


Abstract
Cohesin, which in somatic vertebrate cells consists of SMC1, SMC3, RAD21 and either SA1 or SA2, mediates higher-order chromatin organization. To determine how cohesin contributes to the establishment of tissue-specific transcriptional programs, we compared genome-wide cohesin distribution, gene expression and chromatin architecture in cerebral cortex and pancreas from adult mice. More than one third of cohesin binding sites differ between the two tissues and these show reduced overlap with CCCTC-binding factor (CTCF) and are enriched at the regulatory regions of tissue-specific genes. Cohesin/CTCF sites at active enhancers and promoters contain, at least, cohesin-SA1. Analyses of chromatin contacts at the Protocadherin (Pcdh) and Regenerating islet-derived (Reg) gene clusters, mostly expressed in brain and pancreas, respectively, revealed remarkable differences that correlate with the presence of cohesin. We could not detect significant changes in the chromatin contacts at the Pcdh locus when comparing brains from wild-type and SA1 null embryos. In contrast, reduced dosage of SA1 altered the architecture of the Reg locus and decreased the expression of Reg genes in the pancreas of SA1 heterozygous mice. Given the role of Reg proteins in inflammation, such reduction may contribute to the increased incidence of pancreatic cancer observed in these animals.
INTRODUCTION
Spatio-temporal control of gene expression in higher eukaryotes is essential for development and differentiation (1–3). Increasing evidence from the use of newly developed Chromosome Conformation Capture (3C)-based techniques indicates that such control relies on the ability of a gene to interact with cis-regulatory elements that can be located up to 1 Mb away (4,5). These interactions usually occur within megabase-sized topologically associated domains (TAD)s, which are largely invariant between cell types (6–9). Two of the architectural elements involved in chromatin folding and DNA looping are the CCCTC-binding factor (CTCF) and cohesin (10–13). CTCF is a protein highly conserved in higher eukaryotes which has a central DNA-binding domain with 11 zinc fingers and has been classically considered an insulator (14). Cohesin is a ring-shaped complex conserved from yeast to human that is composed of four subunits: SMC1, SMC3, RAD21/SCC1 and SA/STAG/SCC3. Cohesin is well known for mediating sister chromatid cohesion (15). Its role in transcriptional regulation, first reported in Drosophila (16), became more evident when its genome-wide distribution revealed a large overlap with CTCF both in mouse and human cells (17–19). Cohesin was then shown to be required for the formation or stabilization of chromatin loops at certain loci, together with CTCF or other factors such as the coactivator Mediator (20–27). Although the common and distinct roles of cohesin and CTCF are still far from being understood, recent results suggest that maintenance of TADs’ boundaries mostly relies on CTCF whilst both cohesin and CTCF contribute to chromatin contacts within the TADs (28–30).
In somatic vertebrate cells, the SA subunit of cohesin can be either SA1 or SA2 (31,32). Despite the high degree of homology between the two proteins, cohesin complexes carrying one or the other subunit show some functional specificity. Cohesin-SA1 and cohesin-SA2 mediate telomere and centromere cohesion, respectively, and both contribute to cohesion along chromosome arms (33,34). The two complexes display a similar genome-wide distribution in mouse cells. However, in the absence of SA1, a fraction of cohesin-SA2 repositions to new sites with reduced overlap with CTCF sites and promoters, and gene expression is altered (35). This result suggests that cohesin-SA1 could have a more prominent role in transcriptional regulation than cohesin-SA2.
Mutations in cohesin and in its loader, NIPBL, cause a human developmental syndrome known as Cornelia de Lange. Transcriptional dysregulation of genes sensitive to cohesin dosage has been proposed to underlie the aetiology of the syndrome, at least in part (36–38). Developmental genes could be particularly sensitive to cohesin (39–41). Cohesin mutations have also been recently identified in several tumour types although it is unclear which dysfunction of the complex contributes most to the disease, cohesion or gene regulation (42–45). Understanding the role of cohesin in transcriptional control may thus provide important clues to improve diagnosis and treatment of the affected patients. Recent studies indicate the existence of ubiquitous and tissue-specific cohesin positions (23,46–47). It has been proposed that tissue-specific cohesin binding sites could help define cell-type-specific transcriptional programs (48). To explore this possibility, we have analysed genome-wide cohesin distribution and gene expression in two different mouse tissues, brain and pancreas, and its contribution to defining active cis-regulatory regions. Changes in cohesin distribution in a given locus between the two tissues correlate not only with differences in gene expression but also with distinct chromatin architecture, as judged by chromosome conformation capture combined with high-throughput sequencing (4C-seq) analyses. Moreover, mice deficient in SA1 show altered gene expression in both tissues. Altogether, our results support a prominent contribution of cohesin to tissue-specific gene expression through the establishment of the required chromatin contacts. Moreover, we present evidence suggesting that reduced cohesin-SA1 levels lead to gene expression changes in pancreas that increase cancer susceptibility in SA1 heterozygous mice.
MATERIALS AND METHODS
Animals
Tissues for the experiments described below were obtained from adult mice or embryos from a colony of mice heterozygous for SA1 (34). They were housed in a pathogen-free animal facility following the animal care standards of the institution.
ChIP sequencing (Chip-seq)
Chromatin immunoprecipitation (ChIP) was performed in tissues (cerebral cortex and pancreas of 8–10-week-old C57Bl/6 mice, and whole brain of E17.5 embryos) with custom made rabbit polyclonal antibodies against SA1 and SMC1 (34), RNApolIISer2P (AB5095, Abcam), CTCF (07–729, Millipore) and normal rabbit IgG as described (49) with some modifications. Fresh tissues collected in 6-well plates containing cold phosphate buffered saline (PBS) with 1-mM PMSF and protease inhibitor cocktail (Roche) were immediately minced to small fragments of ∼1-mm diameter. Tissue pieces (pooled from three individuals) were centrifuged at 2000 rpm for 2 min at 4ºC, resuspended in 1 ml of fixing solution (1% formaldehyde, 50-mM HEPES-KOH, 100-mM NaCl, 1-mM ethylenediaminetetraacetic acid (EDTA), 0.5-mM EGTA) and incubated 20 min at room temperature (RT) in a rotating wheel. Cross-linking was stopped by adding 1/20 volume of 2.5-M glycine for 5 min at RT. Tissue pieces were washed twice with cold PBS, recovered by centrifugation at 3000 rpm for 7 min at 4ºC, resuspended in 2-ml lysis buffer (1% sodium dodecyl sulphate, 10-mM EDTA, 50-mM Tris-HCl pH 8.1; 2 × 107 cells per ml) and sonicated in Covaris system (shearing time 30 min, 20% duty cycle, intensity 10, 200 cycles per burst and 30 s per cycle). DNA (10 ng of as quantitated by fluorometry) was electrophoresed on agarose gel and fractions corresponding to 100–200 bp in length were excised and purified. ChIP samples were processed into sequencing libraries and analysed according to Illumina's ‘ChIP Sequencing Sample Prep Guide’ (part #11257047 Rev. A), with the exception that gel extraction was replaced with Agencourt AMPure XP (Beckman Coulter) bead purification. Adapter-ligated library was completed by limited-cycle polymerase chain reaction (PCR) with Illumina PE primers (11 cycles). DNA libraries were applied to an Illumina flow cell for cluster generation and sequenced with the Illumina Genome Analyzer IIx (GAIIx) or the Illumina HiSeq2000. Image analysis was performed with Illumina Real Time Analysis software (RTA1.8). Sequence alignment to the reference genome (NCBIm37/mm9, April 2007) was made with Illumina's ELANDv2 algorithm on its ‘eland_extended’ mode from within CASAVA-1.7 package, using default settings. Only reads with unique alignment in the reference genome were used for the peak detection, which was performed using MACS v1.4 setting a P-value cut-off of 10−5 and FDR threshold better than 5% (50). All comparisons were done with the input track as control and each one of the data sets as treatment, using RUbioSeq 2.0 as automation analysis pipeline (51). Genomic interval overlaps and signal distributions were obtained using BEDTools v2.12 (52), PeakAnalyzer v1.3 (53) and custom UNIX shell scripting. All the statistical tests and correlations were calculated using R functions (http://cran.r-project.org).
