


Abstract
In eukaryotes, translation termination is performed by eRF1, which recognizes stop codons via its N-terminal domain. Many previous studies based on point mutagenesis, cross-linking experiments or eRF1 chimeras have investigated the mechanism by which the stop signal is decoded by eRF1. Conserved motifs, such as GTS and YxCxxxF, were found to be important for termination efficiency, but the recognition mechanism remains unclear. We characterized a region of the eRF1 N-terminal domain, the P1 pocket, that we had previously shown to be involved in termination efficiency. We performed alanine scanning mutagenesis of this region, and we quantified in vivo readthrough efficiency for each alanine mutant. We identified two residues, arginine 65 and lysine 109, as critical for recognition of the three stop codons. We also demonstrated a role for the serine 33 and serine 70 residues in UGA decoding in vivo. NMR analysis of the alanine mutants revealed that the correct conformation of this region was controlled by the YxCxxxF motif. By combining our genetic data with a structural analysis of eRF1 mutants, we were able to formulate a new model in which the stop codon interacts with eRF1 through the P1 pocket.
INTRODUCTION
Translation termination is mediated by a complex of class 1 and 2 release factors (RFs) (1). When a stop codon (UAA, UAG or UGA) enters the A site of the ribosome, a class 1 RF decodes it and induces peptide release by stimulating the peptidyl-transferase centre before ribosome recycling (2). Unlike prokaryotes, which use two release factors—RF1 (decoding UAA and UAG) and RF2 (decoding UAA and UGA)—eukaryotes have only one class 1 release factor, eRF1. This protein has three distinct domains: N, M and C (3,4). The N-terminal domain (N) is responsible for stop codon recognition (5); the central domain (M) is involved in peptide hydrolysis, via its methylated GGQ motif (6–8); and the C-terminal domain (C) interacts with the class 2 RF, eRF3, which stimulates termination activity by triggering a change in the conformation of eRF1 (9,10).
The X-ray structures of RF1 or RF2 within the ribosome have provided important information about the recognition of stop codons in prokaryotes (11). In eukaryotes, only medium-resolution CryoEM maps are available and, despite considerable progress in recent years, the exact mechanism by which stop codons are decoded remains unclear (12,13). Regions in the N-terminal domain of eRF1 involved in stop codon decoding have been identified on the basis of biochemical and genetic data. Cross-linking experiments (14,15), mutagenesis (16,17) and biochemical studies (18) implicated the universally conserved TASNIKS motif (positions 58–64, human numbering) in stop codon decoding. Two other motifs YxCxxxF (positions 125–131) and GTS (positions 31–33) have also been shown to be involved in distinguishing the purine bases in the second and third positions of the stop codon (19,20). Much of the available information about the role of the N domain in stop codon recognition came from studies of chimeric eRF1s containing regions of the N domain from organisms using variant genetic codes (18,21–24). In particular, analyses of the N domains of unipotent or bipotent ciliate eRF1s led to the identification of residues critical for stop codon recognition specificity. Determination of the structure of the human eRF1 N domain by nuclear magnetic resonance (NMR) revealed that the GTS loop could adopt different conformations, which were modulated by mutations surrounding this loop, with an impact on stop codon recognition specificity (25). Cross-linking experiments also showed this motif to be proximal to the guanine residues in the second or third position but more distal to the adenine residues in the second or third position of the stop codon. Mutagenesis of the conserved residues in the N-terminal domain of human eRF1 identified amino acids crucial for discrimination between adenine and guanine residues in the second and third positions of the stop codon (26). This in vitro study suggested that residues S33 and S70 would be involved in guanine recognition at the second position, and that the decoding of adenines at this position would involve T32, S36 and F131 (from the YxCxxxF motif). For the decoding of the third base of the stop codon, they suggested that residues E55, Y125 (from the YxCxxxF motif) and N129 would recognize guanines, whereas the valine in position 71 would be involved in the recognition of adenine residues. The current model for stop codon recognition by eRF1, based on the latest data from cross-linking and mutagenesis studies, involves two steps, including a change in the conformation of the termination factor governed by the purine residues present in positions 2 and 3 within the stop codon. Support for this model was provided by the recent observation of changes in the conformation of eRF1 following the dissociation of eRF3 and the binding of ABCE1 (13). Indeed, the authors observed that the eRF1 N domain appeared to be ‘fragmented’ in their cryoEM maps, indicating conformational flexibility. However, the data obtained in the presence of eRF3 were highly consistent with previously obtained cryoEM data and confirmed the proximity of the NIKS, GTS and YxCxxxF motifs to the stop codon (12). Despite the large number of studies carried out and the models proposed, the mechanism of stop codon recognition remains poorly understood. This may be due to the diversity of systems used to assay eRF1 activity and the lack of in vivo data concerning the eRF1 residues involved in stop codon decoding.
