Interestingly, in addition to its well established role in reward-based learning (Kalivas et al., 2005), the NAc, particularly the core subregion of the NAc, has been shown to mediate pain-induced analgesia and control anhedonic symptoms of pain (Gear et al., 1999; Goffer et al., 2013). PL-PFC is known to project strongly to the NAc core (Sesack et al., 1989). Thus, we hypothesized that the analgesic effect of PL-PFC activation is mediated at least in part by its projection to the NAc core.
Four weeks after we injected ChR2-eYFP into the PL-PFC, we began to find robust expression in the NAc core, indicating that ChR2s are expressed in the axon terminals of prefrontal neurons that project to the NAc core (Fig. 6A). These findings are comparable to earlier reports of corticostriatal projections (Sesack et al., 1989). To ensure that ChR2 at the axon terminals of PFC neurons are functional, we performed in vivo and slice recordings. First, in vivo optrode recordings showed that illumination of the NAc core produced spikes, indicating that stimulation of the PL-PFC axons caused local neurotransmitter release to activate postsynaptic medium spiny neurons (MSNs) in the NAc (Fig. 6B). To rule out the possibilities of antidromic activation and/or activation of bypassing fibers, we made whole-cell recordings in slices containing the NAc core. Here, localized photoactivation reliably produced EPSPs in MSNs, and these EPSPs were large enough to trigger action potentials (Fig. 6C,D). Furthermore, light-induced EPSPs were blocked by NBQX (Fig. 6D), indicating that light treatment in the NAc promoted synaptic transmission to activate the MSNs. Together, these immunohistological and electrophysiological results demonstrate that pyramidal neurons in the PL-PFC form monosynaptic connections with MSNs in the NAc core, and that these synapses can be directly targeted by optogenetic stimulation.
Figure 6.

Light treatment in the NAc core selectively activates axon terminals of prefrontal neurons that project to the NAc core. A, ChR2-eYFP expression was found in the axon terminals of PL-PFC neurons in the NAc core 4 weeks after viral injections in the PL-PFC. B, In vivo recordings from the NAc core showed that light treatment in the NAc produced spikes. C, Whole-cell patch-clamp recordings from slices containing the NAc core showed that LED pulses produce EPSPs and spikes in MSNs of the NAc core. D, Light-induced EPSPs in the NAc were blocked by bath application of NBQX.
We next examined the role of this corticostriatal circuit in pain modulation by photoactivation of the prefrontal axons in the NAc core. Thus, we shone light in the NAc core after expressing ChR2-eYFP in PL-PFC neurons (Fig. 7A,D). We found that photoactivation of this corticostriatal projection had significant antiallodynic effects in SNI-treated rats, as it decreased their mechanical and cold hypersensitivity (Fig. 7B,C; B, p < 0.001; C, p < 0.01). In contrast, as expected, light treatment did not have any observable antiallodynic effects on sham-treated rats.
Figure 7.
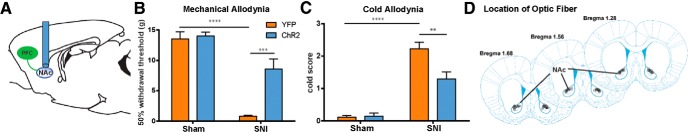
Activation of prefrontal projections to the NAc core has antinociceptive effects in the chronic neuropathic pain state. A, Schematic showing the experimental paradigm. Four weeks after viral injections in the PL-PFC, light treatment was performed in the NAc core to activate prefrontal neurons that project to the NAc during behavior tests. B, Photoactivation of PL-PFC axons in the NAc core reduced mechanical allodynia in SNI-treated rats expressing ChR2-eYFP. Two-way ANOVA, n = 4–7, p < 0.001. C, Activation of PL-PFC axons in the NAc core reduced cold allodynia in SNI-treated rats expressing ChR2-eYFP. Two-way ANOVA, n = 4–8, p < 0.01. D, Schematic showing the locations of optic fibers in the NAc core. Error bars show mean and SEM.
To test whether the activation of this corticostriatal pathway can also relieve the affective component of pain, we next evaluated the impact photoactivation of PL-PFC axons in the NAc core has on the aversive quality and depressive symptoms of pain using the CPP test, the SPT, and the FST (Fig. 8). We injected ChR2 into the PFC, and then activated the corticostriatal circuit by light stimulation in the NAc core during these affective tests (Fig. 8A). First, we found that SNI-operated, but not sham-operated, rats developed a preference for the chamber that is associated with light treatment in the NAc (Fig. 8B; p < 0.01). These data indicate that the PFC–NAc circuit regulates the aversive state of tonic or ongoing pain. Next, we examined whether the activation of this circuit also relieved anhedonia. We found that light activation of this circuit fully restored sucrose preference in SNI-treated rats to control levels (Fig. 8C; p < 0.001). In contrast, light treatment did not have any antidepressant effect on sham-treated rats. We did not find any difference in the amount of total fluid consumption between SNI-treated rats that received light activation and rats that did not (Fig. 8D). Instead, SNI-treated rats that received light activation consumed a higher volume of sucrose solution (Fig. 8E; p < 0.01) and a smaller volume of water during the SPT (Fig. 8F; p < 0.05), further indicating an improvement in their anhedonic symptoms. Next, we found that the activation of this corticostriatal circuit improved symptoms of immobility, suggesting a relief of behavioral despair associated with persistent pain (Fig. 8G; p < 0.05). Interestingly, the effects of corticostriatal activation on the phenotypes on the CPP test, the SPT, and the FST (Fig. 8B,C,G) were in fact similar to the effects of direct activation of the PL-PFC (Fig. 5B,C,G). Together, these results demonstrate that activation of this corticostriatal circuit not only regulates nociception, but it likely also modulates affective symptoms of pain.