RNA-sequencing (RNA-seq)
RNA samples (two independent replicates for condition, with material from four animals in each case) from different tissues were extracted using the guanidine thyocianate buffer, followed by acid phenol chloroform and treated with DNaseI (Ambion). polyA+RNA was purified with the Dynabeads mRNA purification kit (Invitrogen), randomly fragmented and converted to double-stranded cDNA and processed through subsequent enzymatic treatments of end-repair, dA-tailing and ligation to adapters as in Illumina's ‘TruSeq RNA Sample Preparation Guide’ (Part # 15008136 Rev. A). Adapter-ligated library was completed by limited-cycle PCR with Illumina PE primers (eight cycles). The resulting purified cDNA library was applied to an Illumina flow cell for cluster generation (TruSeq cluster generation kit v5) and sequenced on the Genome Analyzer IIx with SBS TruSeq v5 reagents by following manufacturer's protocols. Fastq files (54) with 40-nt single-end sequenced reads were quality-checked with FastQC (S. Andrews, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and aligned to the mouse genome (GRCm37/mm9) with TopHat-2.0.0 (55) using Bowtie 0.12.7 (56) and Samtools 0.1.16 (57) allowing two mismatches and five multihits. Transcript assembly, estimation of their abundances and differential expression were calculated with Cufflinks 1.3.0 (55) using the mouse genome annotation data set GRCm37.v65 from Ensembl (58). For differential expression, genes with FPKM values lower than 0.05 in both conditions were excluded. GO analyses were performed using FatiScan tool available at Babelomics suite (http://www.babelomics.org). To this end, genes were ranked based on Limma's moderated t-statistic and GO enrichment was evaluated by segmentation test. GO terms showing FDR < 0.05 were considered statistically significant.
4C-sequencing (4C-seq)
Preparation of 4C-seq samples was performed as described (59) with some modifications. Freshly collected tissues were incubated with collagenase (0.0065% collagenase I in 10% FBS/PBS for cortex, and 1.33-mg/ml collagenase P in HBSS for pancreas) for 40 min at 37ºC in a shaker. The resulting cell suspension was filtered through a 40-μm cell strainer in order to get a single cell preparation. Cells were centrifuged at 1100 rpm for 5 min at RT and the cell pellet was resuspended in 10 ml of fixing solution containing 2% formaldehyde in 10% FBS/PBS. After 10 min, 0.125-M glycine was added to stop fixation and the mixture was incubated for 5 min at RT. Cells were pelleted and nuclei isolated upon incubation in cold lysis buffer in the following conditions: for the brain, 10 min in 50-mM Tris pH 7.5, 150-mM NaCl, 5-mM EDTA, 0.5% NP-40, 1% TX-100 + protease inhibitors cocktail; for the pancreas, 40 min in 50-mM Tris pH 7.5, 50-mM NaCl, 5-mM EDTA, 1% NP40, 2% TX-100 + protease inhibitors cocktail. Proper lysis was determined by methyl green-pyronin staining. HindIII and CvIQ (in the case of the six-cutter libraries) and DpnII and CvIQ (in the case of the four-cutter libraries) were used as first and second cutters, respectively. 4C-seq libraries were amplified using long primers with 18–21-bp homology to the bait sequence and Illumina paired-end adapter flanks. Primer sequences were chosen to viewpoint sites, which were as close as possible to SMC1/SA1 ChIP-seq peaks (Supplementary Table S7). 4C-seq data analysis and normalization was performed with 4Cseqpipe (60) on the NCBI Build 37 mouse genome assembly.
Microarray analysis of gene expression in pancreas
Pancreas from wild-type and SA1-heterozygous mice (five replicates corresponding to five individuals per genotype) were removed and immediately processed to isolate total RNA using guanidine thyocianate buffer, followed by acid phenol chloroform. Since isolation of high-quality RNA from pancreas is challenging due to the large quantities of RNases, RNA Integrity Numbers were in the range 6.5–8.4 when assayed by Lab-chip technology on an Agilent 2100 Bioanalyzer. Hundred nanograms of RNA per condition were analysed by two-colour hybridization on Whole Mouse Genome DNA microarrays (G4852A; Agilent) and images were quantified with Agilent Feature Extraction Software (v. 10.7). Differentially expressed genes between SA1-heterozygous and wild-type MEFs were obtained by Limma (Smyth GK; Bioconductor project; http://www.bioconductor.org). FDR adjustment was employed to account for multiple testing.
Quantitative RT-PCR and protein analysis
Total RNA extracted as described above was treated with DNaseI (Ambion), and cDNAs were prepared according to the manufacturer's instructions using the Superscript II reverse transcriptase (Invitrogen). qRT-PCR analysis was performed using the SYBR Green PCR Master Mix and an ABI Prism® 7900HT instrument (Applied Biosystems®). Primers (Supplementary Table S8) were designed using OligoPerfect Designer™ (Invitrogen) and reactions were performed in triplicate. Quantifications were normalized to endogenous GAPDH, using the ΔΔCt method. To check cohesin-SA1 levels in pancreas of wild type and SA1 heterozygous mice, extracts were prepared from a piece of tissue that was pulverized in a mortar containing liquid nitrogen and lysed in RIPA buffer. Equal amounts of protein were run in 7.5 Bis/Tris gels followed by western blotting with antibodies against SA1 and Rad21 (34) and GAPDH (Sigma, G8795).
Chromatin states
Chromatin segmentation was performed with ChromHMM (61) including 15 states in the model and setting a bin size of 500 bp for the model learning and segmentation. The model learning was done using adult brain SMC1 and SA1 marks obtained in this study, and marks from LICR-m (9): 8-weeks cerebellum (H3K27Me3) and adult cortex (CTCF, H3K4Me1, H3K4Me3 and H3K27Ac).
Data access
ChIP-seq, RNA-seq and 4C-seq from this study have been submitted to GEO database (GSE59119).
RESULTS
Cohesin distribution is tissue-specific
With the aim to understand the functional role of cohesin in the establishment of tissue-specific transcriptional programs we performed ChIP followed by massive parallel DNA sequencing (ChIP-seq) analysis with antibodies against two cohesin subunits, SMC1 and SA1, in two tissues derived from different mouse embryonic germ layers. We chose cerebral cortex as representative of ectoderm lineage and pancreas as part of the digestive apparatus, derived from the endoderm embryonic layer. Genome-wide analysis of the sequenced tags defined 46 398 and 35 048 binding sites for SMC1 and SA1, respectively, in cortex and 32 381 and 26 896 SMC1 and SA1 binding sites, respectively, in pancreas (cut-off P-value of 10−5 and FDR of 0.05; Figure 1A and Supplementary Table S1). As expected, most SA1 sites are also SMC1 sites. The sites with SMC1 but no SA1 could correspond to cohesin-SA2.
