

Abstract
The demyelinating disease multiple sclerosis (MS) has an early inflammatory phase followed by an incurable progressive phase with subdued inflammation and poorly understood neurodegenerative mechanism. In this study, we identified various parallelisms between progressive MS and the dysmyelinating mouse model Shiverer and then genetically deleted a major neuron-specific mitochondrial anchoring protein Syntaphilin (SNPH) from the mouse. Prevailing evidence suggests that deletion of SNPH is harmful in demyelination. Surprisingly, SNPH deletion produces striking benefits in the Shiverer by prolonging survival, reducing cerebellar damage, suppressing oxidative stress, and improving mitochondrial health. In contrast, SNPH deletion does not benefit clinical symptoms in experimental autoimmune encephalomyelitis (EAE), a model for early-phase MS. We propose that deleting mitochondrial anchoring is a novel, specific treatment for progressive MS.
Keywords: axonal degeneration, mitochondria, multiple sclerosis, oxidative stress, syntaphilin
Introduction
Multiple sclerosis (MS) consists of an initial remitting relapsing MS characterized by inflammatory demyelinating lesions, followed by a noninflammatory, secondary progressive MS (SPMS) with relentless and irreversible neurodegeneration. All current treatments for MS are effective for the early phase and not for the progressive phase (Witte et al., 2014). Here, we hypothesize a novel treatment that works the other way: it is specific for late, but not early phase MS. Our hypothesis is based on studies of a novel compound mutant generated by crossing the well known CNS dysmyelinating mouse Shiverer (Shi) (Inoue et al., 1981; Roach et al., 1983; Molineaux et al., 1986) with a mouse lacking Syntaphilin (SNPH), a major axon-specific mitochondrial anchoring protein (Kang et al., 2008; Sheng, 2014). This study is predicated on several similarities between Shi and progressive MS.
First, Andrews et al. (2006) suggested that Shi is a mouse model for progressive MS. Although MS and Shi have different starting points (myelination vs dysmyelination), in the end, both pathologies face the same crisis of maintaining metabolic matching in axons with chronic myelin loss. Second, Shi apparently provides metabolic matching by increasing the axonal mitochondrial mass (Andrews et al., 2006). This adaptive response is common to various demyelination animal models (Zambonin et al., 2011; Ohno et al., 2014) and in human progressive MS (Mahad et al., 2009). Third, the increase in mitochondrial mass uses SNPH, a key axon-specific mitochondrial docking protein (Kang et al., 2008). Therefore, Mahad et al. (2009) first detected a dramatic upregulation of SNPH in tissues from progressive MS patients over healthy patients. Importantly, a dramatic upregulation of SNPH in late-phase Shi was also observed in our studies. Both Shi and progressive MS appear to use SNPH upregulation to increase stationary energy sites to provide better metabolic match in chronic myelin loss. Fourth, the noninflammatory background of Shi resembles the subdued inflammation seen in progressive MS. Indeed, some investigators have suggested that neurodegeneration, not inflammation, is the predominant pathology in MS and that the real MS is exemplified by 10–20% of MS patients with primary progressive MS (PPMS) lacking significant inflammation throughout the course of disease (Stys et al., 2012; Stys, 2013; Witte et al., 2014). Fifth, recent studies have uncovered a striking age-dependent neurodegeneration in the Shi cerebellum (Loers et al., 2004). This is significant because prominent cerebellar neurodegeneration is also seen in 80% of MS patients (Swingler and Compston, 1992; Thompson et al., 2010; Campbell et al., 2011; Shields et al., 2012).
These parallelisms between Shi and human progressive MS suggest that SNPH-mediated increase in mitochondrial mass is an adaptive response beneficial to both pathologies. We crossed the SNPH knock-out (SNPH-KO) mouse into the Shi, expecting a worsening of symptoms. Unexpectedly, we report here that deletion of SNPH in Shi significantly prolonged survival and reduced cerebellar degeneration. In sharp contrast, SNPH deletion confers no benefits in the experimental autoimmune encephalomyelitis (EAE), a model for early inflammatory MS. Our results suggest that deleting mitochondrial anchoring is potentially a new specific treatment for progressive MS.
Materials and Methods
Generation of compound mutant mice.
All animal usage and protocols were reviewed by Animal Care and Usage Committee and approved by University of Wisconsin- Madison Research Animal Resources Center. Homozygous C3Fe.SWV-Mbpshi/J (Mbp−/−, Shi; The Jackson Laboratory, http://jaxmice.jax.org/strain/001428.html) were crossed with homozygous C57BL/6 SNPH−/− (SNPH-KO) (obtained from the Sheng laboratory at the National Institute of Neurological Disorders and Stroke, National Institutes of Health) mice and double heterozygous (Mbp+/−; SNPH+/−) mice were obtained. The double heterozygous female mice were backcrossed with SNPH−/− for four generations. The double heterozygous mice were interbred to produce compound mutants on a mixed background; wild-type Wt: (Mbp+/+; SNPH+/+), Shi: (Mbp−/−; SNPH+/+), SNPH-KO: (Mbp+/+; SNPH−/−) and Shi-SNPH-KO: (Mbp−/−; SNPH−/−). Alternately, Shi-SNPH-Het (Mbp−/−; SNPH+/−) were interbred to produce Shi: (Mbp−/−; SNPH+/+) and Shi-SNPH-KO: (Mbp−/−; SNPH−/−) littermates. The compound mutant mice were PCR genotyped. To minimize the impact of mixed genetic background, we compared littermates in this study.
Immunohistochemistry.
Mice were anesthetized at different age with intraperitoneal injection of ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight) and then perfused transcardially with PBS and 4% paraformaldehyde (PFA). Cerebellums were dissected out and placed in 4% PFA overnight at 4°C and subsequently processed for paraffin sectioning. The sections were deparaffinized with xylene and hydrated through a series of alcohol washes and water, boiled for 20 min in 10 mm sodium citrate buffer, pH 6, for antigen retrieval, washed with PBS 3 times, and incubated in blocking solution containing 0.3% Triton X-100, 10% normal goat serum, 1% bovine serum albumin (BSA) in PBS for 1 h at room temperature (RT). Sections were then incubated with primary antibodies against SNPH (1:500, Sheng laboratory), neurofilament associated antigen (1:250; Developmental Studies Hybridoma Bank), myelin basic protein (MBP: 1:500; Santa Cruz Biotechnology) VDAC (1:50; NeuroMab), IBA1 (1:400; Daco), neurofilament L (1:250), nonphosphorylated neurofilament antibody (1:100; BioLegend), and SDHA (1:50; Cell Signaling Technology), MTCO1/Cox1 (1:100; Boster Immunoleader) overnight at 4°C. The following day, sections were washed with PBS and incubated with Alexa Fluor-labeled secondary antibodies (1:250; Invitrogen) for 1 h at RT. Sections were washed five times with PBS and mounted with prolong antifade reagent (Invitrogen). Fluorescent images were acquired using a Nikon A1 confocal microscope with 60× and 100× 1.4 numerical aperture plan apochromatic oil-immersion objective at 1024 × 1024 resolution. The optical thickness was set at 0.5 μm and 3D volume scans (z-series) images were collected from randomly selected fields. All image acquisition settings were kept identical for different samples to ensure the comparability between experimental groups. Image analysis was accomplished using Nikon Elements software.
Histochemistry of mitochondrial respiratory chain functions.
Cerebellums from compound mutants were snap frozen in liquid nitrogen/dry ice and embedded in optimal cutting temperature (OCT) medium and 10 μm thin frozen sections were obtained. Mitochondrial cytochrome oxidase (Cox) and succinate dehydrogenase (SDH) histochemistry was performed using a previously described method (Mahad et al., 2009; Campbell et al., 2011; Tanji and Bonilla, 2001). In Cox activity, the 3,3′-diaminobenzidine tetrahydrochloride (DAB) acts as an electron donor for cytochrome C. Active Cox oxidizes DAB, producing brown, insoluble indamine polymer that correlates directly to the Cox activity. Similarly, the reduction of nitro-blue tetrazolium produces blue color that correlates directly to the SDH activity. The intensity of brown and blue color was quantifyed using ImageJ software. Frozen sections were air dried for 10 min at RT and incubated with Cox medium (100 mm cytochrome c, 4 mm DAB, and 20 mg/ml catalase in 0.2 m phosphate buffer, pH 7.0, at 37°C for 45 min. Adjacent section were incubated with SDH medium (130 mm sodium succinate, 1.5 mm nitroblue tetrazolium, 200 mm phenazine methosulphate, 1 mm sodium azide, in 0.2 m phosphate buffer, pH 7.0) at 37°C for 60 min. Sections were washed with PBS, dehydrated through graded alcohol and mounted using Harleco Krystalon mounting media (Millipore). Image analysis was performed using measure tool of ImageJ software.
Western blotting.