Figure 8.

Activation of prefrontal projections to the NAc core relieves the affective symptoms associated with chronic pain. A, Schematic of the light treatment. B, Light activation of the corticostriatal circuit induced chamber preference in SNI-treated, but not sham-treated, rats. Two-way ANOVA with repeated measures and Bonferroni's post-test, n = 8, p < 0.01 for the SNI group; n = 7, p > 0.05 for the sham group. C, Photoactivation of the prefrontal projection to the NAc core improved sucrose preference in SNI-treated rats. Two-way ANOVA, n = 6–7, p < 0.001. D, Photoactivation of the PL–NAc circuit had no effect on the total fluid consumption of SNI-treated rats during the SPT. Student's t test, p > 0.05. E, Activation of the PL–NAc circuit increased sucrose consumption in SNI-treated rats during the SPT. Student's t test, p < 0.01. F, Activation of the PL–NAc circuit decreased water consumption in SNI-treated rats during the SPT. Student's t test, p < 0.05. G, Activation of the prefrontal projection to the NAc core decreased immobility on the FST in SNI-treated rats. Student's t test, n = 5–6, p < 0.05. Error bars show mean and SEM.
To further verify that the light effect is mediated through synapses between prefrontal neurons and MSNs, we examined whether the administration in the NAc core of NBQX, an AMPA receptor blocker that inhibits synaptic transmission, could block the pain-relieving effects of PL-PFC photoactivation (Fig. 9A,E). We found that rats that received NBQX, compared with control (saline-treated) rats, displayed a marked increase in mechanical and cold sensitivity (Fig. 9B,C; B, p < 0.01; C, p < 0.05). These results show that NBQX in the NAc core blocked the antiallodynic effects of PFC activation. Next, we examined whether NBQX could also block the relief of pain-induced depression by PFC activation. We found that NBQX-treated rats, compared with control (saline-treated) rats, displayed a significant decrease in sucrose preference (Fig. 9D; p < 0.05). Together, these data provide strong evidence that activation of the PL-PFC to NAc core circuit can inhibit neuropathic pain.
Figure 9.

NBQX in the NAc blocks the pain-relieving effects of photoactivation of the PL-PFC. A, To test the role of the corticostriatal circuit in pain regulation, NBQX was injected into the NAc core before photoactivation of the PL-PFC and behavior tests. Tests were performed on SNI-treated rats that expressed ChR2. B, NBQX infusion in the NAc blocked the effect of PFC activation on mechanical allodynia in SNI-treated rats. Student's t test, n = 9–11, p < 0.01. C, NBQX infusion in the NAc blocked the effect of PL-PFC activation on cold allodynia in SNI-treated rats. Student's t test, n = 8–10, p < 0.05. D, NBQX infusion in the NAc blocked the effect of light treatment in the PL-PFC on sucrose preference in SNI-treated rats. Student's t test, n = 4–5, p < 0.05. E, Schematic showing the locations of NBQX infusion in the NAc core. Error bars show mean and SEM.
Discussion
In this study, we have identified a novel neural circuit for pain modulation. We find that activation of the PL-PFC to NAc circuit produces strong antinociceptive effects, and it can also regulate affective symptoms of pain.
Human imaging and animal studies have shown that the PFC undergoes structural and functional changes in chronic pain states (Apkarian et al., 2004, 2005; Metz et al., 2009; Ji et al., 2010; Li et al., 2010; Ji and Neugebauer, 2011; Moayedi et al., 2011; Obara et al., 2013; Hung et al., 2014; Kucyi et al., 2014). Earlier studies using electrical stimulation and opioid-receptor activation suggest that prefrontal activation can produce antinociceptive effects (Cooper, 1975; Hardy and Haigler, 1985; Hardy, 1985). Compared with pharmacologic and electrical stimulation, optogenetic stimulation has the advantage of greater temporal specificity and the ability to activate a larger population of neurons, respectively. Thus, our approach allowed direct assessment of the prefrontal activity in pain regulation.
The PL-PFC has been shown to inhibit fear conditioning as well as related anxiety-type and depression-type behaviors in rodents (Corcoran and Quirk, 2007; Sotres-Bayon et al., 2012). Thus, our data demonstrating that PL-PFC activities can treat both sensory and affective pain symptoms are compatible with the established role of this region in mood regulation. An area of the PFC in humans, DL-PFC, is involved in emotional regulation and can play a protective role against depression and anxiety (Tucker et al., 1978; Pascual-Leone et al., 1996; Schlaepfer et al., 2003; Bishop et al., 2004; Balconi and Ferrari, 2012). Recent evidence indicates that the DL-PFC undergoes gray matter loss during chronic pain (Apkarian et al., 2004). Putting our data into this context, decreased activity in the PFC can be speculated to account (at least partially) for the persistence of pain.