Figure 1.
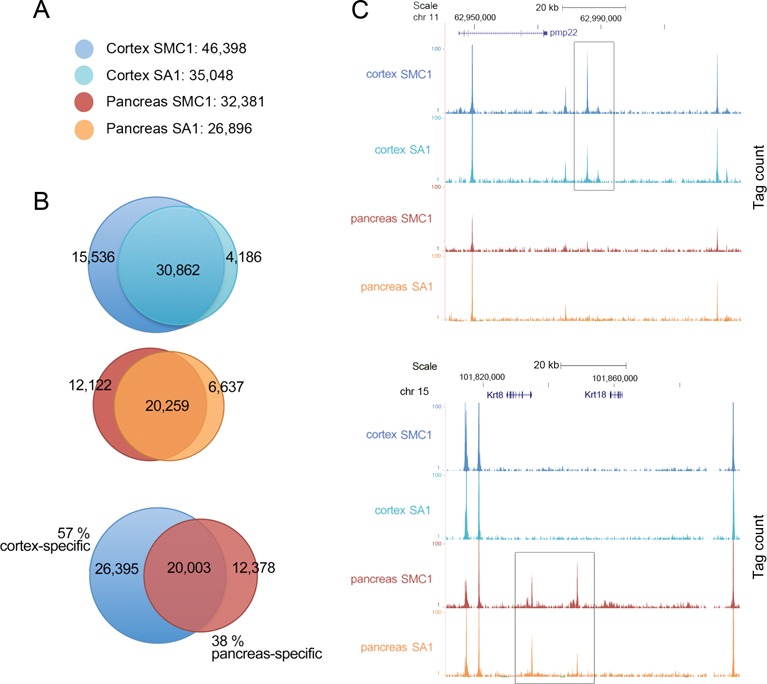
A large fraction of genomic cohesin positions are tissue-specific. (A) Number of cohesin SMC1 and SA1 positions in cerebral cortex and pancreas from 10 week-old mice obtained by ChIP-seq. Two replicates corresponding to independent experiments containing tissue from at least three individuals were performed. (B) Venn diagrams showing the overlap between SMC1 and SA1 positions in each tissue and between tissues. For cohesin and CTCF peaks (see Supplementary Figure S1), overlap is defined as reciprocal coincidence over at least 50% of peak length. (C) UCSC genome browser images illustrating cortex-specific (upper panel) and pancreas-specific (lower panel) cohesin positions located in chromosomes 11 and 15, respectively. The values for ChIP-seq data (y-axis) are normalized to input.
We next compared cohesin distribution in the two tissues and found that 20 003 SMC1 binding sites are common (43% of the cortex and 62% of the pancreas positions) thus leaving an important number of tissue-specific cohesin positions (26 395 in cortex and 12 378 in pancreas; Figure 1B and C). If we consider cohesin positions as the sum of SMC1 and SA1 positions, the overlap increases slightly to 48% of cortex and 63% of pancreas (Supplementary Figure S1A). Ubiquitous and tissue-specific cohesin positions are similarly distributed amongst gene-associated and intergenic regions (Supplementary Figure S1A). Consistent with previous reports of a large overlap between cohesin and CTCF, 64% of cohesin binding sites that we identified in cortex coincide with CTCF sites reported in the same tissue (9). These cohesin/CTCF positions represent 86% of ubiquitous cohesin sites but only 41% of cortex-specific sites (Supplementary Figure S1B). The fraction of cohesin/CTCF sites is reduced to 39% in pancreas due to the smaller number of CTCF sites identified in this tissue (17 625) compared to cohesin sites (38 002), but they represent 56% of ubiquitous cohesin sites and only 10% of pancreas-specific sites (Supplementary Figure S1C). Thus, cohesin/CTCF sites are more invariant between tissues than cohesin/non-CTCF sites.
We observed that cohesin distributes throughout the body of highly expressed genes encoding pancreatic enzymes, such as chemotrypsinogen or amylase, same as the elongating form of RNA polymerase II (RNA pol II; Supplementary Figure S2A). Cohesin can also be found throughout the body of some of the genes most highly expressed in cortex, although its accumulation is not as high as in the pancreatic genes described above (Supplementary Figure S2B and C). Cohesin could be promoting efficient transcription by RNA pol lI in these genes, as recently described in Drosophila (62).
Cohesin distribution correlates with tissue-specific transcription
We next analysed cohesin distribution around the transcription start site (TSS) of the genes that are expressed specifically in each tissue. To assess tissue-specific gene expression RNA-seq was performed in the same samples used for ChIP (Supplementary Tables S2 and S3). We defined tissue-specific genes as those differentially expressed when compared with the other tissue (log2FC > 4, FDR < 0.05), in which the expression should be low, i.e. Fragments Per Kilobase of exon per Million fragments mapped (FPKM) <3. Using these criteria, we selected 2126 cortex-specific genes and 514 pancreas-specific genes. Gene ontology analyses verified the functional specificity of the gene sets (Figure 2A and Supplementary Table S4). We found that cohesin binding sites (both SMC1 and SA1) are enriched around the TSS of tissue-specific genes mainly in the tissue in which they are expressed (Figure 2B).
Figure 2.
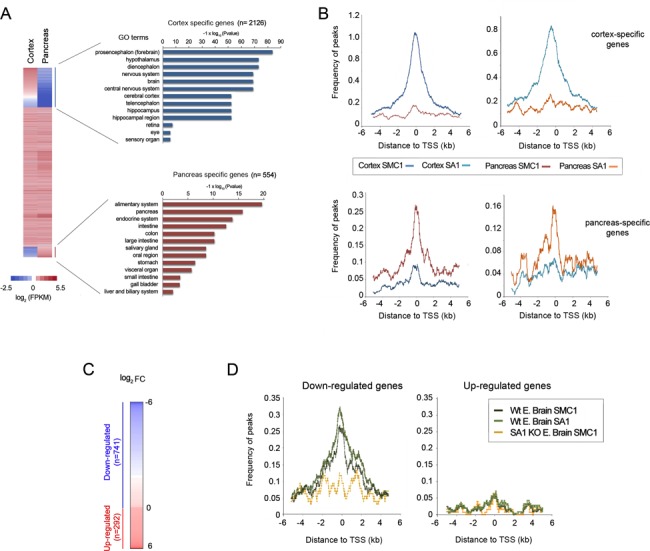
Cohesin is enriched at the promoters of actively transcribed genes. (A) Transcriptional profiles from the cortex (left) and pancreas (right) of 10-week-old mice were determined by RNA-seq. Tissue-specific genes were selected for each tissue (see main text for details) and further confirmed by Gene Ontology analysis. (B) Cohesin distribution around the TSS (±6 kb) of genes expressed specifically in cortex (upper panels) and pancreas (lower panels) defined as peak frequency (%). Both SMC1 and SA1 distributions are shown. (C) Gene expression changes between wild-type and SA1 null brains (KO) from E17.5 embryos were characterized by RNA-seq (FDR < 0.05). (D) Cohesin distribution around the TSS of genes found to be down-regulated (left) and up-regulated (right) in SA1 null embryonic brains.