Snap-frozen cerebellar tissue from different genotypes were lysed in protein extraction buffer consisting of the following (in mm): 20 Na2HPO4, 50 NaF, 10 Na3PO4, 150 NaCl, 5 EGTA, 5 EDTA, along with 2% Triton X-100 and 0.5% deoxycholate; Na3VO4 (1 mm) and Sigma protease inhibitor mixture were added to the extraction buffer immediately before use. Tissue lysates were briefly sonicated and centrifuged at 14,000 rpm for 10 min at 4°C. Supernatant was used for protein estimation by Lowry's method. Then, 20 μg of protein was loaded in each well of 10–12% SDS-PAGE at 150 V. Proteins were subsequently transferred to nitrocellulose at 100 V for 1 h. Membranes were blocked with 5% nonfat dry milk in pH 7.4 1× Tris buffered saline (TBS) with 0.05% Tween 20 (1× TBST) at RT for 1 h. Blots were incubated with anti-SNPH (1:5000, 5% BSA in TBST), anti-VDAC (1:1000, 5% nonfat dry milk in TBST), and anti-IBA1 (1:5000, 5% BSA in TBST) overnight at 4°C, washed with 1× TBST, and then incubated with the appropriate secondary antibodies for 1 h at room temperature. After washing, protein bands were visualized with Super Signal West Pico. Blots were subsequently stripped and reprobed for β-actin as a loading control.
Morphometric analysis of cerebellar Purkinje cells.
The axonal pathology in the cerebellum was evaluated using Bielschowsky silver impregnation kit (Loers et al., 2004) obtained from Newcomer Supply. The sections were deparafinized with xylene and hydrated through a series of alcohol washes and water. The staining was performed according to the protocol provided by manufacturer. The sections were dehydrated through graded alcohol and xylenes and mounted using Harleco Krystalon mounting media (Millipore). The axonal spheroids/torpedoes were counted in the granular layer. Total area of cerebellar granular layer was calculated using a 1 mm2 grid on glass slide (Nikon) and spheroid count was calculated in units per square millimeter of granular layer. For H&E staining, cerebellar sections were deparaffinized and hydrated through gradient alcohol and placed in Hematoxylin (Thermo Fisher Scientific WI) for 5 min and rinsed with tap water for 3 min followed by incubation in eosin (1%) for 1 min. After eosin staining, sections were thoroughly washed with tap water and dehydrated through alcohol and xylene and mounted using Harleco Krystalon mounting media (Millipore). Bright-field images were collected with a Nikon E300 microscope using a 20× objective.
In situ superoxide detection.
Superoxide levels in cerebellar sections were measured using specific fluorescent probe dihydroethidium (DHE) as described previously (Quick and Dugan, 2001; Iwai et al., 2004; Shichinohe et al., 2004). The cerebellum was removed and immediately frozen in liquid nitrogen, mounted in OCT medium, and 10 μm sections were obtained and stained for DHE dissolved in DMSO and diluted in PBS, pH 7.4. Next, 10 μm DHE was added directly onto the sections, followed by incubation in a humidified chamber for 30 min at 37°C. DHE gets oxidized by superoxide radicals into ethidium, which binds to DNA and remains stable after formaldehyde fixation. After DHE labeling, sections were washed five times with PBS and fixed with 4% PFA for 30 min at RT and stained for calbindin antibody with similar procedure described for immunohistochemistry. DHE fluorescence was collected at 515/540 nm excitation and emission, respectively. The analysis of DHE fluorescence was performed in ImageJ. The regions of interest (ROIs) were collected from PC soma (green channel) and added to the ROI manager and these ROIs were located in DHE fluorescence (red channel) by clicking individual ROIs in the ROI manager; fluorescence intensity was measured using measure tool.
Transmission electron microscopic analysis.
Mice were anesthetized at different ages with intraperitoneal injection of ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight) and perfused transcardially with PBS and 4% PFA. A small piece of cerebellum was sectioned and postfixed in a mixture of 2.5% glutaraldehyde, 2% PFA buffered in 0.1 m sodium phosphate buffer (PB) at RT, washed 5 times with PB and postfixed in 1% osmium tetroxide, 1% potassium ferrocyanide in 0.1 m PB for 1 h at RT. The tissue samples were washed five times with PB and dehydrated through alcohol gradient. Fully dehydrated tissue samples were infiltrated in increasing concentrations of Poly/Bed 812 (Polysciences) and propylene oxide mixtures and embedded in fresh Poly/Bed 812 for 48 h at 60°C. The samples were sectioned on a Leica EM UC6 ultramicrotome at 90 nm. The sections were collected on Cu, pioloform/carbon coated 2 × 1 oval slot grids (EMS) and poststained in uranyl acetate and lead citrate. The sectioned samples were viewed at 80 kV on a Philips CM120 transmission electron microscope equipped with MegaView III camera (Olympus).
EAE induction.
C57BL/6 mice were immunized by subcutaneous injection of 200 μg of MOG35-55 peptide (Genemed Synthesis) in complete Freund's adjuvant (Sigma-Aldrich) containing 5 mg/ml mycobacterium tuberculosis H37Ra (BD Biosciences). Then, 200 ng of pertussis toxin was injected intraperitoneally on day 0 and 2. Animals were in scored on a 0–5 scale as follows: 0 = no clinical symptoms, 1 = limp/flaccid tail, 2 = hindlimb weakness with incomplete paralysis, 3 = complete hindlimb paralysis, 4 = paraplegia, and 5 = moribund or dead. Mice were anesthetized and perfused with PBS and 4% PFA and processed for Immunohistochemistry analysis.
Statistical analysis.
Survival statistics were analyzed by Kaplan–Meier survival curves by SigmaPlot (Systat Software). Data are represented as mean ± SEM and statistical significance was calculated using one-way ANOVA followed by Student–Newman–Keuls analysis.
Results
Dysmyelinated Shi mouse does not show inflammation in cerebellum
Although the Shi model has been suggested to be noninflammatory, there are reports of modest inflammation in certain regions such as the spinal cord (Loers et al., 2004). Further, our goal for using Shi is to study the effect of eliminating mitochondrial anchoring on animal health without the effect being complicated by a prior, inflammation-induced damage on mitochondria. We therefore first validated the absence of inflammation in cerebellum of dysmyelinated Shi mice by examining the activation of microglia, the resident macrophage cells in CNS. Thin sections from cerebellum were stained for IBA1, a marker for resting and activated microglia. Figure 1 shows the low-magnification image of neurofilament (green) and IBA1 (red) immunoreactivities in the cerebellum of the 4.5-month-old compound mutants. Sporadic microglial population was present throughout the molecular (ML), Purkinje (PL), and granular layers (GL). The IBA1 reactivity did not change significantly among these mutants. Moreover, IBA1-positive cells in all mutants revealed a ramified morphology suggesting that they are resting microglia. On the contrary, cerebellar sections from EAE mice, a positive control for inflammatory microglial activation, showed an increased number of activated microglia with rounded or amoeboid shape (Fig. 1N, closed arrow) over resting microglia with ramified morphology (Fig. 1N, open arrow). IBA1 expression in the whole cerebellum of compound mutants was also evaluated by Western blotting. Actin was used as a loading control. As shown in Figure 1P, IBA1 expression did not change among the mutants.
Figure 1.