In contrast to the pain-relieving role of the PL-PFC, the anterior cingulate cortex (ACC), an adjacent brain area, has been shown to promote the aversive quality of pain (Rainville et al., 1997; Sawamoto et al., 2000; Johansen and Fields, 2004; Bissière et al., 2008; De Felice et al., 2013), and in some cases to enhance pain sensitivity (Li et al., 2010). In addition, another component of the PFC, the infralimbic PFC (IL-PFC), has also been implicated in pain regulation (Ji and Neugebauer, 2014). Although the exact role of the IL-PFC in pain states remains unknown, optogenetic activation of this region has been shown to inhibit firing of neurons in the PL-PFC while increasing IL-PFC activities in pain models (Ji and Neugebauer, 2012). Thus, the interplay between the PL-PFC and IL-PFC may strongly influence pain modulation. Overall, current literature suggests that different regions within the PFC likely play distinct roles in pain regulation. At the circuit level, both the PL-PFC and the ACC project to the NAc core (Beckstead and Norgren, 1979; Sesack et al., 1989; Sesack and Pickel, 1992; Brog et al., 1993; Vertes, 2004). The IL-PFC, in contrast, has a weaker input to the shell subregion of the NAc. Thus, future functional mapping of distinct microcircuits within the corticostriatal projection can further elucidate how the interplay between the cortex and striatum controls pain.
The most important contribution of our study is the identification of a corticostriatal circuit in pain regulation. One of the primary prefrontal targets is the NAc (Beckstead and Norgren, 1979; Sesack et al., 1989; Brog et al., 1993; Ishikawa et al., 2008). The PFC–NAc projection provides top-down control of sensory and affective processes (Kalivas et al., 2005; Goff et al., 2008; Ishikawa et al., 2008; Mena et al., 2013; Shen et al., 2014). Recent human imaging studies showing altered connections between the PFC and NAc in chronic pain states emphasize the importance of this corticostriatal circuit (Baliki et al., 2010, 2012). Our behavioral studies indicate that this corticostriatal circuit can be a key mechanism for the brain to exert top-down control for pain.
Central pain regulation can be achieved through descending modulation of the spinal dorsal horn via the PAG–RVM circuit (Fields et al., 1983; Morgan et al., 1989). Pain can also be modulated by supraspinal mechanisms (Morgan et al., 1989). The NAc has been suggested to project both to the PAG and to the brainstem (Yu and Han, 1990; Gear et al., 1999; Becerra et al., 2001; Magnusson and Martin, 2002; Becerra and Borsook, 2008; Geha et al., 2008; Gear and Levine, 2009; Baliki et al., 2010; Goffer et al., 2013). Thus, the corticostriatal projection in the current study can potentially provide descending pain inhibition via the PAG–RVM pathway.
The NAc also projects to the ventral pallidum and substantia nigra. These projections terminate in the ventral anterior, dorsal, and lateral thalamus, which in turn projects to the motor cortex as well as to parts of the PFC and ACC. Through this striatothalamocortical circuit, the NAc can influence behavioral outcomes in the presence of rewards (Russo and Nestler, 2013). It is plausible that the NAc can also use this striatothalamocortical pathway to regulate pain, particularly the affective dimension of pain, as a supraspinal mechanism.
Anatomic and functional changes in the PFC–NAc circuit, including prefrontal gray matter density loss and increased corticostriatal connectivity, have been found in chronic pain patients (Apkarian et al., 2004; Geha et al., 2008; Baliki et al., 2010, 2012; Moayedi et al., 2011; Kucyi et al., 2014). Our results on the Hargreaves test suggest that PFC activation can reduce acute nociception, whereas data on allodynia and CPP tests show that activation of the corticostriatal circuit can also relieve evoked as well as ongoing chronic pain. Thus, alterations in this corticostriatal connectivity in chronic pain states could clearly affect pain sensitivity. Future studies, however, are needed to fully dissect the role of this corticostriatal circuit in acute nociception versus central plasticity resulting from chronic pain.
Our results on the CPP test, the SPT, and the FST demonstrate that the PFC–NAc circuit can regulate the aversive quality and depressive symptoms of pain. Aversion and depression are important affective expression of pain (Dworkin and Gitlin, 1991; Fishbain et al., 1997; Bair et al., 2003; King et al., 2009; Miller and Cano, 2009; Boyce-Rustay et al., 2010; Wang et al., 2011; Kim et al., 2012; De Felice et al., 2013; Goffer et al., 2013; Le, 2014). Thus, the corticostriatal circuit can modulate certain affective components of pain, and it will be interesting to further examine the role of this circuit in the full spectrum of affective pain behaviors, including anxiety, depression, pain-associated negative reinforcement, and even addiction.