In order to demonstrate a causal relationship between the presence of cohesin at promoters and gene expression in vivo, we used SA1 deficient mice that we previously generated. Homozygous deletion of the gene encoding SA1 is embryonic lethal but a low percentage of embryos survive to late developmental stages (35). We therefore performed ChIP-seq and RNA-seq analysis in brains obtained from E17.5 wild-type and SA1 null embryos (Supplementary Figure S3 and Supplementary Tables S2 and S3). Consistent with our previous results in mouse embryo fibroblasts, lack of cohesin-SA1 had a profound effect on gene expression also in the brain, featured by changes in hundreds of genes (Figure 2C and Supplementary Table S4). Genes down-regulated in the SA1 null brains have cohesin around their TSSs in wild-type tissue but little is left in the mutant (Figure 2D and Supplementary Table S5). In contrast, genes up-regulated in the SA1 null brains show no cohesin enrichment around their promoters. In this case, the spatial organization of the loci containing the affected genes may be altered without involving discrete promoter–enhancer DNA looping.
Cohesin contributes to defining the functionality of the chromatin landscape
To further explore the relationship between cohesin and functional genomic elements, we performed chromatin profiling combining our own data for cohesin SMC1 and SA1 in cortex with available data from chromatin marks associated with enhancers (H3K4me1), promoters (H3K4me3), active elements (H3K27Ac), repressed regions (H3K27me3) and CTCF in the same tissue ((9); Supplementary Figure S4). Thirteen chromatin states obtained from the analysis were given a candidate annotation—as in (63)—and their functional enrichment in tissue-specific promoters and enhancers was assessed (Figure 3A). Active promoters can have no cohesin (AP), cohesin but no CTCF (APC) or both cohesin and CTCF (APCC). Cohesin-SA1 is likely to be present at most promoters featured by the APCC state, since the probability of having SA1 is similar to SMC1 (83% and 94%, respectively). In the APC state, the difference is more prominent (55% SA1 and 86% SMC1), suggesting that cohesin at some of these promoters could be cohesin-SA2. The promoters of the genes more transcribed in cortex (FPKM > 6) are more often defined by the APC state than by the APCC state (Figure 3B). Moreover, the difference in enrichment in cortex-specific promoters compared to pancreas-specific promoters is also higher for the APC state than for the APCC (Figure 3A, right). These results further support the involvement of cohesin in tissue-specific transcription.
Figure 3.
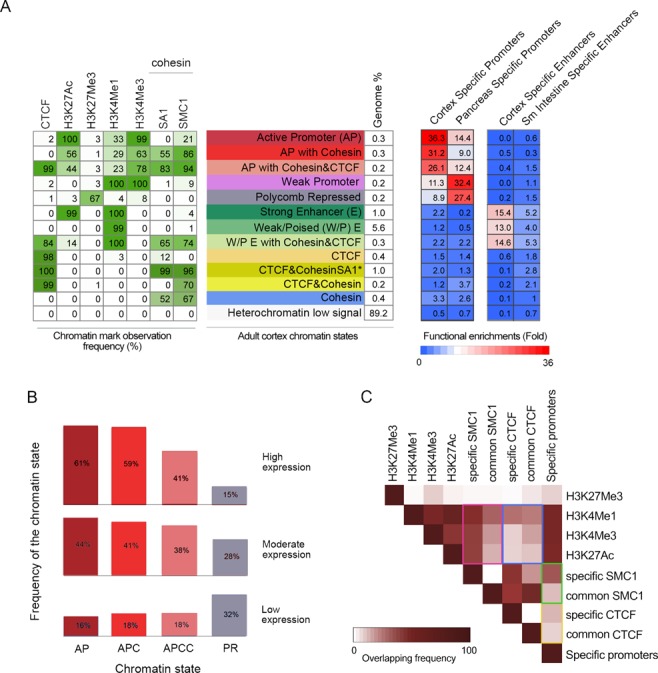
Cohesin is present at active promoters and enhacers. (A) Chromatin profiling in adult cortex by means of Hidden Markov Model (HMM) led to define 13 chromatin states (shown in different colours) according to the observed frequency of different chromatin marks (H3K4Me1, H3K4Me3, H3K27Me3, H3K27Ac), CTCF and cohesin SMC1 and SA1. The numbers in the table on the left correspond to the frequency with which a given mark is found at genomic positions corresponding to the chromatin state, annotated on the middle and assigned a colour code. Green shading indicates intensity (0–100). Genome% indicates the percentage of the genome corresponding to each chromatin state. The columns on the right show the functional enrichment of each chromatin state in cortex and pancreas-specific promoters (as selected in Figure 2) and cortex and small-intestine enhancers (as defined in (9)). (*) The sate named ‘CTCF&CohesinSA1’ contains, at least, cohesin-SA1 but it may also contain cohesin-SA2. (B) Genes were classified into quartiles according to their expression levels, obtained by RNA-seq, as follows: Q1, FPKM = 0 (n = 18 361), no expression; Q2, 0<FPKM≤0.296312 (n = 7316), low expression; Q3, 0.296312<FPKM≤6.032485 (n = 9315), moderate expression; Q4, FPKM>6.032485 (n = 9316), high expression. The frequency of the indicated chromatin states is shown for the quartiles containing expressed genes (Q1–Q3). AP, active promoters; APC, active promoters with cohesin; APCC, active promoters with cohesin and CTCF; PR, Polycomb repressed. (C) Heat-map visualization of the pairwise overlap between cerebral cortex chromatin marks and cortex-specific and non-specific (‘common’) cohesin and CTCF positions. Comparisons mentioned in the main text are highlighted by coloured rectangles.
Inactive promoters or repressed regions (defined by the presence of H3K27Me3, chromatin state labelled in grey in Figure 3A) do not contain cohesin or CTCF. Cohesin and CTCF are also absent from strong enhancers (chromatin state labelled in dark green in Figure 3A), whereas they co-occur at a fraction of poised/weak enhancers (labelled in light green in Figure 3A). This suggests that the activity of weak enhancers could rely on the ability of cohesin/CTCF to stabilize genomic interactions with their target gene promoters whereas this function could be dispensable in the case of strong enhancers. As in the case of APCC, most of these cohesin/CTCF sites have, at least, cohesin-SA1. Finally, there are four chromatin states that have CTCF and/or cohesin in the absence of any of the other chromatin marks. Consistent with the overlap between cohesin and CTCF, the CTCF-only state occupies 0.4% of the genome whilst the two CTCF/cohesin states, which differ in the probability of having SA1, together encompass 1.2% of the genome. The functional distinction between these three states is unclear at the moment. A state with CTCF and no chromatin marks has been previously annotated as ‘Insulator’ (63). The cohesin-only state is the most enriched at gene promoters, in particular those expressed specifically in cortex (Figure 3A and Supplementary Figure S5). Thus, the presence of cohesin at a promoter is a strong indicator of its activity and may predict gene expression in cases where classical active promoter-associated chromatin marks (e.g. H3K27Ac) are not detected.
We next assessed the overlap between cortex-specific positions for cohesin and CTCF with the chromatin marks of this tissue. For this, we defined cortex-specific CTCF sites as those present in cortex but not in pancreas (Supplementary Figure S6). Cortex-specific cohesin binding sites display a better overlap with enhancer and promoter marks (H3K4Me1 and H3K4Me3) than ubiquitous cohesin binding sites (Figure 3C, purple rectangle). This is not the case when comparing the overlap of cortex-specific and common CTCF positions with those same chromatin marks (Figure 3C, blue rectangle). Moreover, cortex-specific cohesin is more frequently found at the promoters of cortex-specific genes than cortex-specific CTCF (Figure 3C, compare green and yellow rectangles). These observations indicate that cohesin could have a more prominent role in controlling tissue-specific transcription whilst CTCF would be more involved in the transcription of non tissue-specific genes.