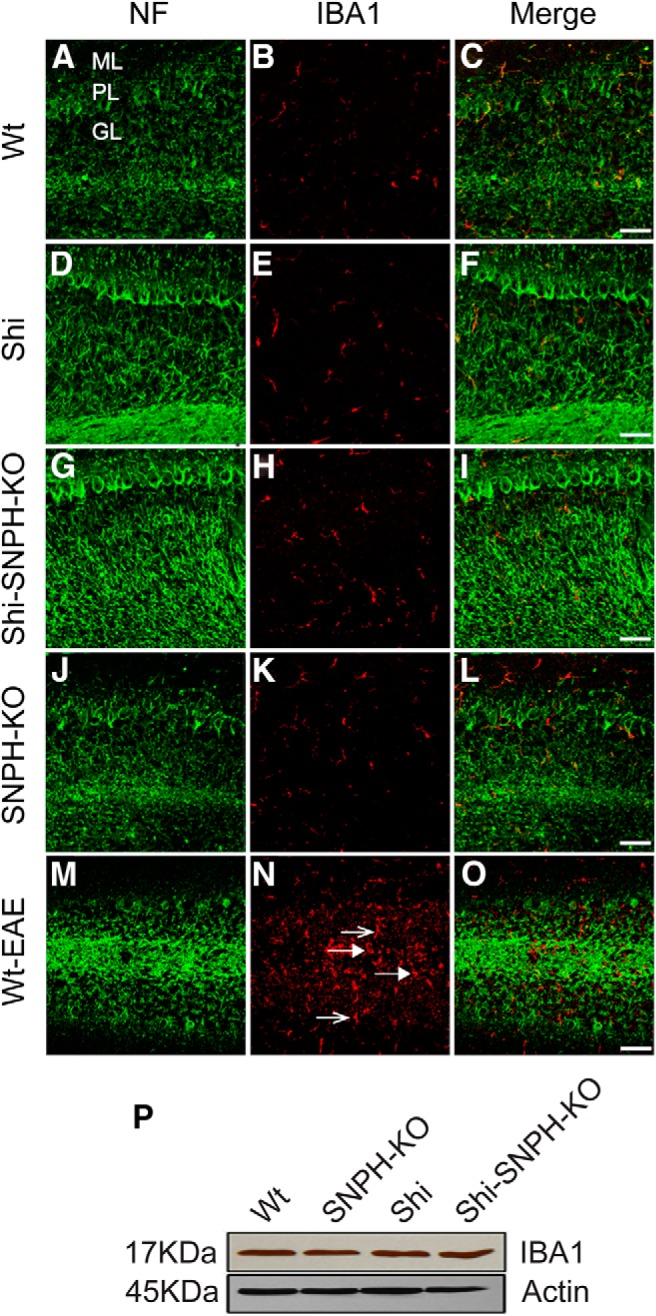
Absence of inflammation in cerebellum of Shi. IBA1 immunohistochemistry was performed in the cerebellar sections from 4.5-month-old compound mutants. B, E, H, and K represent the IBA1 immunoreactivity in the cerebellum of Wt, Shi, Shi-SNPH-KO, and SNPH-KO, respectively. Cerebellar section from EAE mice was stained for IBA1 as positive control for microglial activation (N). The amoeboid morphology of microglia (closed arrow) was observed near PCs and throughout the granular layer in EAE cerebellum. A few ramified microglias were also identified (open arrow). NFs were labeled to locate axons in the granular and molecular layer shown in panels A, D, G, J, and M. Merges of IBA1 and NF for each genotype are shown in far right panels C, F, I, L, and O. ML, Molecular layer; PL, Purkinje layer; GL, granular layer. The microglial morphology and IBA1 expression was consistent in three independent observations. Scale bar, 50 μm. P, Western blot analysis of IBA1 expression in tissue lysate from cerebellum of respective mutants did not show any change. Actin served as internal control for equal protein loading for each mutant.
SNPH expression and mitochondrial number are elevated in Shi cerebellum
Recent studies demonstrated that SNPH is dramatically elevated in postmortem samples of patients diagnosed with SPMS or PPMS (Mahad et al., 2009). We therefore examined whether SNPH is elevated in the Shi cerebellum, the region with strong degeneration as the animal ages (Loers et al., 2004). Western blot of 4.5-month-old cerebellum shows a significant increase in SNPH in the Shi over Wt (Fig. 2M). This SNPH elevation in cerebellum is further confirmed by immunohistochemistry (Fig. 2). SNPH is elevated in PCs and their axons in the Shi over Wt (Fig. 2D). Specificity of SNPH labeling is confirmed by absence of immunoreactivity in the SNPH-KO (Fig. 2G,J). The significant upregulation of SNPH in late-phase Shi is strikingly similar to that seen in human SPMS (see Fig. 5 of Mahad et al. (2009)).
Figure 2.

Upregulation of SNPH in dysmyelinated Shi. Sections from cerebellum of 4.5-month-old mice were stained for SNPH (green) and NF (red). A, D, G, and J represent the SNPH immunoreactivity in Wt, Shi, Shi-SNPH-KO, and SNPH-KO mice, respectively. The NF immunoreactivity for respective genotype is shown in panels B, E, H, and K, while merges of SNPH and NF are shown in panels C, F, I, and L. Shown here are representative images of independent observations from three mice. Scale bar, 20 μm. Arrow in F indicates innervation of GABAergic terminal regions on PCs. M, Western blot analysis in tissue lysate from cerebellum of respective mutants shows upregulation of SNPH in Shi. Actin served as internal control for equal protein loading for each mutant.
Figure 5.
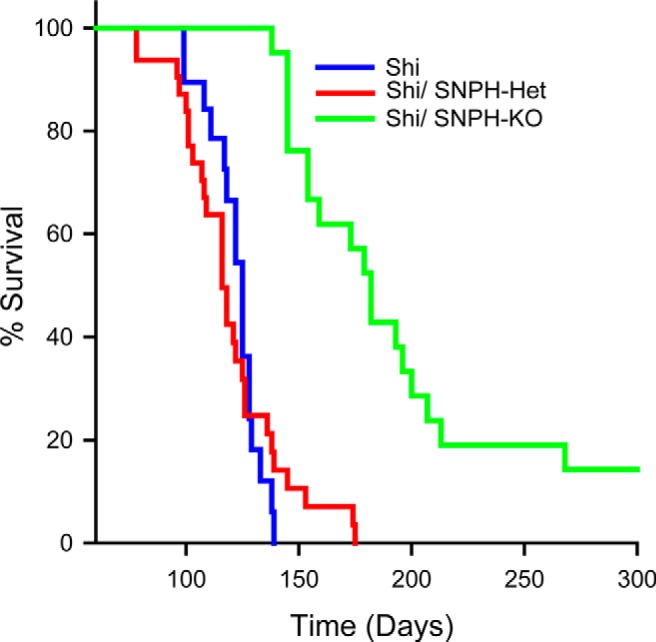
Deletion of SNPH expands the lifespan of dysmyelinated Shi. Shown are Kaplan–Meier survival curves for Shi (blue line), Shi-SNPH-Het (red line), and Shi-SNPH-KO (green line) mice. Most of the Shi mice fail to survive beyond 140 d (n = 20), whereas Shi-SNPH-KO littermates (n = 21) lived longer and some of the Shi-SNPH-KO mice lived beyond ∼270 d (p < 0.001). Shi-SNPH-Het followed a similar life expectancy curve as Shi. The median survival days for Shi and Shi-SNPH-KO mice were 125 and 182, respectively.
Having established an elevation of SNPH in Shi, we next investigated whether mitochondrial number is also elevated in Shi as reported previously (Andrews et al., 2006). We started using Western blotting of whole cerebellar lysates with anti-VDAC to monitor possible changes in mitochondrial mass (Fig. 3M). The VDAC signal is elevated in the Shi over the Wt, suggesting an increase in mitochondrial mass. Deleting SNPH in Shi (Shi-SNPH-KO) causes a modest reduction in mitochondrial mass after correcting for loading using actin (Fig. 3M). Although interesting, these VDAC blots contain both neurons and non-neuronal cells. We next examined possible changes in cellular mitochondrial distribution in the various compound mutants. We used immunohistochemistry and VDAC staining in cerebellar sections (Fig. 3). For the Shi, we observed an apparent increase in VDAC intensity in the PCs at both the soma and the axon level (Fig. 3E). The increase in mitochondrial number in the Shi PC axons is likely caused by an elevation of SNPH. An important question is whether deletion of SNPH causes a reduction in axonal mitochondrial number when mitochondria travel back to the soma. Studies in cultured neurons have demonstrated that SNPH deletion causes a reduction in axonal mitochondrial number (Kang et al., 2008). To address this issue in vivo, we used EM analysis. In EM cerebellar sections, we counted the mitochondrial density both in the PC soma (Fig. 4A–D) and in randomly selected granular axons (Fig. 4Ai,Bi,Ci,Di) in compound mutant mice. We only selected longitudinal axons in the granular layer in close proximity to the PCs soma and counted the average number of mitochondria per unit micrometer length. Mitochondrial numbers in the PC soma and granular axon of Shi are both increased compared with the Wt (Fig. 4E). This is consistent with the VDAC Western blots and VDAC immunohistochemistry showing an increase in the Shi over Wt. Interestingly, when SNPH is deleted from Shi (Shi-SNPH-KO), the biggest change in the mitochondrial number occurs in the axons and not in the soma. Therefore, deleting SNPH from the Shi only causes a modest reduction in the somal mitochondrial density (Fig. 4E). However, deleting SNPH from Shi causes a significant reduction in granular axon mitochondrial density (Fig. 4F). We acknowledged that the granular layer contains mixed axons and that it is not possible to distinguish PC axons from other axons when PC axons are dysmyelinated. However, there are thin sheaths around these axons (Fig. 4Bi,Ci), suggesting that they are PC axons that are normally myelinated. Further, the fact that we randomly selected granular axons for analysis in both Shi and Shi-SNPH-KO suggests that there is a reduction in mitochondrial density in Shi PC axons after SNPH deletion. This is consistent with studies in cultured neurons showing that SNPH deletion similarly caused a reduction in axonal mitochondrial density (Kang et al., 2008).
Figure 3.

Increased mitochondrial mass in cerebellum of dysmyelinated Shi. Cerebellar sections from 4.5-month-old compound mutants were stained for VDAC (green) and NF (red) immunoreactivity. A, D, G, and J represent the VDAC immunoreactivity in Wt, Shi, Shi-SNPH-KO, and SNPH-KO, respectively, while NF immunoreactivity for these genotypes is shown in panels B, E, H, and K. Merged image of VDAC and NF is shown in panels C, F, I, and L. Arrow indicates the increased VDAC expression in swollen axons. Shown here are representative images of independent observation from three mice. Scale bar, 20 μm. M, Western blot analysis in cerebellar lysate from compound mutants shows upregulation of VDAC in Shi.