Although optogenetic stimulation typically induces short-term temporally regulated neuronal activation (Tye and Deisseroth, 2012), photostimulation has been occasionally associated with long-term plasticity (Chaudhury et al., 2013). It is yet unclear whether stimulation of the PFC can result in long-term potentiation or depression at NAc synapses. Such long-term plasticity, however, would suggest that brief neuromodulation of this circuit could provide enduring pain relief.
A limitation of our study is that we did not distinguish between glutamatergic pyramidal neurons and GABAergic interneurons in the PFC by selective optogenetic targeting. However, two experiments in our study suggest pyramidal neurons likely play an important role. First, when we photostimulated the NAc after expressing ChR2 in the PFC (Fig. 6), light activation most likely occurred in the axon terminals of pyramidal neurons that project to the NAc. Thus, the strong antinociceptive and antidepressant effects we observed with photostimulation suggest that activation of the projection from pyramidal neurons in the PFC to the NAc has pain-relieving properties (Figs. 7, 8). The second, even stronger, evidence comes from our experiment using NBQX in the NAc to block glutamatergic transmissions from the PFC to the NAc (Fig. 9). NBQX, by blocking AMPA receptors in the NAc, can only block transmission from glutamatergic pyramidal neurons and not GABAergic interneurons. We found that blocking glutamatergic transmissions from these pyramidal neurons to the NAc (at least partially) blocked the analgesic effect of light activation within the PFC (Fig. 9). Together, these two experiments provide indirect but strong evidence that pyramidal neurons in the PFC project to the NAc core to regulate pain. However, these experiments do not rule out the possible role of interneurons in pain modulation, and in the future a promotor-specific strategy can directly dissect the pain-modulatory roles of pyramidal neurons versus interneurons.
In conclusion, we have identified a novel corticostriatal circuit in the regulation of chronic neuropathic pain. The projection from the PL-PFC to the NAc provides important antinociceptive effects and can regulate affective symptoms of pain. There is active research to apply deep brain stimulation and transcranial magnetic stimulation to treat depression and other mood disorders (Fox et al., 2012; Holtzheimer et al., 2012; Riva-Posse et al., 2013; Liston et al., 2014). Neuromodulation therapy for pain remains limited to spinal or peripheral nerve stimulation (Deer et al., 2014). Our study, however, raises the possibility of targeting the PFC–NAc circuit in the brain for treating intractable pain.
Footnotes
This work was supported by the National Institute for General Medical Sciences (GM102691) and the Anesthesia Research Fund of New York University Department of Anesthesiology.
The authors declare no competing financial interests.
References
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, Gitelman DR. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci. 2004;24:10410–10415. doi: 10.1523/JNEUROSCI.2541-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Arai A, Kessler M, Xiao P, Ambros-Ingerson J, Rogers G, Lynch G. A centrally active drug that modulates AMPA receptor gated currents. Brain Res. 1994;638:343–346. doi: 10.1016/0006-8993(94)90669-6. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Wang MJ, Paspalas CD. Neuromodulation of thought: flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron. 2012;76:223–239. doi: 10.1016/j.neuron.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bair MJ, Robinson RL, Katon W, Kroenke K. Depression and pain comorbidity: a literature review. Arch Intern Med. 2003;163:2433–2445. doi: 10.1001/archinte.163.20.2433. [DOI] [PubMed] [Google Scholar]
- Balconi M, Ferrari C. rTMS stimulation on left DLPFC increases the correct recognition of memories for emotional target and distractor words. Cogn Affect Behav Neurosci. 2012;12:589–598. doi: 10.3758/s13415-012-0090-1. [DOI] [PubMed] [Google Scholar]
- Baliki MN, Chialvo DR, Geha PY, Levy RM, Harden RN, Parrish TB, Apkarian AV. Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci. 2006;26:12165–12173. doi: 10.1523/JNEUROSCI.3576-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Geha PY, Fields HL, Apkarian AV. Predicting value of pain and analgesia: nucleus accumbens response to noxious stimuli changes in the presence of chronic pain. Neuron. 2010;66:149–160. doi: 10.1016/j.neuron.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, Fields HL, Apkarian AV. Corticostriatal functional connectivity predicts transition to chronic back pain. Nat Neurosci. 2012;15:1117–1119. doi: 10.1038/nn.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra L, Borsook D. Signal valence in the nucleus accumbens to pain onset and offset. Eur J Pain. 2008;12:866–869. doi: 10.1016/j.ejpain.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra L, Breiter HC, Wise R, Gonzalez RG, Borsook D. Reward circuitry activation by noxious thermal stimuli. Neuron. 2001;32:927–946. doi: 10.1016/S0896-6273(01)00533-5. [DOI] [PubMed] [Google Scholar]