Tissue-specific transcription correlates with tissue-specific architecture
Cohesin has been proposed to stabilize cis-interactions between distal genomic elements involved in gene regulation. To ascertain the correlation between tissue-specific transcription and chromatin architecture mediated by cohesin, we performed 4C-seq analyses in the Protocadherin (Pcdh) clusters, a genomic region containing genes expressed specifically in the brain (Figure 4 and Supplementary Table S6). Pcdh alpha, beta and gamma clusters encode more than 50 protein isoforms as a result of stochastic promoter choice and alternative splicing (64). Mouse models deficient for CTCF, for the cohesin loader Nipbl or for cohesin-SA1, present altered expression of the Pcdh genes (35,65–67). Moreover, 3C analyses in mouse and human neuroblastoma cell lines indicate that cohesin and CTCF mediate DNA looping between enhancers located 3′ of the Pcdh alpha cluster and individual upstream promoters (26,27). We first chose one of these enhancers, HS5–1, as anchoring point to identify long-range interactions both in cortex and pancreas (Figure 4B, top). In cortex, the Pcdh alpha locus is folded in such a way that the HS5–1 enhancer is spatially close to most Pcdh alpha gene promoters. Interacting regions correlate with both cohesin distribution and Pcdh alpha gene expression (see RNA-seq track at the bottom of Figure 4B). Importantly, such spatial conformation is tissue specific, since none of the described interactions can be detected in pancreas.
Figure 4.
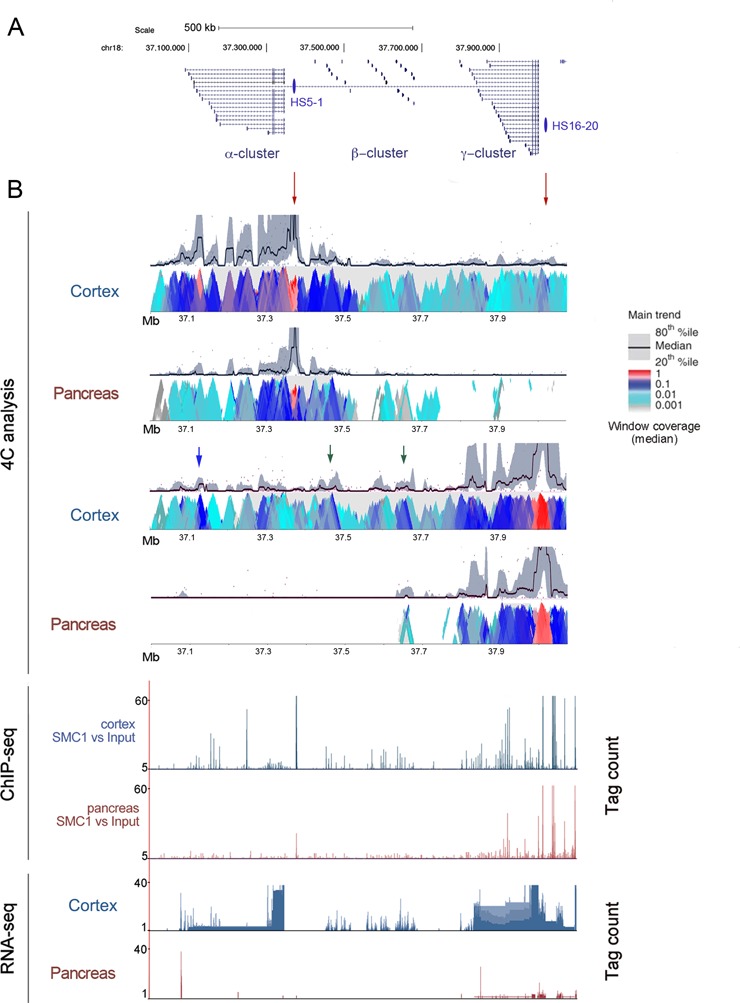
Chromatin architecture of the Pcdh locus in adult cortex and pancreas. (A) Details of the chromosome 18qB3 region containing the mouse Pcdh alpha, beta and gamma clusters. HS5–1 and HS16–20 enhancer regions are shown. (B) Upper panel: chromatin interactions detected by 4C-seq in cerebral cortex and pancreas using a 20-kb window size in the main trend subpanel. Red arrowheads indicate viewpoint positions. Middle and bottom panels show cohesin positions (defined by ChIP-seq) and gene expression (obtained by RNA-seq), respectively.
There is another enhancer located downstream of the Pcdh gamma cluster, HS16–20 (68). 4C-seq analysis using as viewpoint a strong cohesin binding site present in this enhancer revealed long-range interactions with several genomic regions located within the Pcdh beta locus and even in the Pcdh alpha locus (green and blue arrowheads in Figure 4, middle, respectively). These results can explain why deletion of the HS16–20 region in mice leads to a nearly complete loss of expression across the Pcdh beta cluster, whilst a much less drastic effect is observed for the genes of the immediately adjacent Pcdh gamma cluster (68). These interactions could not be detected in pancreas.
We next repeated the 4C-seq analysis using brains from wild-type and SA1 null embryos. The chromatin contacts between HS5–1 enhancer and the Pcdh alpha promoters were analogous to those described in adult brain, consistent with the presence of similar cohesin-SA1 peaks. Unexpectedly, most contacts were also maintained in SA1 null brains (Supplementary Figure S7). Quantitation of SMC1 binding to Pcdha4 and Pcdha6 promoters revealed only a 2-fold reduction in the absence of SA1, suggesting that this remaining cohesin could be sufficient to support the observed chromatin contacts (Supplementary Figure S7, bottom). The contacts between the HS16–20 enhancer and the Pcdh beta cluster were decreased in the wild-type embryonic brain compared with the adult and even further reduced in the SA1 null brain. This was accompanied by decreased SA1 peak height at some positions that could be more relevant for the chromatin contact (blue arrowhead in Supplementary Figure S7, right). Both binding of SMC1 and transcription of genes in this region were reduced in brains of SA1 null embryos compared to wild-type (Pcdhb17, 20 and 21; Figure 7B and C in (35)). Thus, in this particular locus, a better correlation exists between cohesin-SA1 levels, chromatin organization and gene expression.
Cohesin-SA1 contribution to chromatin architecture in the Reg locus
We recently reported increased incidence of pancreatic tumours in SA1 heterozygous mice (34). As judged by immunoblot analysis, SA1 protein levels are reduced in the pancreas of SA1 heterozygous animals compared to wild-type (Figure 5A). Microarray analyses carried out in pancreas obtained from healthy 10-week-old wild-type and SA1 heterozygous mice detected significant changes in the expression of only four genes (FDR < 0.15; Figure 5B and C). Three of them, Reg2, Reg3a and Reg3b (from Regenerating islet-derived), belong to a gene family strongly associated with pancreatitis (69–71). Interestingly, Reg genes are organized in a cluster, consistent with the idea that cohesin may be important to coordinate the expression of spatially related genes (72). They are highly expressed in pancreas and not in cortex. In pancreas, cohesin bound abundantly within the Reg gene cluster but was absent from the surrounding regions. The opposite was observed in cortex (Supplementary Figure S8A). In agreement with reported Hi-C data (7), analysis of chromatin contacts by 4C-seq using a viewpoint located in Reg1 (VP1 in Supplementary Figure S8A) revealed that this region is more densely packed in adult cortex than in pancreas. Some differential interactions could be detected more than 1 Mb away from the viewpoint in the two tissues. This architecture suggests that the presence of cohesin at the Reg locus in pancreas might prevent chromatin compaction and positively regulate gene expression. Consistent with this possibility, the amount of cohesin bound to three sites inside the Reg locus was clearly reduced in the pancreas of SA1 heterozygous animals compared to wild-type (Figure 5D). Moreover, a high-resolution 4C analysis performed in pancreas from wild-type and SA1 heterozygous mice using two additional viewpoints within the Reg locus showed different interaction patterns (VP2 in Figure 5E and VP3 in Supplementary Figure S8B). An increased number of chromatin contacts were observed in SA1 deficient pancreas, reminiscent of the higher compaction observed in cortex. The Reg locus could therefore be particularly sensitive to a decrease in cohesin levels. It is tempting to speculate that altered gene expression of Reg genes contributes to tumourigenesis in SA1 deficient mice.