Figure 4.

Increased mitochondrial density in cerebellum of dysmyelinated Shi. Electron micrographs of ultrathin sections from cerebellum of compound mutants were analyzed to quantify the mitochondrial numbers in single PC soma and granular axon. A–D, Representative images from PC of Wt, Shi, Shi-SNPH-KO, and SNPH-KO, respectively. Ai, Bi, Ci, and Di show the mitochondria in axons from cerebellar granular layer of respective mutants. For axonal analysis, we only selected the longitudinal axons in the granular layer close to the PCs. The mitochondrion in each panel is indicated by arrow. For the PC analysis, 50 images from each genotype were collected from randomly selected regions. Then mitochondria in a total of 350 PCs from 5–7 mice of each genotype were counted and represented as mean ± SEM. Scale bar, 5 μm. Mitochondrial number was significantly increased in both Shi and Shi-SNPH-KO compared with Wt and SNPH-KO (E, *p < 0.05). Quantification of mitochondrial number in granular axons is shown in F. Mitochondria in a total of 45–50 axons from three mice of each genotype were counted and represented as mitochondria/μm axonal length (mean ± SEM). Scale bar, 2 μm. Mitochondrial numbers in Shi axons were significantly increased compared with Wt and SNPH-KO (*p < 0.05), which was decreased significantly upon deletion of SNPH (#p < 0.05).
Elimination of SNPH prolongs the lifespan of Shi
Having established a noninflammatory background in the Shi cerebellum and documented changes in cellular distribution of mitochondria in the cerebellum after SNPH deletion, we next investigated whether deleting mitochondrial anchoring is detrimental or beneficial to Shi survival. Surprisingly, deleting SNPH is beneficial to Shi survival (Fig. 5). Shi mice (blue line) has a mean survival of 125 d (range 99–140, n = 20). The Shi-SNPH-KO (n = 21) littermates (green line) outlived Shi significantly (mean 182 d, p < 0.001). The life expectancy of Shi-SNPH-Het (n = 28) followed a similar lifespan as observed in Shi (red line), suggesting that a complete deletion of SNPH is required to improve the survival of Shi. The life expectancy of these mice was independent of their sex and we did not observe a significant reduction in the body weights of respective sexes at any age (data not shown). These results suggest that increased mitochondrial anchoring in Shi mice is detrimental and that SNPH elimination is protective.
SNPH deletion protects gray and white matter damage in Shi cerebellum
Cerebellum in Shi has been known to display prominent white matter injuries as the animal ages (Loers et al., 2004). Further, recent studies suggest that gray matter damage is also significant in various demyelination (MacKenzie-Graham et al., 2009; Hulst and Geurts, 2011; Inglese et al., 2011). We therefore examined cerebellar white and gray matter protection in Shi upon SNPH elimination in compound mutants at 2.5, 3.5, and 4.5 months of age. For gray matter analysis, thin cerebellar sections were stained with H&E to facilitate counting of PC densities. For white matter analysis, Bielschowsky's silver impregnation was used to quantitate the axonal injuries by counting torpedoes (axonal swelling), as described in previous studies (Griffiths et al., 1998; Trapp et al., 1998; Loers et al., 2004; Pitt et al., 2010; Haider et al., 2011). A representative image of gray matter analysis in compound mutants at 4.5 months is shown in Figure 6, A–D. Densely packed PCs were observed in Wt (Fig. 6A) and SNPH-KO (Fig. 6D) mice, whereas a loss of PCs was observed in Shi (Fig. 6B). Interestingly, higher PC counts were observed in Shi-SNPH-KO (Fig. 6C) compared with Shi, suggesting partial gray matter protection. Developmental data did not show significant changes in PC number in the Wt and SNPH-KO from 2.5 to 4.5 months (Fig. 6E). In contrast, Shi displays a gradual developmental loss of PCs that was partially prevented in the Shi-SNPH-KO (Fig. 6E). Compared with Wt, the PC loss at 3.5 months was 12% for Shi and 9% for Shi-SNPH-KO (p < 0.05) and at 4.5 months the loss was 37% for Shi and 19% for Shi-SNPH-KO (p < 0.01; Fig. 6E). Representative image of white matter analysis with silver staining in 4.5 months is shown for Wt (Fig. 6F), Shi (Fig. 6G), Shi-SNPH-KO (Fig. 6H), and SNPH-KO (Fig. 6I). Axonal torpedoes were observed in the granular layer in close proximity to PCs of Shi and Shi-SNPH-KO (arrows in Fig. 6G,H). The torpedoes were observed either as swollen axonal end bulbs or detached spherical bodies, representing ongoing degeneration to complete axonal transection. Quantitative analysis of torpedoes density (per square millimeter of cerebellar granular layer) revealed that torpedoes are rare in Wt and SNPH-KO mice at all age groups (Fig. 6J). In Shi, torpedoes are minimal at 2.5 months, but dramatically increased from 3.5 to 4.5 months (Fig. 6J). At each Shi age, SNPH-KO reduced torpedoes count by 60% at 3.5 months (p < 0.05) and by 44% at 4.5 months (p < 0.05), respectively (Fig. 6J).
Figure 6.

Elimination of mitochondrial anchoring protects cerebellar gray and white matter in dysmyelinated Shi. Cerebellar sections from compound mutant were stained with H&E. A–D show representative images of H&E staining in Wt, Shi, Shi-SNPH-KO, and SNPH-KO, respectively, at 4.5 months. PCs were counted in unit millimeters of Purkinje layer and normalized to Wt. Bar chart in E represents the mean ± SEM of normalized results obtained from three mice of each group. Similarly, Bielschowsky's silver impregnation analysis was performed to visualize axonal swelling in Wt, Shi, Shi-SNPH-KO, and SNPH-KO is shown in F–I, respectively. Torpedoes (indicated by arrows) in the granular layer (GL) were observed in Shi (G) and Shi-SNPH-KO (H). Histogram in J represents the mean ± SEM of torpedoes in unit square millimeter of granular layer from each group. A total of six serial sections from three mice of each group were analyzed. *p < 0.05 in E and J shows the significance between Wt and Shi; #p < 0.05 between Shi and SNPH-KO.
Specificity of white matter injury in Shi cerebellum to PC axons
Bielschowsky's silver impregnation is a convenient method to identify axonal torpedoes, but cannot resolve the identity of the axons undergoing injury. Although torpedoes lie close to the PCs and are likely from PC axons, the granular layer of cerebellum contains many other types of axons (climbing fibers and axons from granule cells and Golgi cells) all of which could be damaged in the Shi. Therefore, we used a PC-cell-specific marker to determine whether the torpedoes are specific to PC axons. The entire axonal population was labeled with neurofilament; calbindin was used to specifically label PC axons. Figure 7 shows a representative image of calbindin (Fig. 7A) and neurofilament (Fig. 7B) labeling in the cerebellar section of a 4.5-month-old Shi. Merged image is shown in Figure 7C and high-magnification images from the respective panels are shown in Figure 7, Ai, Bi, Ci, respectively. Similar to Bielschowsky's silver impregnation, torpedoes were observed in calbindin-positive (green) axons in the granular layer. Strikingly, of the larger set of neurofilament-positive axons, only the smaller subset of calbindin-positive axons shows torpedoes. Therefore, axonal damage in the granular layer is specific to PCs (indicated by arrows in Fig. 7Bi). Moreover, the calbindin-positive axons in Shi showed increased SNPH immunoreactivity (yellow in Fig. 7E) compared with Wt (Fig. 7D). The SNPH-filled axons in Wt and Shi can be observed through different angles in 3D reconstruction (Fig. 7Di,Dii and Ei,Eii, respectively). Fluorescence intensity analysis of SNPH immunoreactivity in axons revealed 1.5- and 2-fold increased expression in Shi axons and torpedoes than Wt axons (Fig. 7F). Together, these data suggest that PC axons are selectively damaged in the Shi and are partially protected by SNPH deletion. Why are PC axons in the granular layer selectively vulnerable to damage? One possibility is that PCs are unusually metabolically active due to their high resting firing and are more susceptible to metabolic stress.
Figure 7.

Specificity of torpedoes in Purkinje axons. Cerebellar PCs in 4.5-month-old Shi were labeled with calbindin (A) and NF (B) antibodies. Merge image is shown in C. High-magnification image from granular layer showing axonal swelling in PCs (Ai). Panel Bi represents the NF staining of all the axons in granular layer from the same region as in Ai. Axonal swelling in Bi is marked by arrow. Merged image of calbindin and NF is shown in Ci. Scale bar, 20 μm. D and E, 3D reconstruction of SNPH immunoreactivity in calbindin-labeled axons in granular layer of Wt and Shi, respectively. Arrows in E indicate torpedo in Shi. The same axon is shown from two different angles in Di, Dii and Ei, Eii, respectively. F, Quantitative analysis of SNPH intensity (mean ±SEM) in axons from Wt, Shi, and torpedoes from Shi mice. * (p < 0.05).