- Beckstead RM, Norgren R. An autoradiographic examination of the central distribution of the trigeminal, facial, glossopharyngeal, and vagal nerves in the monkey. J Comp Neurol. 1979;184:455–472. doi: 10.1002/cne.901840303. [DOI] [PubMed] [Google Scholar]
- Bishop S, Duncan J, Brett M, Lawrence AD. Prefrontal cortical function and anxiety: controlling attention to threat-related stimuli. Nat Neurosci. 2004;7:184–188. doi: 10.1038/nn1173. [DOI] [PubMed] [Google Scholar]
- Bissière S, Plachta N, Hoyer D, McAllister KH, Olpe HR, Grace AA, Cryan JF. The rostral anterior cingulate cortex modulates the efficiency of amygdala-dependent fear learning. Biol Psychiatry. 2008;63:821–831. doi: 10.1016/j.biopsych.2007.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourquin AF, Süveges M, Pertin M, Gilliard N, Sardy S, Davison AC, Spahn DR, Decosterd I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain. 2006;122:14.e1–14. doi: 10.1016/j.pain.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Zhong C, Kohnken R, Baker SJ, Simler GH, Wensink EJ, Decker MW, Honore P. Comparison of mechanical allodynia and the affective component of inflammatory pain in rats. Neuropharmacology. 2010;58:537–543. doi: 10.1016/j.neuropharm.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Brog JS, Salyapongse A, Deutch AY, Zahm DS. The patterns of afferent innervation of the core and shell in the “accumbens” part of the rat ventral striatum: immunohistochemical detection of retrogradely transported fluoro-gold. J Comp Neurol. 1993;338:255–278. doi: 10.1002/cne.903380209. [DOI] [PubMed] [Google Scholar]
- Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci. 2013;14:502–511. doi: 10.1038/nrn3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso-Cruz H, Sousa M, Vieira JB, Lima D, Galhardo V. Prefrontal cortex and mediodorsal thalamus reduced connectivity is associated with spatial working memory impairment in rats with inflammatory pain. Pain. 2013;154:2397–2406. doi: 10.1016/j.pain.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, Ferguson D, Tsai HC, Pomeranz L, Christoffel DJ, Nectow AR, Ekstrand M, Domingos A, Mazei-Robison MS, Mouzon E, Lobo MK, Neve RL, Friedman JM, Russo SJ, Deisseroth K, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–536. doi: 10.1038/nature11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SJ. Anaesthetisation of prefrontal cortex and response to noxious stimulation. Nature. 1975;254:439–440. doi: 10.1038/254439a0. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Quirk GJ. Activity in prelimbic cortex is necessary for the expression of learned, but not innate, fears. J Neurosci. 2007;27:840–844. doi: 10.1523/JNEUROSCI.5327-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard T, Larsen DB, Hansen SL, Grayson B, Neill JC, Plath N. Positive modulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors reverses sub-chronic PCP-induced deficits in the novel object recognition task in rats. Behav Brain Res. 2010;207:144–150. doi: 10.1016/j.bbr.2009.09.048. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Deer TR, Mekhail N, Provenzano D, Pope J, Krames E, Thomson S, Raso L, Burton A, DeAndres J, Buchser E, Buvanendran A, Liem L, Kumar K, Rizvi S, Feler C, Abejon D, Anderson J, Eldabe S, Kim P, Leong M, et al. The appropriate use of neurostimulation: avoidance and treatment of complications of neurostimulation therapies for the treatment of chronic pain. Neuromodulation. 2014;17:571–598. doi: 10.1111/ner.12206. [DOI] [PubMed] [Google Scholar]
- De Felice M, Eyde N, Dodick D, Dussor GO, Ossipov MH, Fields HL, Porreca F. Capturing the aversive state of cephalic pain preclinically. Ann Neurol. 2013;74:257–265. doi: 10.1002/ana.23922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin RH, Gitlin MJ. Clinical aspects of depression in chronic pain patients. Clin J Pain. 1991;7:79–94. doi: 10.1097/00002508-199106000-00004. [DOI] [PubMed] [Google Scholar]
- Edwards RR, Haythornthwaite JA, Smith MT, Klick B, Katz JN. Catastrophizing and depressive symptoms as prospective predictors of outcomes following total knee replacement. Pain Res Manag. 2009;14:307–311. doi: 10.1155/2009/273783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011;34:389–412. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Bry J, Hentall I, Zorman G. The activity of neurons in the rostral medulla of the rat during withdrawal from noxious heat. J Neurosci. 1983;3:2545–2552. doi: 10.1523/JNEUROSCI.03-12-02545.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbain DA, Cutler R, Rosomoff HL, Rosomoff RS. Chronic pain-associated depression: antecedent or consequence of chronic pain? A review. Clin J Pain. 1997;13:116–137. doi: 10.1097/00002508-199706000-00006. [DOI] [PubMed] [Google Scholar]