Figure 5.
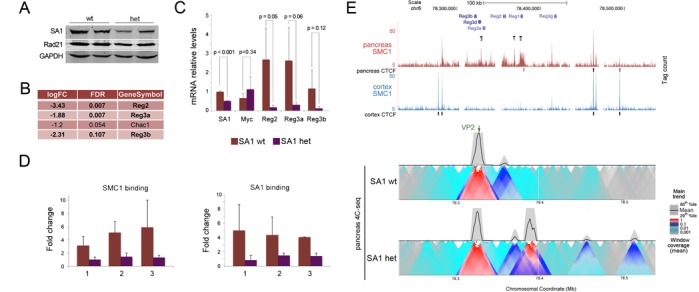
Chromatin architecture of the Reg locus in adult cortex and pancreas. (A) Levels of cohesin were assayed by immunoblot with SA1 and Rad21 antibodies in pancreas from wild-type and SA1 heterozygous mice (two individuals per genotype). GAPDH serves as loading control. (B) Table showing the four genes differentially expressed (FDR < 0.15) in pancreas from 10-week-old wild-type and SA1 heterozygous mice. (C) Validation of transcriptional changes in Reg genes by RT-qPCR. At least three individuals per condition were analysed. A two-tailed Student's t-test was applied. Data are shown as mean ± SEM (standard error of the mean). (D) The amount of cohesin SA1 and SMC1 at the sites in the Reg locus marked in (E) was analysed by ChIP-qPCR in the pancreas of wild-type and SA1 heterozygous mice. At least four animals per condition were used for the analysis. (E) Cohesin distribution across the Reg locus in cortex and pancreas, as determined by ChIP-seq. CTCF binding sites are also indicated. Black lines specify the position of primer pairs (1, 2 and 3) used in (D). The lower panel shows the analysis of the three-dimensional organization of the Reg locus in pancreas from wild-type and SA1 heterozygous mice by 4C-seq. Main trend corresponds to a 5-kb window size. The position of the viewpoint (VP2) is indicated (green arrow).
DISCUSSION
Previous efforts to identify tissue-specific cohesin signatures have compared cohesin binding sites with those of ubiquitous and tissue-specific transcriptional regulators in a single tissue (47) or have examined cohesin distribution in human cancer cell lines (46). We here report the first comparison of cohesin distribution in vivo in two different tissues from the same individuals and its correlation with gene expression and chromatin architecture at selected loci. Consistent with previous results, we found that a significant proportion of cohesin positions are only identified in the tissue under analysis. Such tissue-specific binding sites are present not only at gene promoters but appear to mark other functional cis-regulatory elements. In the mouse and human genomes, cohesin positions largely overlap with those of CTCF (17–19,23,35). We combined chromatin marks recently characterized in an 8-week-mouse-old brain (9) with our data on cohesin distribution to get a comprehensive idea of cohesin contribution to define the genomic landscape in cerebral cortex. Contrary to the conclusion of (47) that cohesin/CTCF sites show minimal signs of transcriptional activity and that CTCF is absent from most genomic elements featured by enhancer marks, we find cohesin and CTCF together at active promoters and enhancers. However, it is true that promoters with cohesin/CTCF belong to genes less transcribed than those containing cohesin and that CTCF/no cohesin sites do not co-occur with active promoter marks. Our analysis further reveals that cohesin complexes that co-occur with CTCF at active promoters and enhancers most likely contain the SA1 subunit. Consistent with previous reports, we also observe that cohesin has a more prominent role in controlling tissue-specific transcription, whilst CTCF is more relevant for the expression of common/housekeeping genes (9,47).
Cohesin contributes to transcriptional regulation in several ways. One is by facilitating long-range interactions. Our analysis of chromatin contacts at the Pcdh clusters in adult pancreas, adult cortex and embryonic brain by means of 4C-seq reveals remarkable differences between the two adult tissues and, to a lesser extent, between two developmental stages of the same tissue. These architectural differences are accompanied by changes both in gene expression and cohesin distribution. Assessing the role of cohesin in the establishment and/or maintenance of the observed chromatin contacts is not trivial, since cohesin is essential for proliferation. Nevertheless, conditional gene ablation or forced cleavage of cohesin in cells leading to notable—but incomplete—removal of the complex from chromatin results in changes in chromatin architecture and gene expression (13,29–30). Although we could detect some changes in chromatin contacts in the Pcdh clusters of embryonic brains upon complete ablation of cohesin-SA1, they were less dramatic than expected. We had previously reported that cohesin-SA1 is more prevalent in this locus than cohesin-SA2 and that cohesin occupancy is clearly reduced in SA1 null brains (35). Here we show that the reduction in bound cohesin may not be so dramatic at other Pcdha promoters and therefore the remaining cohesin, necessarily cohesin-SA2, could be sufficient to maintain locus architecture in the absence of cohesin-SA1. Finally, whilst in the aforementioned studies the effect of removing cohesin is assessed within hours of the elimination, the SA1 null embryos develop from the zygote in the absence of cohesin-SA1 and those that survive to 17.5 dpc may have found ways to cope with this loss.
In contrast to the Pcdh locus, the Reg locus appears to be sensitive to a decrease in cohesin levels. A different set of chromatin contacts can be observed in the Reg locus in pancreas from wild-type and SA1 heterozygous mice. We favor the hypothesis that cohesin is one of the several elements promoting a particular chromatin contact, including CTCF, transcription factors or nearby transcriptional activity. The combination of such contributions would dictate the probability of the contact and therefore, the dependency on cohesin would be variable amongst different genomic loci. A 2-fold reduction in the amount of SA1 protein in pancreas leads to a clear decrease in cohesin binding in the Reg locus. We propose that this reduction, in turn, alters chromatin architecture to make it more similar to the architecture of the Reg locus in cortex, where the expression of Reg genes is around three orders of magnitude lower than in pancreas.