We previously identified two pockets (P1 and P2) involved in termination efficiency, by screening hyperactive mutants of eRF1 (27). In this study, we aimed to characterize the role of the P1 cavity, which is located close to the NIKS motif, in more detail. We performed an alanine scanning experiment on all the residues of P1 and carried out an NMR analysis of proteins with a mutated N domain, to identify the residues of P1 involved in stop codon recognition (Figure 1). We identified two positions (R65 and K109, human numbering) for which mutation to yielded alanine residues greatly decreased termination efficiency, for all stop codons. Two other mutations, the S33A and S70A mutations, decreased termination efficiency specifically for UGA stop codons. We then carried out an NMR analysis of the structure of the N-terminal domain of human eRF1 mutants, for the characterization of these mutants with particular termination phenotypes. Replacing the serine 70 residue with an alanine residue had almost no impact on the local conformation of the N domain of human eRF1, whereas the S33A, R65A and K109A mutations induced local changes in the conformation of the N domain, with a particularly strong impact on the highly conserved YxCxxxF motif. Our genetic data and the NMR analysis revealed that some P1 pocket residues interacted directly with the stop codon, whereas others helped to position the mRNA correctly. These findings led us to propose a two-step model, in which the stop codon on the mRNA interacts directly with eRF1 via the P1 cavity.
Figure 1.
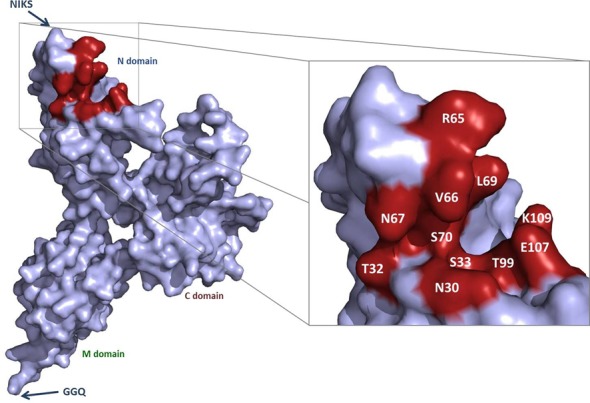
P1 pocket residues mutated to alanine. Representation of the structure of eRF1, with the three domains and the conserved NIKS and GGQ motifs indicated. The magnified view shows the surface of the P1 pocket. The residues mutated to alanines are shown in red.
MATERIALS AND METHODS
Strains and media
The Rosetta (DE3) pLysS Escherichia coli strain (F− ompT hsdSB(RB− mB−) gal dcm λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) pLysSRARE (CamR)) is a derivative of strain BL21. It was used for production of the heRF1 N-terminal domain. This strain was grown in M9-based minimal medium supplemented with 15N-ammonium sulphate as the nitrogen source for protein labelling.
The FS1ΔSUP45 Saccharomyces cerevisiae strain (MATα ΔSUP45::ADE2 ade2-592 lys2Δ201 leu2-3,112 his3Δ200 ura3-52) is a derivative of FS1 (MATα ade2-592 lys2Δ201 leu2-3,112 his3Δ200 ura3-52). It was used for the production of mutant forms of Sup45p (the yeast eRF1) (27). As SUP45 is essential in yeast, this strain also carries a wild-type copy of the gene on a plasmid, pFL44L (p2μ, SUP45, URA3). We used 5FOA at a concentration of 1 mg/ml to select for the loss of URA3 plasmids. This strain was used to quantify the efficiency of the readthrough conferred by Sup45p mutants.