SNPH deletion rescues mitochondrial respiratory functions in the Shi
We monitored the mitochondrial respiratory chain functions in the cerebellum of compound mutants by measuring the activity of complex IV (Cox) and complex II (SDH) activity using a histochemical method. The genes for Cox are encoded by mitochondrial DNA, whereas those for SDH are encoded by nuclear genome. The ratio of Cox/SDH were normalized to Wt and represented as index of mitochondrial metabolic health. A decrease in this ratio reflects Cox-deficient mitochondria. We first analyzed Cox and SDH activity in PC soma by measuring the intensity of Cox (brown color) and SDH (blue color) reaction product. Representative images of Cox activity in cerebellar sections from 4.5-month-old compound mutants are shown in Figure 8, A–D, whereas SDH activity is shown in Figure 8, Ai–Di. PCs in Shi displayed a decreased brown Cox reaction product (arrows in Fig. 8B) compared with other three genotypes, whereas the blue SDH reaction product did not differ among these mutants. Quantitative analysis of brown and blue color intensity revealed that the Cox/SDH ratio in Shi was reduced to 80% (p < 0.05) compared with Wt, showing respiratory deficiency (Fig. 8K). Importantly, deleting SNPH from Shi (Shi-SNPH-KO) restores Shi respiratory function to 107.2% (p < 0.01) relative to the Wt level (Fig. 8K). Note that this apparent full restoration of Shi respiration functionally at the PC level by SNPH deletion does not mean full restoration at each mitochondrion level because there are likely more mitochondria per PC in the Shi over Wt. We therefore used the PC soma mitochondrial density data (Fig. 4E) to calculate respiration changes per mitochondrion. We calculated that, at each mitochondrion level, the Shi respiratory function was reduced to 45% of Wt and was partially restored to 64% of Wt after deleting SNPH from Shi. Put differently, even though SNPH deletion only partially restores respiration per mitochondrion, because Shi PCs have more mitochondria, the net effect of SNPH deletion is a full restoration of respiration per PC.
Figure 8.

Deletion of SNPH restores loss of mitochondrial respiratory chain activity in dysmyelinated Shi. Fresh-frozen sections from cerebellum of 4.5-month-old compound mutants were incubated with Cox and SDH reaction buffer and mitochondrial electron transport chain activity was monitored. A–D, Cox activity in Wt, Shi, Shi-SNPH-KO, and SNPH-KO. SDH activity in these mutants is shown in Ai, Bi, Ci, and Di respectively. E–H, Cox1 expression in compound mutants. SDHA expression in these mutants is shown in Ei, Fi, Gi, and Hi. Scale bar, 20 μm. Arrow in B and F indicates reduced Cox activity and Cox1 expression in Shi. I and J, Cox-1- and SDHA-labeled mitochondria (green) in NF-labeled axons (red) from Wt, Shi, Shi-SNPH-KO, and SNPH-KO mice. Intensities of brown and blue color were quantitated and mitochondrial metabolic health was represented as Cox/SDH ratio. Quantitative analysis of normalized COX/SDH ratios in PC soma and Cox1/SDHA in axonal regions of compound mutants are shown in K and L. Data shown are mean ± SEM from three mice of each mutants. *p < 0.05 between Wt and Shi; #p < 0.05 between Shi and Shi-SNPH-KO.
These results were further verified by immunohistochemical analysis of the mitochondrial cytochrome C oxidase subunit 1 (Cox-1) and succinate dehydrogenase subunit A (SDHA) subunits. The expression of Cox1 and SDH subunits has been reported to correlate with their enzymatic activities (Kageyama et al., 2012). Figure 8, E–H, show the Cox1 expression in PC soma of 4.5-month-old Wt, Shi, Shi-SNPH-KO, and SNPH-KO, respectively, whereas SDHA expression in these mice is shown in Figure 8, Ei, Fi, Gi, Hi, respectively. The Cox1 expression was reduced in Shi PCs (arrow in Fig. 8F). Due to difficulties in resolving Cox and SDH reaction products in axons, we used Cox-1 and SDHA immunoreactivities to analyze mitochondrial health in axons colabeled with neurofilaments (NFs). The healthier mitochondria show a good expression of above subunits and turn yellow when colocalized with neurofilament (yellow over red axons in Fig. 8I,J). The Cox1 expression was decreased in Shi compared with other mutants (Fig. 8I), whereas SDHA expression appeared to be similar in all mutants (Fig. 8J). Fluorescence intensity of individual mitochondria in axons was quantitated and the metabolic health index was calculated as the Cox1/SDHA intensity ratio (Fig. 8L). Cerebellar axons in Shi showed a 30% reduction in ratio (p < 0.05) that was rescued upon deletion of SNPH (Fig. 8L). The ratio in Shi-SNPH-KO was increased by 57% (p < 0.01) compared with Shi (Fig. 8L). Interestingly, the Cox/SDH ratio was significantly reduced in the torpedoes of both Shi and Shi-SNPH-KO by 40% and 66%, respectively, compared with the ratios in their axons (Fig. 8L).
Together, these results show that the respiratory function of mitochondria is reduced in Shi but increased in Shi-SNPH-KO, consistent with an improvement in the quality of mitochondria.
SNPH deletion decreases superoxide stress in Shi
The lower mitochondrial complex IV activity in Shi could produce more free radicals via leak of electrons from the respiratory chain. We therefore measured the superoxide levels in cerebellar sections of compound mutants using DHE that specifically detects superoxide levels. PCs were identified by calbindin antibody. Representative images of DHE fluorescence in fresh frozen cerebellar sections of compound mutants are shown in Figure 9, B (Wt), E (Shi), H (Shi-SNPH-KO), and K (SNPH-KO) and the corresponding calbindin labeling is shown in Figure 9, A, D, G, and J, respectively. Wt and SNPH-KO (Fig. 9B,K) showed very low DHE fluorescence and both Shi and Shi-SNPH-KO showed an increase in DHE fluorescence (Fig. 9E,H). It was noticed that the granular cells show higher DHE fluorescence than PCs in compound mutants. Quantitative analysis of DHE fluorescence is shown in Figure 9 for PC soma (Fig. 9M), cerebellar granular layer (Fig. 9N), and molecular layer (Fig. 9O) of compound mutants. The PC soma DHE fluorescence was measured by averaging the red fluorescence within the cell body outlined by calbindin (see Materials and Methods). The DHE signal was increased in the PCs (Fig. 9M) of Shi over the Wt by 2.75-fold (p < 0.01). Importantly, SNPH deletion from Shi reduced the Shi DHE signal by 34% (p < 0.05; Fig. 9M). The fluorescence intensities between Wt, Shi-SNPH-KO, and SNPH-KO did not differ significantly (Fig. 9M). Similarly, the DHE fluorescence was significantly elevated in the granular (2.38-fold, p < 0.05, Fig. 9N) and molecular layers (2.83-fold, p < 0.01, Fig. 9O) of Shi cerebellum that was decreased by deleting SNPH in Shi (36.5%, p < 0.05 in granular and 49%, p < 0.05 in molecular layer). Notice that cells in the granular layer have elevated DHE signal in the Shi that is reduced by SNPH-KO (Fig. 9E,H). One possibility is that this is a secondary effect of primary damage in the PC axons. However, we cannot rule out the possibility of a direct effect of SNPH deletion on alleviating oxidative stress in these granular cells. Together, these results provide evidence of increased oxidative stress in Shi that was reduced by deleting SNPH.
Figure 9.

Deletion of SNPH attenuates increase in oxidative stress in Shi cerebellum. Fresh-frozen sections from cerebellum of 4.5-month-old mutants were incubated with dihydrethidium (DHE) and stained for calbindin. DHE fluorescence in Wt, Shi, Shi-SNPH-KO, and SNPH-KO is shown in B, E, H, and K, respectively; calbindin staining in these mutants is shown in A, D, G, and J. Merged image of calbindin and DHE for each mutant is shown in panels C, F, I, and L. A magnified region of PC in each panel is shown in the insert. A representative image is shown from three mice from each genotype. Scale bar, 50 μm. Quantitative analysis of mean ± SEM DHE fluorescence in PCs, granular layer, and molecular layer is shown in M–O, respectively. *p < 0.05 between Wt and Shi; # p < 0.05 between Shi and Shi-SNPH-KO.