Impaired homeostasis of the pancreas due to altered expression of the Reg genes could make SA1 heterozygous mice prone to pancreatic cancer (34). Four out of five human Reg genes are present in a single locus at chromosome 2 (2p12) that is syntenic to the murine locus described in this study (6qC3). Cohesin positions can be found within the human locus in a cell line of pancreatic origin but not in glioblastoma cells (our own unpublished data). Thus, the chromatin architecture that we have described for the Reg gene cluster in mice could be conserved in humans and affect Reg gene expression. Interestingly, 20% of pancreatic cancer cases analysed by the ICGC (90/450) harbour mutations in the SA1 gene. Even though the actual impact of most of these mutations in SA1 expression and/or function is unclear, their incidence seems to be particularly high in this type of tumour. Our current results suggest that altered chromatin architecture leading to transcriptional dysregulation may be one of the mechanisms underlying the role of cohesin mutations in human cancer (45,73)
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We wish to thank G. Gómez-López of the Bioinformatics Unit for the peak calling analysis of CTCF in pancreas, O. Domínguez and his team in the Genomics Unit for help with ChIP-seq and M. Serrano for his continuous support.
Footnotes
Present address: Silvia Remeseiro, Developmental Biology Unit, European Molecular Biology Laboratory, 69117 Heidelberg, Germany.
FUNDING
The Spanish Ministry of Economy and Competitiveness (MINECO) [BFU2013-48481-R to A.L.]; ‘Ramón y Cajal’ Contract [RYC-2010-06122 to A.C.]; Fundación La Caixa [PhD Fellowship to S.R.]. Funding for open access charge: The Spanish Ministry of Economy and Competitiveness (MINECO) [BFU2013-48481-R].
Conflict of interest statement. None declared.
REFERENCES
- 1.Fraser P., Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature. 2007;447:413–417. doi: 10.1038/nature05916. [DOI] [PubMed] [Google Scholar]
- 2.Spitz F., Furlong E.E. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 3.Cavalli G., Misteli T. Functional implications of genome topology. Nat. Struct. Mol. Biol. 2013;20:290–299. doi: 10.1038/nsmb.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li G., Ruan X., Auerbach R.K., Sandhu K.S., Zheng M., Wang P., Poh H.M., Goh Y., Lim J., Zhang J., et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanyal A., Lajoie B.R., Jain G., Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen Y., Yue F., McCleary D.F., Ye Z., Edsall L., Kuan S., Wagner U., Dixon J., Lee L., Lobanenkov V.V., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibcus J.H., Dekker J. The hierarchy of the 3D genome. Mol. Cell. 2013;49:773–782. doi: 10.1016/j.molcel.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Handoko L., Xu H., Li G., Ngan C.Y., Chew E., Schnapp M., Lee C.W., Ye C., Ping J.L., Mulawadi F., et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMare L.E., Leng J., Cotney J., Reilly S.K., Yin J., Sarro R., Noonan J.P. The genomic landscape of cohesin-associated chromatin interactions. Genome Res. 2013;23:1224–1234. doi: 10.1101/gr.156570.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phillips-Cremins J.E., Sauria M.E., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S., Ong C.T., Hookway T.A., Guo C., Sun Y., et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ong C.T., Corces V.G. CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014;15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nasmyth K., Haering C.H. Cohesin: its roles and mechanisms. Annu. Rev. Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 16.Dorsett D., Eissenberg J.C., Misulovin Z., Martens A., Redding B., McKim K. Effects of sister chromatid cohesion proteins on cut gene expression during wing development in Drosophila. Development. 2005;132:4743–4753. doi: 10.1242/dev.02064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubio E.D., Reiss D.J., Welcsh P.L., Disteche C.M., Filippova G.N., Baliga N.S., Aebersold R., Ranish J.A., Krumm A. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8309–8314. doi: 10.1073/pnas.0801273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wendt K.S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T., et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 19.Parelho V., Hadjur S., Spivakov M., Leleu M., Sauer S., Gregson H.C., Jarmuz A., Canzonetta C., Webster Z., Nesterova T., et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 20.Hadjur S., Williams L.M., Ryan N.K., Cobb B.S., Sexton T., Fraser P., Fisher A.G., Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460:410–413. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nativio R., Wendt K.S., Ito Y., Huddleston J.E., Uribe-Lewis S., Woodfine K., Krueger C., Reik W., Peters J.M., Murrell A. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009;5:e1000739. doi: 10.1371/journal.pgen.1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mishiro T., Ishihara K., Hino S., Tsutsumi S., Aburatani H., Shirahige K., Kinoshita Y., Nakao M. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 2009;28:1234–1245. doi: 10.1038/emboj.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S., et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degner S.C., Verma-Gaur J., Wong T.P., Bossen C., Iverson G.M., Torkamani A., Vettermann C., Lin Y.C., Ju Z., Schulz D., et al. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc. Natl. Acad. Sci. U.S.A. 2011;108:9566–9571. doi: 10.1073/pnas.1019391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou C., Dale R., Dean A. Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc. Natl. Acad. Sci. U.S.A. 2010;107:3651–3656. doi: 10.1073/pnas.0912087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo Y., Monahan K., Wu H., Gertz J., Varley K.E., Li W., Myers R.M., Maniatis T., Wu Q. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc. Natl. Acad. Sci. U.S.A. 2012;109:21081–21086. doi: 10.1073/pnas.1219280110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monahan K., Rudnick N.D., Kehayova P.D., Pauli F., Newberry K.M., Myers R.M., Maniatis T. Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of Protocadherin-alpha gene expression. Proc. Natl. Acad. Sci. U.S.A. 2012;109:9125–9130. doi: 10.1073/pnas.1205074109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seitan V.C., Faure A.J., Zhan Y., McCord R.P., Lajoie B.R., Ing-Simmons E., Lenhard B., Giorgetti L., Heard E., Fisher A.G., et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013;23:2066–2077. doi: 10.1101/gr.161620.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sofueva S., Yaffe E., Chan W.C., Georgopoulou D., Vietri Rudan M., Mira-Bontenbal H., Pollard S.M., Schroth G.P., Tanay A., Hadjur S. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 2013;32:3119–3129. doi: 10.1038/emboj.2013.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuin J., Dixon J.R., van der Reijden M.I., Ye Z., Kolovos P., Brouwer R.W., van de Corput M.P., van de Werken H.J., Knoch T.A., van Ijcken W.F., et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. U.S.A. 2014;111:996–1001. doi: 10.1073/pnas.1317788111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Losada A., Yokochi T., Kobayashi R., Hirano T. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J. Cell Biol. 2000;150:405–416. doi: 10.1083/jcb.150.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sumara I., Vorlaufer E., Gieffers C., Peters B.H., Peters J.M. Characterization of vertebrate cohesin complexes and their regulation in prophase. J. Cell Biol. 2000;151:749–762. doi: 10.1083/jcb.151.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Canudas S., Smith S. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J. Cell Biol. 2009;187:165–173. doi: 10.1083/jcb.200903096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remeseiro S., Cuadrado A., Carretero M., Martínez P., Drosopoulos W.C., Cañamero M., Schildkraut C.L., Blasco M.A., Losada A. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. 2012;31:2076–2089. doi: 10.1038/emboj.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Remeseiro S., Cuadrado A., Gómez-López G., Pisano D.G., Losada A. A unique role of cohesin-SA1 in gene regulation and development. EMBO J. 2012;31:2090–2102. doi: 10.1038/emboj.