- Fox MD, Buckner RL, White MP, Greicius MD, Pascual-Leone A. Efficacy of transcranial magnetic stimulation targets for depression is related to intrinsic functional connectivity with the subgenual cingulate. Biol Psychiatry. 2012;72:595–603. doi: 10.1016/j.biopsych.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JM. Cortex and memory: emergence of a new paradigm. J Cogn Neurosci. 2009;21:2047–2072. doi: 10.1162/jocn.2009.21280. [DOI] [PubMed] [Google Scholar]
- Gear RW, Levine JD. Rostral ventral medulla cholinergic mechanism in pain-induced analgesia. Neurosci Lett. 2009;464:170–172. doi: 10.1016/j.neulet.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gear RW, Aley KO, Levine JD. Pain-induced analgesia mediated by mesolimbic reward circuits. J Neurosci. 1999;19:7175–7181. doi: 10.1523/JNEUROSCI.19-16-07175.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geha PY, Baliki MN, Harden RN, Bauer WR, Parrish TB, Apkarian AV. The brain in chronic CRPS pain: abnormal gray-white matter interactions in emotional and autonomic regions. Neuron. 2008;60:570–581. doi: 10.1016/j.neuron.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Lamberti JS, Leon AC, Green MF, Miller AL, Patel J, Manschreck T, Freudenreich O, Johnson SA. A placebo-controlled add-on trial of the Ampakine, CX516, for cognitive deficits in schizophrenia. Neuropsychopharmacology. 2008;33:465–472. doi: 10.1038/sj.npp.1301444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffer Y, Xu D, Eberle SE, D'amour J, Lee M, Tukey D, Froemke RC, Ziff EB, Wang J. Calcium-permeable AMPA receptors in the nucleus accumbens regulate depression-like behaviors in the chronic neuropathic pain state. J Neurosci. 2013;33:19034–19044. doi: 10.1523/JNEUROSCI.2454-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves L, Silva R, Pinto-Ribeiro F, Pêgo JM, Bessa JM, Pertovaara A, Sousa N, Almeida A. Neuropathic pain is associated with depressive behaviour and induces neuroplasticity in the amygdala of the rat. Exp Neurol. 2008;213:48–56. doi: 10.1016/j.expneurol.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Hao JX, Shi TJ, Xu IS, Kaupilla T, Xu XJ, Hökfelt T, Bartfai T, Wiesenfeld-Hallin Z. Intrathecal galanin alleviates allodynia-like behaviour in rats after partial peripheral nerve injury. Eur J Neurosci. 1999;11:427–432. doi: 10.1046/j.1460-9568.1999.00447.x. [DOI] [PubMed] [Google Scholar]
- Hardy SG. Analgesia elicited by prefrontal stimulation. Brain Res. 1985;339:281–284. doi: 10.1016/0006-8993(85)90093-9. [DOI] [PubMed] [Google Scholar]
- Hardy SG, Haigler HJ. Prefrontal influences upon the midbrain: a possible route for pain modulation. Brain Res. 1985;339:285–293. doi: 10.1016/0006-8993(85)90094-0. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Holtzheimer PE, Kelley ME, Gross RE, Filkowski MM, Garlow SJ, Barrocas A, Wint D, Craighead MC, Kozarsky J, Chismar R, Moreines JL, Mewes K, Posse PR, Gutman DA, Mayberg HS. Subcallosal cingulate deep brain stimulation for treatment-resistant unipolar and bipolar depression. Arch Gen Psychiatry. 2012;69:150–158. doi: 10.1001/archgenpsychiatry.2011.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Doods H, Treede RD, Ceci A. Depression-like behaviour in rats with mononeuropathy is reduced by the CB2-selective agonist GW405833. Pain. 2009;143:206–212. doi: 10.1016/j.pain.2009.02.018. [DOI] [PubMed] [Google Scholar]
- Hung KL, Wang SJ, Wang YC, Chiang TR, Wang CC. Upregulation of presynaptic proteins and protein kinases associated with enhanced glutamate release from axonal terminals (synaptosomes) of the medial prefrontal cortex in rats with neuropathic pain. Pain. 2014;155:377–387. doi: 10.1016/j.pain.2013.10.026. [DOI] [PubMed] [Google Scholar]
- Ishikawa A, Ambroggi F, Nicola SM, Fields HL. Dorsomedial prefrontal cortex contribution to behavioral and nucleus accumbens neuronal responses to incentive cues. J Neurosci. 2008;28:5088–5098. doi: 10.1523/JNEUROSCI.0253-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KB, Kosek E, Wicksell R, Kemani M, Olsson G, Merle JV, Kadetoff D, Ingvar M. Cognitive behavioral therapy increases pain-evoked activation of the prefrontal cortex in patients with fibromyalgia. Pain. 2012;153:1495–1503. doi: 10.1016/j.pain.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Pain-related deactivation of medial prefrontal cortical neurons involves mGluR1 and GABA(A) receptors. J Neurophysiol. 2011;106:2642–2652. doi: 10.1152/jn.00461.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Modulation of medial prefrontal cortical activity using in vivo recordings and optogenetics. Mol Brain. 2012;5:36. doi: 10.1186/1756-6606-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. CB1 augments mGluR5 function in medial prefrontal cortical neurons to inhibit amygdala hyperactivity in an arthritis pain model. Eur J Neurosci. 2014;39:455–466. doi: 10.1111/ejn.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Sun H, Fu Y, Li Z, Pais-Vieira M, Galhardo V, Neugebauer V. Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. J Neurosci. 2010;30:5451–5464. doi: 10.1523/JNEUROSCI.0225-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Fields HL. Glutamatergic activation of anterior cingulate cortex produces an aversive teaching signal. Nat Neurosci. 2004;7:398–403. doi: 10.1038/nn1207. [DOI] [PubMed] [Google Scholar]