SNPH deletion does not benefit EAE symptoms
To determine whether SNPH benefits the early phase of MS, we used EAE, a model for the early inflammatory phase of MS. EAE was induced in Wt and SNPH-KO mice and clinical scores were monitored up to 28 d after EAE induction (Fig. 10A). Presence or absence of SNPH did not significantly affect the overall shape of the clinical score profiles (Fig. 10A), suggesting that SNPH deletion provides little or no benefit to EAE. We next performed cellular analysis to determine whether SNPH deletion protects EAE-induced axonal injuries (torpedoes) at 28 d after immunization in cerebellum and spinal cord dorsal column. Tissue sections were labeled with microglial marker (IBA1), myelin antibody (MBP), and nonphosphorylated NFs. Z-stack confocal images were collected from randomly selected regions of these tissues. Figure 11 shows representative fluorescence images of IBA1, MBP, and NF labeling in different regions of spinal cord dorsal column and cerebellum from immunized Wt and SNPH-KO mice. Because EAE induces focal demyelination and heterogeneous regions of inflammation in axons, we divided our torpedoes analysis into two axonal regions, those with (Fig. 11A,B,E,F) and those without (Fig. 11C,D,G,H) inflammation. Inflammation was identified by an increase in IBA1 immunoreactivity and demyelination by a reduction in MBP immunoreactivity. The torpedoes in inflammatory and noninflammatory regions of Wt and SNPH-KO mice were counted by an observer blinded to the genotypes of the mice under analysis. As expected, torpedoes were increased in the inflammation regions (Fig. 11Aii,Bii,Eii,Fii) over the non-inflammation regions (Fig. 11Cii,Dii,Gii,Hii). In the inflammation regions, SNPH deletion did not reduce torpedoes significantly in dorsal column (Fig. 10C; p > 0.05) and cerebellum (Fig. 10D; p > 0.05). This is in contrast to the Shi mouse, in which SNPH deletion significantly reduced torpedoes (Fig. 6J). Interestingly, there were torpedoes in the noninflammation region that were also not reduced by SNPH deletion (Fig. 10C,D; p > 0.05). We did not observe a difference in the PC numbers in Wt and SNPH-KO (Fig. 10B). Collectively, this suggests that SNPH does not protect torpedoes in early-phase demyelination. Interestingly, at an earlier time point of 20 d after immunization (Fig. 10A), deletion of SNPH produces slightly worse symptoms (p < 0.05) over Wt mice. The striking contrast between significant benefits to the Shi model and unremarkable effects on the EAE model both rendered SNPH deficient gives credence to the hypothesis that anchoring deletion is a selective treatment for late-phase MS.
Figure 10.

SNPH deletion does not benefit EAE mice. Wt and SNPH-KO mice were immunized with MOG35-55 peptide in complete Freund's adjuvant (CFA) and clinical symptoms were monitored for 28 d after immunization. A, Clinical scores of Wt (n = 21) and SNPH-KO (n = 13) mice. Cerebellar sections from immunized mice (n = 7 of each genotype) were stained with H&E and PCs were counted. PCs in Wt and SNPH-KO mice did not differ significantly (B). Thin sections from cerebellum and spinal cord dorsal column from Wt (n = 6) and SNPH-KO (n = 5) mice were stained for IBA1, MBP, and NF to identify inflammatory and noninflammatory demyelination in these tissues (Fig. 11). Axonal torpedoes in dorsal column (C) and cerebellum (D) were quantified from inflammatory and noninflammatory regions and shown as mean ± SEM torpedoes in 2.5 × 105 μm3 volumes.
Figure 11.
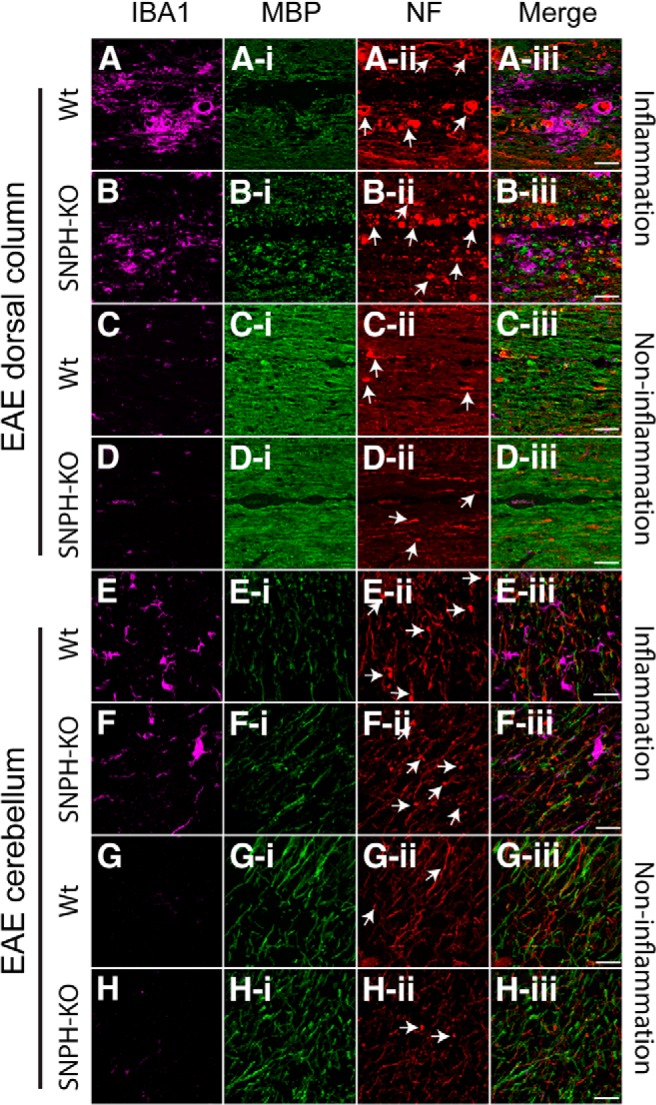
Immunohistochemical analysis of inflammation and axonal torpedoes in EAE mice with or without SNPH. Cerebellum and dorsal column from Wt and SNPH-KO at 28 d after immunization were analyzed for microglial expression and myelin and axonal degeneration (torpedoes) using antibodies IBA1, MBP, and NF, respectively. Regions for analysis were separated into inflammation and noninflammation regions according to elevated or nonelevated IBA1 expression. Some examples of torpedoes are shown by arrows in Aii–Hii. A and B, Inflammation regions in dorsal column of Wt and SNPH-KO mice. Increased IBA1 reactivity with activated microglia (rounded/amoeboid shape), decreased MBP reactivity (Ai, Bi), and increased axonal torpedoes (Aii, Bii) were observed in these inflammatory regions of Wt and SNPH-KO. Merged images from the above panels are shown in far right panels (Aiii, Biii). The noninflammatory regions with less IBA1 reactivity are shown in C and D, respectively. MBP and NF immunoreactivity in these regions are shown in Ci, Di and Cii, Dii, respectively. Ciii and Diii show the merged images. Axonal torpedoes were reduced in noninflammatory regions of both Wt and SNPH-KO. Scale bar, 50 μm. Inflammatory regions from the cerebellum of Wt and SNPH-KO mice with activated microglia are shown in E and F, respectively. Reduced MBP (Ei, Fi) and increased axonal torpedoes (Eii, Fii) were observed in both Wt and SNPH-KO mice. Merged images of these regions are shown in Eiii and Fiii. The noninflammatory regions are shown in G and H. The corresponding MBP and NF immunoreactivity for these regions are shown in Gi, Hi and Gii, Hii, respectively. Giii and Hiii show the merged image of these regions. Scale bar, 20 μm.
Discussion
Once MS has entered the progressive phase (SPMS), the disease becomes resistant to all known treatments (for review, see Lassmann et al., 2012; Witte et al., 2014). In this work, we identify various parallelisms between the Shi mouse and human progressive MS and hypothesize a novel treatment specific for progressive MS based on studies on a new Shi compound mutant deficient in SNPH. The idea for a late-phase specific MS therapy has been emerging in recent years in animal studies. Even though the traditional EAE model is monophasic, biphasic EAE models [i.e., inducing EAE in nonobese diabetic (NOD) mice] have been developed that exhibit a secondary progressive phase resembling progressive MS. Using this biphasic EAE-NOD mouse model, Basso et al. (2008) demonstrated that an anti-oxidant/anti-excitotoxic therapy is effective only for the secondary progressive phase but not the early phase. Our study adds mitochondrial anchoring as a therapeutic locus specific for late-phase MS. The potentials and limitations of this work for progressive MS are discussed below.
Mechanisms for neuroprotection upon SNPH deletion
Whereas this study establishes that SNPH deletion provides significant benefits to Shi, the mechanisms for neuroprotection remain to be elucidated. Several neuroprotective mechanisms are possible. First, SNPH deletion enhances the axon → soma movement of poor mitochondria in axons. Mitochondria are under metabolic stress at dysmyelinated sites due to strong energy demand. Excessive SNPH expression would impede stress-induced, dysfunctional mitochondria from traveling back to the soma for degradation (mitophagy). Without proper degradation of unhealthy mitochondria, the overall quality of mitochondria and their ATP production capacity at dysmyelinated axons will decline, contributing to axonal degeneration. This impediment is removed by deleting SNPH, allowing the overall health of mitochondria to increase. Second, SNPH deletion enhances soma → axon movement of fresh mitochondria. In this case, mitophagy can occur locally in axons (Ashrafi et al., 2014) to degrade poor mitochondria. The soma will then supply axons with fresh mitochondria. Excessive SNPH expression is detrimental by preventing healthy mitochondria in the soma from trafficking to the dysmyelinated axons to replenish mitochondria. Deletion of SNPH confers neuroprotection by improving this soma → axon replenishment. Consistent with either hypothesis, Cox and superoxide labeling demonstrate an increase in mitochondrial quality (Fig. 8) and reduction in oxidative stress (Fig. 9) in Shi upon SNPH deletion. Other neuroprotective mechanisms unrelated to mitochondrial mobility are possible. The output of a PC is controlled by the balance between excitatory (glutamatergic) and inhibitory (GABAergic) synaptic inputs. Strikingly, SNPH appears to be highly elevated at the GABAergic terminal region innervating the PC soma (white arrow in Fig. 2F). Because overexpression of SNPH has been shown to inhibit synaptic release of neurotransmitters in vitro (Lao et al., 2000), overexpression of SNPH in the basket cell terminals might inhibit GABAergic transmission, tilting the balance toward excitatory glutamatergic transmission that ultimately overexcites and kills PCs. Deleting SNPH is protective because it tilts the synaptic balance back toward normalcy.