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J., Zhang Z., Bando M., Itoh T., Deardorff M.A., Clark D., Kaur M., Tandy S., Kondoh T., Rappaport E., et al. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009;7:e1000119. doi: 10.1371/journal.pbio.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Remeseiro S., Cuadrado A., Kawauchi S., Calof A.L., Lander A.D., Losada A. Reduction of Nipbl impairs cohesin loading locally and affects transcription but not cohesion-dependent functions in a mouse model of Cornelia de Lange Syndrome. Biochim. Biophys. Acta. 2013;1832:2097–2102. doi: 10.1016/j.bbadis.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuin J., Franke V., van Ijcken W.F., van der Sloot A., Krantz I.D., van der Reijden M.I., Nakato R., Lenhard B., Wendt K.S. A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet. 2014;10:e1004153. doi: 10.1371/journal.pgen.1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rollins R.A., Korom M., Aulner N., Martens A., Dorsett D. Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Mol. Cell. Biol. 2004;24:3100–3111. doi: 10.1128/MCB.24.8.3100-3111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horsfield J.A., Anagnostou S.H., Hu J.K., Cho K.H., Geisler R., Lieschke G., Crosier K.E., Crosier P.S. Cohesin-dependent regulation of Runx genes. Development. 2007;134:2639–2649. doi: 10.1242/dev.002485. [DOI] [PubMed] [Google Scholar]
- 41.Berlivet S., Paquette D., Dumouchel A., Langlais D., Dostie J., Kmita M. Clustering of tissue-specific sub-TADs accompanies the regulation of HoxA genes in developing limbs. PLoS Genet. 2013;9:e1004018. doi: 10.1371/journal.pgen.1004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Solomon D.A., Kim T., Diaz-Martinez L.A., Fair J., Elkahloun A.G., Harris B.T., Toretsky J.A., Rosenberg S.A., Shukla N., Ladanyi M., et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333:1039–1043. doi: 10.1126/science.1203619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balbas-Martinez C., Sagrera A., Carrillo-de-Santa-Pau E., Earl J., Marquez M., Vazquez M., Lapi E., Castro-Giner F., Beltran S., Bayes M., et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat. Genet. 2013;45:1464–1469. doi: 10.1038/ng.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kon A., Shih L.Y., Minamino M., Sanada M., Shiraishi Y., Nagata Y., Yoshida K., Okuno Y., Bando M., Nakato R., et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013;45:1232–1237. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]
- 45.Losada A. Cohesin in cancer: chromosome segregation and beyond. Nat. Rev. Cancer. 2014;14:389–393. doi: 10.1038/nrc3743. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt D., Schwalie P.C., Ross-Innes C.S., Hurtado A., Brown G.D., Carroll J.S., Flicek P., Odom D.T. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 2010;20:578–588. doi: 10.1101/gr.100479.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faure A.J., Schmidt D., Watt S., Schwalie P.C., Wilson M.D., Xu H., Ramsay R.G., Odom D.T., Flicek P. Cohesin regulates tissue-specific expression by stabilising highly occupied cis-regulatory modules. Genome Res. 2012;22:2163–2175. doi: 10.1101/gr.136507.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merkenschlager M., Odom D.T. CTCF and cohesin: linking gene regulatory elements with their targets. Cell. 2013;152:1285–1297. doi: 10.1016/j.cell.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 49.Cuadrado A., Corrado N., Perdiguero E., Lafarga V., Munoz-Canoves P., Nebreda A.R. Essential role of p18Hamlet/SRCAP-mediated histone H2A.Z chromatin incorporation in muscle differentiation. EMBO J. 2010;29:2014–2025. doi: 10.1038/emboj.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rubio-Camarillo M., Gomez-Lopez G., Fernandez J.M., Valencia A., Pisano D.G. RUbioSeq: a suite of parallelized pipelines to automate exome variation and bisulfite-seq analyses. Bioinformatics. 2013;29:1687–1689. doi: 10.1093/bioinformatics/btt203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salmon-Divon M., Dvinge H., Tammoja K., Bertone P. PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics. 2010;11:415. doi: 10.1186/1471-2105-11-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cock P.J., Fields C.J., Goto N., Heuer M.L., Rice P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767–1771. doi: 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flicek P., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2014. Nucleic Acids Res. 2013;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van de Werken H.J., de Vree P.J., Splinter E., Holwerda S.J., Klous P., de Wit E., de Laat W. 4C technology: protocols and data analysis. Methods Enzymol. 2012;513:89–112. doi: 10.1016/B978-0-12-391938-0.00004-5. [DOI] [PubMed] [Google Scholar]
- 60.van de Werken H.J., Landan G., Holwerda S.J., Hoichman M., Klous P., Chachik R., Splinter E., Valdes-Quezada C., Oz Y., Bouwman B.A., et al. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods. 2012;9:969–972. doi: 10.1038/nmeth.2173. [DOI] [PubMed] [Google Scholar]
- 61.Ernst J., Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schaaf C.A., Kwak H., Koenig A., Misulovin Z., Gohara D.W., Watson A., Zhou Y., Lis J.T., Dorsett D. Genome-wide control of RNA polymerase II activity by cohesin. PLoS Genet. 2013;9:e1003382. doi: 10.1371/journal.pgen.1003382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morishita H., Yagi T. Protocadherin family: diversity, structure, and function. Curr. Opin. Cell Biol. 2007;19:584–592. doi: 10.1016/j.ceb.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 65.Hirayama T., Tarusawa E., Yoshimura Y., Galjart N., Yagi T. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep. 2012;2:345–357. doi: 10.1016/j.celrep.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 66.Kawauchi S., Calof A.L., Santos R., Lopez-Burks M.E., Young C.M., Hoang M.P., Chua A., Lao T., Lechner M.S., Daniel J.A., et al. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/-) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet. 2009;5:e1000650. doi: 10.1371/journal.pgen.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Remeseiro S., Cuadrado A., Kawauch S., Calo A.L., Lander A.D., Losada A. Reduction of Nipbl impairs cohesin loading locally and affects transcription but not cohesion-dependent functions in a mouse model of Cornelia de Lange Syndrome. Biochim. Biophys. Acta. 2013;1832:2097–2102. doi: 10.1016/j.bbadis.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yokota S., Hirayama T., Hirano K., Kaneko R., Toyoda S., Kawamura Y., Hirabayashi M., Hirabayashi T., Yagi T. Identification of the cluster control region for the protocadherin-beta genes located beyond the protocadherin-gamma cluster. J. Biol. Chem. 2011;286:31885–31895. doi: 10.1074/jbc.M111.245605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Orelle B., Keim V., Masciotra L., Dagorn J.C., Iovanna J.L. Human pancreatitis-associated protein. Messenger RNA cloning and expression in pancreatic diseases. J. Clin. Invest. 1992;90:2284–2291. doi: 10.1172/JCI116115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Narushima Y., Unno M., Nakagawara K., Mori M., Miyashita H., Suzuki Y., Noguchi N., Takasawa S., Kumagai T., Yonekura H., et al. Structure, chromosomal localization and expression of mouse genes encoding type III Reg, RegIII alpha, RegIII beta, RegIII gamma. Gene. 1997;185:159–168. doi: 10.1016/s0378-1119(96)00589-6. [DOI] [PubMed] [Google Scholar]
- 71.Gironella M., Folch-Puy E., LeGoffic A., Garcia S., Christa L., Smith A., Tebar L., Hunt S.P., Bayne R., Smith A.J., et al. Experimental acute pancreatitis in PAP/HIP knock-out mice. Gut. 2007;56:1091–1097. doi: 10.1136/gut.2006.116087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cuadrado A., Remeseiro S., Gomez-Lopez G., Pisano D.G., Losada A. The specific contributions of cohesin-SA1 to cohesion and gene expression: Implications for cancer and development. Cell Cycle. 2012;11:2233–2238. doi: 10.4161/cc.20318. [DOI] [PubMed] [Google Scholar]
- 73.Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.