- Jørum E, Warncke T, Stubhaug A. Cold allodynia and hyperalgesia in neuropathic pain: the effect of N-methyl-d-aspartate (NMDA) receptor antagonist ketamine—a double-blind, cross-over comparison with alfentanil and placebo. Pain. 2003;101:229–235. doi: 10.1016/S0304-3959(02)00122-7. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–650. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Kanju PM, Parameshwaran K, Sims C, Bahr BA, Shonesy BC, Suppiramaniam V. Ampakine CX516 ameliorates functional deficits in AMPA receptors in a hippocampal slice model of protein accumulation. Exp Neurol. 2008;214:55–61. doi: 10.1016/j.expneurol.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Kim H, Chen L, Lim G, Sung B, Wang S, McCabe MF, Rusanescu G, Yang L, Tian Y, Mao J. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J Clin Invest. 2012;122:2940–2954. doi: 10.1172/JCI61884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucyi A, Moayedi M, Weissman-Fogel I, Goldberg MB, Freeman BV, Tenenbaum HC, Davis KD. Enhanced medial prefrontal-default mode network functional connectivity in chronic pain and its association with pain rumination. J neurosci. 2014;34:3969–3975. doi: 10.1523/JNEUROSCI.5055-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011;70:855–862. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le AM, Lee M, Su C, Zou A, Wang J. AMPAkines have novel analgesic properties in rat models of persistent neuropathic and inflammatory pain. Anesthesiology. 2014;121:1080–1090. doi: 10.1097/ALN.0000000000000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Ko HG, Chen T, Descalzi G, Koga K, Wang H, Kim SS, Shang Y, Kwak C, Park SW, Shim J, Lee K, Collingridge GL, Kaang BK, Zhuo M. Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science. 2010;330:1400–1404. doi: 10.1126/science.1191792. [DOI] [PubMed] [Google Scholar]
- Liston C, Chen AC, Zebley BD, Drysdale AT, Gordon R, Leuchter B, Voss HU, Casey BJ, Etkin A, Dubin MJ. Default mode network mechanisms of transcranial magnetic stimulation in depression. Biol Psychiatry. 2014;76:517–526. doi: 10.1016/j.biopsych.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson JE, Martin RV. Additional evidence for the involvement of the basal ganglia in formalin-induced nociception: the role of the nucleus accumbens. Brain Res. 2002;942:128–132. doi: 10.1016/S0006-8993(02)02489-7. [DOI] [PubMed] [Google Scholar]
- Mena JD, Selleck RA, Baldo BA. Mu-opioid stimulation in rat prefrontal cortex engages hypothalamic orexin/hypocretin-containing neurons, and reveals dissociable roles of nucleus accumbens and hypothalamus in cortically driven feeding. J Neurosci. 2013;33:18540–18552. doi: 10.1523/JNEUROSCI.3323-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz AE, Yau HJ, Centeno MV, Apkarian AV, Martina M. Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc Natl Acad Sci U S A. 2009;106:2423–2428. doi: 10.1073/pnas.0809897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LR, Cano A. Comorbid chronic pain and depression: who is at risk? J Pain. 2009;10:619–627. doi: 10.1016/j.jpain.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Moayedi M, Weissman-Fogel I, Crawley AP, Goldberg MB, Freeman BV, Tenenbaum HC, Davis KD. Contribution of chronic pain and neuroticism to abnormal forebrain gray matter in patients with temporomandibular disorder. Neuroimage. 2011;55:277–286. doi: 10.1016/j.neuroimage.2010.12.013. [DOI] [PubMed] [Google Scholar]
- Morgan MM, Sohn JH, Liebeskind JC. Stimulation of the periaqueductal gray matter inhibits nociception at the supraspinal as well as spinal level. Brain Res. 1989;502:61–66. doi: 10.1016/0006-8993(89)90461-7. [DOI] [PubMed] [Google Scholar]
- Morgan MM, Gold MS, Liebeskind JC, Stein C. Periaqueductal gray stimulation produces a spinally mediated, opioid antinociception for the inflamed hindpaw of the rat. Brain Res. 1991;545:17–23. doi: 10.1016/0006-8993(91)91264-2. [DOI] [PubMed] [Google Scholar]
- Navratilova E, Porreca F. Reward and motivation in pain and pain relief. Nat Neurosci. 2014;17:1304–1312. doi: 10.1038/nn.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson A, Schneider DM, Takatoh J, Sakurai K, Wang F, Mooney R. A circuit for motor cortical modulation of auditory cortical activity. J Neurosci. 2013;33:14342–14353. doi: 10.1523/JNEUROSCI.2275-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13:1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara I, Goulding SP, Hu JH, Klugmann M, Worley PF, Szumlinski KK. Nerve injury-induced changes in Homer/glutamate receptor signaling contribute to the development and maintenance of neuropathic pain. Pain. 2013;154:1932–1945. doi: 10.1016/j.pain.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohayon MM, Schatzberg AF. Using chronic pain to predict depressive morbidity in the general population. Arch Gen Psychiatry. 2003;60:39–47. doi: 10.1001/archpsyc.60.1.39. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Morimura K, Porreca F. Descending pain modulation and chronification of pain. Curr Opin Support Palliat Care. 2014;8:143–151. doi: 10.1097/SPC.0000000000000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Leone A, Rubio B, Pallardó F, Catalá MD. Rapid-rate transcranial magnetic stimulation of left dorsolateral prefrontal cortex in drug-resistant depression. Lancet. 1996;348:233–237. doi: 10.1016/S0140-6736(96)01219-6. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Anton G, Blavet N, Jalfre M. Behavioural despair in rats: a new model sensitive to antidepressant treatments. Eur J Pharmacol. 1978;47:379–391. doi: 10.1016/0014-2999(78)90118-8. [DOI] [PubMed] [Google Scholar]