Role of mitochondria in MS
Over the last 5–7 years, significant progress has been made toward establishing a pathologic role for mitochondria in MS. In general, these studies focus on mitochondria as targets of damage from upstream inflammation and toxic molecules. These include reactive oxygen species (ROS) and nitric oxide released by activated microglia and macrophages. Recently, genetic inactivation of cyclophilin D and p66ShcA (components of mPTP) in EAE (Forte et al., 2007; Su et al., 2012) and pretreatment of EAE mice with a synthetic antioxidant Mito Q have been shown to be neuroprotective (Davies et al., 2013; Mao et al., 2013). In addition, cumulative mtDNA damage from patients in late-phase MS has been suggested to result from inflammation insult from the early phase (Campbell et al., 2011).
These studies are significant in suggesting therapies to protect mitochondria from inflammation-induced damage. However, our work is novel in showing how mitochondria, in a noninflammatory background, could initiate damage when they are excessively anchored. This aspect of our work is particularly germane to mitochondrial therapy in progressive MS. We conclude by proposing a novel hypothesis for a biphasic role of SNPH in MS (Fig. 12A,B) to stimulate future thinking toward designing an optimal SNPH-based therapy to reverse late-phase degeneration (Fig. 12C,D).
Figure 12.

Hypothesis for biphasic role of SNPH in MS and optimal timing of SNPH-KO therapy. A and B, Evolution of SNPH function from adaptive in early phase (A) to maladaptive in the late progressive phase (B). A, Early phase. Mitochondria anchored by SNPH have larger volume than unanchored ones, producing more ATP to match energy demand of demyelinated axons (Ohno et al., 2014). Simultaneous to this positive functional impact of SNPH, a slight negative functional impact occurs when anchoring modestly impedes either unhealthy mitochondria from returning to the soma for mitophagy (left pointing green dashed arrow) or fresh mitochondria from the soma to travel down the axon to replenish the immobilized pool (right pointing green dashed arrow). Overall, the metabolic benefit of SNPH-mediated increase in mitochondrial size outweighs negativity. Axonal health increases. B, Late phase. Chronic anchoring of mitochondria to increase mitochondrial mass to match energy demand turns maladaptive. Quality of congested mitochondria is compromised, presumably resulting from cumulative failure either of mitochondria in early phase to return to the soma for mitophagy (left pointing green arrow) or fresh mitochondria from the soma to reach axons to replenish the immobilized pool (right pointing green arrow). The resultant poor mitochondria also produce ROS to fuel a vicious cycle. A further, a yet-to-be determined signal might also dramatically trigger a steep upregulation of SNPH in the late phase, exacerbating the problem. Excessive SNPH anchoring over time kills neural tissues in late phase or SPMS. C and D, Idealized scheme for optimal therapy by selective SNPH-KO in the late phase. C, Early phase remains unaffected in this therapy. D, Late phase. SNPH-KO therapy applied selectively to late-phase by conditional techniques. Anchorless mitochondria become smaller in size, producing less ATP per mitochondrion. However, these smaller mitochondria are healthier because removal of anchoring impediment results either in better somal mitophagy (left pointing green arrow) or better replenishment of the immobilized pool by fresh mitochondria from the soma (right pointing green arrow). The reduction in axonal mitochondrial density after SNPH deletion (Fig. 4F) suggests that SNPH deletion predominantly enhances the axon → soma mitochondrial movement (D). The net effect is the maintenance of a good ratio of healthy to unhealthy mitochondria, leading to a better energy support to keep the axon alive. We hypothesize that SNPH deletion produces the best metabolic mode for survival in SPMS by using highly mobile and smaller mitochondria. Note that, in this study, SNPH is deleted from both phases in the Shi and we predict that the protection achieved in this work (Figs. 5, 6) is not yet optimal.
Hypothesis for a biphasic role of SNPH in MS
Interestingly, a recent study by Ohno et al. (2014) demonstrated that deletion of SNPH is counterproductive to axonal survival in acute demyelination. They proposed that SNPH serves an adaptive function by creating large-volume, stationary mitochondria to better fit the energy demands of acutely demyelinated axons and help them survive. In contrast, our studies demonstrate that SNPH deletion in chronic dysmyelination (Shi) dramatically prolongs survival and prevents neural damage. It is clear that these two studies address different phases of MS. We propose a biphasic model for the functional impact of SNPH during MS: mitochondrial anchoring by SNPH is adaptive in the early phase (Fig. 12A) and maladaptive in the late phase (Fig. 12B).
During the early phase of acute myelin loss, as in the study of Ohno et al. (2014), SNPH is modestly elevated to arrest motile mitochondria to meet metabolic demands to keep axons alive (Fig. 12A). Deletion of SNPH at this stage is detrimental to axons. However, as chronic myelin loss persists (Fig. 12B), increasing energy demand leads to increasing mitochondrial mass through anchoring. As discussed above, one mechanism for excessive SNPH to degrade health is by impeding two pathways for mitochondrial flow. Excessive SNPH may impede either the axon → soma return of poor mitochondria for degradation (green arrow pointing left), or the soma → axon replenishment with good mitochondria (green arrow point right). The result is ROS production fueling a vicious cycle. In addition, a yet-to-be identified signal might also trigger a steep, late-phase upregulation of SNPH, further exacerbating the problem. Whereas this model predicts that SNPH deletion is protective in the late phase by enhancing the movement of mitochondria in these two pathways, more work is needed to sort out which pathway is predominant in protection. We observed that SNPH deletion in the Shi causes a reduction in mitochondrial density in the granular axons (Fig. 4F). This suggests that SNPH deletion mediates protection by predominantly enhancing the axon → soma return of mitochondria for degradation (Fig. 12D). In fact, in preliminary EM studies (our unpublished data), we observed that deletion of SNPH in Shi also enhances mitophagy activities in the PC soma. This tentatively favors the axon → soma pathway as underlying neuroprotection: SNPH deletion releases immobilized mitochondria in the axon to return to the soma, which triggers enhanced mitophagy activity. The overall result is better circulation of mitochondria, leading to an increased ratio of good to bad mitochondria through degradation. Clearly, more work is required to test this mechanism. Regardless of which pathways for neuroprotection is correct, our hypothesis predicts that disrupting mitochondrial anchoring plays opposing roles during progression of axon dysmyelination: it is harmful in the early phase but neuroprotective in the late phase.
This hypothesis is supported by the sharp contrast between marked protection on the Shi and the unremarkable effects on EAE (an early-phase model) when SNPH is rendered deficient in both models. Indeed, our biphasic model predicts that SNPH deletion should harm EAE. Interestingly, compared with wild-type mice, inducing EAE in SNPH-KO mice slightly hastened the onset of clinical scores and produced a modest worsening of symptoms at 20 d after immunization (Fig. 10A). However, the clinical scores overlapped thereafter, and cellular analysis at 28 d after immunization shows no significant difference in neural damage in EAE with or without SNPH (Fig. 10C,D). This suggests that any harmful effects of SNPH deletion on EAE in the early period are transient. In the Shi, we have a long window of 4–6 months from birth when SNPH deletion covers both an early adaptive and a late maladaptive phase. We focused our analysis on late-phase Shi and did not address the possible harmful effects of SNPH deletion in the early-phase Shi. However, we observed insignificant torpedoes at 2.5 months in Shi with or without SNPH (Fig. 6J). We cannot rule out the possibility that SNPH deletion produces subthreshold damages (such as elevated calcium) in the early-phase Shi before 2.5 months. The fact that there is a net gain in benefits when SNPH is deleted from both phases suggests that the maladaptive phase determinates the disease outcome and must be the priority site for intervention. In fact, any subthreshold harm done to the early-phase Shi in the Shi-SNPH-KO (such as elevated calcium) might spill into the late phase and work against the emerging protective effect of SNPH deletion. If correct, this hypothesis predicts that an optimal strategy for neuroprotection in progressive MS is a conditional deletion of SNPH timed at conversion of the disease into the late phase (Fig. 12D).