- Rainville P, Duncan GH, Price DD, Carrier B, Bushnell MC. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science. 1997;277:968–971. doi: 10.1126/science.277.5328.968. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Mayberg HS. Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nat Neurosci. 2007;10:1116–1124. doi: 10.1038/nn1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds SM, Berridge KC. Emotional environments retune the valence of appetitive versus fearful functions in nucleus accumbens. Nat Neurosci. 2008;11:423–425. doi: 10.1038/nn2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva-Posse P, Holtzheimer PE, Garlow SJ, Mayberg HS. Practical considerations in the development and refinement of subcallosal cingulate white matter deep brain stimulation for treatment-resistant depression. World Neurosurg. 2013;80:S2.e25–34. doi: 10.1016/j.wneu.2012.11.074. [DOI] [PubMed] [Google Scholar]
- Romano JM, Turner JA. Chronic pain and depression: does the evidence support a relationship? Psychol Bull. 1985;97:18–34. doi: 10.1037/0033-2909.97.1.18. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamoto N, Honda M, Okada T, Hanakawa T, Kanda M, Fukuyama H, Konishi J, Shibasaki H. Expectation of pain enhances responses to nonpainful somatosensory stimulation in the anterior cingulate cortex and parietal operculum/posterior insula: an event-related functional magnetic resonance imaging study. J Neurosci. 2000;20:7438–7445. doi: 10.1523/JNEUROSCI.20-19-07438.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer TE, Kosel M, Nemeroff CB. Efficacy of repetitive transcranial magnetic stimulation (rTMS) in the treatment of affective disorders. Neuropsychopharmacology. 2003;28:201–205. doi: 10.1038/sj.npp.1300038. [DOI] [PubMed] [Google Scholar]
- Schneider DM, Nelson A, Mooney R. A synaptic and circuit basis for corollary discharge in the auditory cortex. Nature. 2014;513:189–194. doi: 10.1038/nature13724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Pickel VM. Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol. 1992;320:145–160. doi: 10.1002/cne.903200202. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Deutch AY, Roth RH, Bunney BS. Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol. 1989;290:213–242. doi: 10.1002/cne.902900205. [DOI] [PubMed] [Google Scholar]
- Shen HW, Gipson CD, Huits M, Kalivas PW. Prelimbic cortex and ventral tegmental area modulate synaptic plasticity differentially in nucleus accumbens during cocaine-reinstated drug seeking. Neuropsychopharmacology. 2014;39:1169–1177. doi: 10.1038/npp.2013.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Sierra-Mercado D, Pardilla-Delgado E, Quirk GJ. Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron. 2012;76:804–812. doi: 10.1016/j.neuron.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Amata M, Sakaue G, Nishimura S, Inoue T, Shibata M, Mashimo T. Experimental neuropathy in mice is associated with delayed behavioral changes related to anxiety and depression. Anesth Analg. 2007;104:1570–1577. doi: 10.1213/01.ane.0000261514.19946.66. table of contents. [DOI] [PubMed] [Google Scholar]
- Tucker DM, Antes JR, Stenslie CE, Barnhardt TM. Anxiety and lateral cerebral function. J Abnorm Psychol. 1978;87:380–383. doi: 10.1037/0021-843X.87.8.380. [DOI] [PubMed] [Google Scholar]
- Tye KM, Deisseroth K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat Rev Neurosci. 2012;13:251–266. doi: 10.1038/nrn3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- Wang J, Goffer Y, Xu D, Tukey DS, Shamir DB, Eberle SE, Zou AH, Blanck TJ, Ziff EB. A single subanesthetic dose of ketamine relieves depression-like behaviors induced by neuropathic pain in rats. Anesthesiology. 2011;115:812–821. doi: 10.1097/ALN.0b013e31822f16ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IB, Steinberg EE, Lee SY, Davidson TJ, Zalocusky KA, Brodsky M, Yizhar O, Cho SL, Gong S, Ramakrishnan C, Stuber GD, Tye KM, Janak PH, Deisseroth K. Recombinase-driver rat lines: tools, techniques, and optogenetic application to dopamine-mediated reinforcement. Neuron. 2011;72:721–733. doi: 10.1016/j.neuron.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu LC, Han JS. Habenula as a relay in the descending pathway from nucleus accumbens to periaqueductal grey subserving antinociception. Int J Neurosci. 1990;54:245–251. doi: 10.3109/00207459008986640. [DOI] [PubMed] [Google Scholar]