In conclusion, we have generated a double mutant mouse line, Shi-SNPH-KO, for studying neuroprotection in MS research. The robust protective phenotype of the Shi-SNPH-KO mutant suggests that elimination of mitochondrial anchoring is potentially a new specific treatment for progressive MS. Future studies are required to elucidate how SNPH deletion produces protection in MS, in particular the time point in disease progression at which SNPH turns from adaptive to maladaptive. Our hypothesis, if substantiated by further studies, will stimulate a pharmaceutic screen for small-molecule inhibitors of SNPH to treat MS patients during transitions into the late phase.
Footnotes
This work was supported by the National Institutes of Health (Grants RO1NS073743 and R21NS082725 to S.Y.C.) and by the Intramural Research Program of NINDS, NIH (Z.-H.S). We thank Phil Smith, Lance Rodenkirch (UW Keck imaging facility), and Randall Massey (UW medical school electron microscopy facility) for help in data collection and Timothy Gomez and Miles Epstein for helpful discussion during this work.
The authors declare no competing financial interests.
References
- Andrews H, White K, Thomson C, Edgar J, Bates D, Griffiths I, Turnbull D, Nichols P. Increased axonal mitochondrial activity as an adaptation to myelin deficiency in the Shiverer mouse. J Neurosci Res. 2006;83:1533–1539. doi: 10.1002/jnr.20842. [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206:655–670. doi: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso AS, Frenkel D, Quintana FJ, Costa-Pinto FA, Petrovic-Stojkovic S, Puckett L, Monsonego A, Bar-Shir A, Engel Y, Gozin M, Weiner HL. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest. 2008;118:1532–1543. doi: 10.1172/JCI33464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM, Mahad DJ. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol. 2011;69:481–492. doi: 10.1002/ana.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AL, Desai RA, Bloomfield PS, McIntosh PR, Chapple KJ, Linington C, Fairless R, Diem R, Kasti M, Murphy MP, Smith KJ. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann Neurol. 2013;74:815–825. doi: 10.1002/ana.24006. [DOI] [PubMed] [Google Scholar]
- Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Rahder M, Stem K, Bernardi P, Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc Natl Acad Sci U S A. 2007;104:7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Haider L, Fischer MT, Frischer JM, Bauer J, Höftberger R, Botond G, Esterbauer H, Binder CJ, Witztum JL, Lassmann H. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–1924. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulst HE, Geurts JJ. Gray matter imaging in multiple sclerosis: what have we learned? BMC Neurol. 2011;11:153. doi: 10.1186/1471-2377-11-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese M, Oesingmann N, Casaccia P, Fleysher L. Progressive multiple sclerosis and gray matter pathology: an MRI perspective. Mt Sinai J Med. 2011;78:258–267. doi: 10.1002/msj.20247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Nakamura R, Mikoshiba K, Tsukada Y. Fine structure of the central myelin sheath in the myelin deficient mutant Shiverer mouse, with special reference to the pattern of myelin formation by oligodendroglia. Brain Res. 1981;219:85–94. doi: 10.1016/0006-8993(81)90269-9. [DOI] [PubMed] [Google Scholar]
- Iwai M, Liu HW, Chen R, Ide A, Okamoto S, Hata R, Sakanaka M, Shiuchi T, Horiuchi M. Possible inhibition of focal cerebral ischemia by angiotensin II type 2 receptor stimulation. Circulation. 2004;110:843–848. doi: 10.1161/01.CIR.0000138848.58269.80. [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, Sesaki H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. 2012;197:535–551. doi: 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao G, Scheuss V, Gerwin CM, Su Q, Mochida S, Rettig J, Sheng ZH. Syntaphilin: a syntaxin-1 clamp that controls SNARE assembly. Neuron. 2000;25:191–201. doi: 10.1016/S0896-6273(00)80882-X. [DOI] [PubMed] [Google Scholar]
- Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8:647–656. doi: 10.1038/nrneurol.2012.168. [DOI] [PubMed] [Google Scholar]
- Loers G, Aboul-Enein F, Bartsch U, Lassmann H, Schachner M. Comparison of myelin, axon, lipid, and immunopathology in the central nervous system of differentially myelin-compromised mutant mice: a morphological and biochemical study. Mol Cell Neurosci. 2004;27:175–189. doi: 10.1016/j.mcn.2004.06.006. [DOI] [PubMed] [Google Scholar]
- MacKenzie-Graham A, Tiwari-Woodruff SK, Sharma G, Aguilar C, Vo KT, Strickland LV, Morales L, Fubara B, Martin M, Jacobs RE, Johnson GA, Toga AW, Voskuhl RR. Purkinje cell loss in experimental autoimmune encephalomyelitis. Neuroimage. 2009;48:637–651. doi: 10.1016/j.neuroimage.2009.06.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, Lassmann H, Turnbull DM. Mitochondrial changes within axons in multiple sclerosis. Brain. 2009;132:1161–1174. doi: 10.1093/brain/awp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Manczak M, Shirendeb UP, Reddy PH. MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim Biophys Acta. 2013;1832:2322–2331. doi: 10.1016/j.bbadis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineaux SM, Engh H, de Ferra F, Hudson L, Lazzarini RA. Recombination within the myelin basic protein gene created the dysmyelinating shiverer mouse mutation. Proc Natl Acad Sci U S A. 1986;83:7542–7546. doi: 10.1073/pnas.83.19.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N, Chiang H, Mahad DJ, Kidd GJ, Liu L, Ransohoff RM, Sheng ZH, Komuro H, Trapp BD. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc Natl Acad Sci U S A. 2014;111:9953–9958. doi: 10.1073/pnas.1401155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt D, Gonzales E, Cross AH, Goldberg MP. Dysmyelinated axons in shiverer mice are highly vulnerable to alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor-mediated toxicity. Brain Res. 2010;1309:146–154. doi: 10.1016/j.brainres.2009.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick KL, Dugan LL. Superoxide stress identifies neurons at risk in a model of ataxia-telangiectasia. Ann Neurol. 2001;49:627–635. [PubMed] [Google Scholar]
- Roach A, Boylan K, Horvath S, Prusiner SB, Hood LE. Characterization of cloned cDNA representing rat myelin basic protein: absence of expression in brain of shiverer mutant mice. Cell. 1983;34:799–806. doi: 10.1016/0092-8674(83)90536-6. [DOI] [PubMed] [Google Scholar]
- Sheng ZH. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J Cell Biol. 2014;204:1087–1098. doi: 10.1083/jcb.201312123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichinohe H, Kuroda S, Yasuda H, Ishikawa T, Iwai M, Horiuchi M, Iwasaki Y. Neuroprotective effects of the free radical scavenger Edaravone (MCI-186) in mice permanent focal brain ischemia. Brain Res. 2004;1029:200–206. doi: 10.1016/j.brainres.2004.09.055. [DOI] [PubMed] [Google Scholar]
- Shields SD, Cheng X, Gasser A, Saab CY, Tyrrell L, Eastman EM, Iwata M, Zwinger PJ, Black JA, Dib-Hajj SD, Waxman SG. A channelopathy contributes to cerebellar dysfunction in a model of multiple sclerosis. Ann Neurol. 2012;71:186–194. doi: 10.1002/ana.22665. [DOI] [PubMed] [Google Scholar]
- Stys PK. Pathoetiology of multiple sclerosis: are we barking up the wrong tree? F1000Prime Rep. 2013;5:20. doi: 10.12703/P5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Zamponi GW, van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci. 2012;13:507–514. doi: 10.1038/nrn3275. [DOI] [PubMed] [Google Scholar]
- Su KG, Savino C, Marracci G, Chaudhary P, Yu X, Morris B, Galipeau D, Giorgio M, Forte M, Bourdette D. Genetic inactivation of the p66 isoform of ShcA is neuroprotective in a murine model of multiple sclerosis. Eur J Neurosci. 2012;35:562–571. doi: 10.1111/j.1460-9568.2011.07972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swingler RJ, Compston DA. The morbidity of multiple sclerosis. Q J Med. 1992;83:325–337. [PubMed] [Google Scholar]
- Tanji K, Bonilla E. Optical imaging techniques (histochemical, immunohistochemical, and in situ hybridization staining methods) to visualize mitochondria. Methods Cell Biol. 2001;65:311–332. doi: 10.1016/S0091-679X(01)65019-2. [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Toosy AT, Ciccarelli O. Pharmacological management of symptoms in multiple sclerosis: current approaches and future directions. Lancet Neurol. 2010;9:1182–1199. doi: 10.1016/S1474-4422(10)70249-0. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Witte ME, Mahad DJ, Lassmann H, van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med. 2014;20:179–187. doi: 10.1016/j.molmed.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Zambonin JL, Zhao C, Ohno N, Campbell GR, Engeham S, Ziabreva I, Schwarz N, Lee SE, Frischer JM, Turnbull DM, Trapp BD, Lassmann H, Franklin RJ, Mahad DJ. Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain. 2011;134:1901–1913. doi: 10.1093/brain/awr110. [DOI] [PMC free article] [PubMed] [Google Scholar]