Plasmids
Sequences encoding heRF1 proteins with mutated N domains and an N-terminal 6xhis tag were inserted into pET29 (KanR). They were expressed under the control of T7 promoters, following induction with IPTG (isopropyl β-D-1-thiogalactopyranoside).
The mutant SUP45 (encoding the yeast eRF1) and ETF1 (encoding the human eRF1) sequences were inserted into the PvuII restriction site of pHS8, a derivative of pFL61 (p2μ, HIS3). The various mutant SUP45 and ETF1 sequences were expressed under control of the SUP45 promoter.
Western blot
Protein samples were subjected to electrophoresis in sodium dodecyl sulphate–polyacrylamide gels and the resulting bands were then electroblotted onto PVDF membrane at 16 V for 45 min. Monoclonal antibodies raised against eRF1 (a gift from Valérie Heurgué-Hamard) were hybridized to the membranes and primary antibody binding was detected by incubation with alkaline phosphatase-conjugated secondary antibodies and NBT/BCIP staining.
Quantification of readthrough efficiency
The pAC derivative vectors were constructed by inserting the fragment of interest into the single MscI site of pAC99 (28). Luciferase and β-galactosidase activities were assayed in the same crude extract, as previously described (29). Data are reported as box plots, with median values for at least five independent measurements. The significance of differences was evaluated in nonparametric Mann–Whitney tests.
Purification of the heRF1 N domain
For heRF1 N domain purification, the Rosetta (DE3) pLysS strain was transformed with the various pET29 constructs. IPTG (1 mM) was added to a 2-l culture at OD600 of 0.4 to induce expression of the protein at 23°C overnight. The culture was centrifuged to obtain a cell pellet, which was resuspended in Tris buffer containing 20-mM Tris pH 7.5, 200-mM NaCl, 10-mM imidazole, 6-mM β-mercaptoethanol and 1× Complete ethylenediaminetetraacetic acid-free protease inhibitor cocktail. Cells were disrupted with a French press and by sonication, and the resulting whole-cell extract was applied to a 1-ml HisTrap HP column (GE Healthcare). The N domain was purified with an AKTA purifier10 FPLC system, in which heRF1-Nter was bound to the resin in the presence of resuspension buffer without protease inhibitors. Proteins were eluted from the affinity column with a Tris-imidazole buffer containing 20-mM Tris pH7.5, 200-mM NaCl, 500-mM imidazole and 6-mM β-mercaptoethanol. Non-specific contaminants were removed from the samples in a second purification step. Centrifugal filters (Sartorius Vivaspin) were used to replace the buffer of the sample eluted from the HisTrap column with a buffer containing 100-mM NaCl but no imidazole, for purification on an anion exchange column (Q FF from GE Healthcare). The sample was applied to a 1-ml Q FF column in a Tris-NaCl buffer (20-mM Tris pH 7.5, 100-mM NaCl and 6-mM β-mercaptoethanol) and the proteins were eluted with a 1-M NaCl gradient.
NMR studies of the heRF1 N domain
NMR experiments were carried with a 4-channel, 5-amplifier 14.1 T (600 MHz 1H) Bruker AVANCE 3 spectrometer equipped with a 5-mm QCI-F cryoprobe and Bruker TopSpin software. All experiments were conducted at 298 K, with Shigemi NMR tubes (BMS-005B) and referencing based on the resonance of water and the relative gyromagnetic ratio for 15N [13]. 1H-15N HSQC spectra were collected with 2048 and 256 points in F2 and F1, respectively, and a spectral width of 16 ppm for 1H and 18.5 ppm for 15N. The data were processed with NMRPipe [16] and analysed with the CCPN Analysis package [17, 18]. Chemical shift assignments were determined by comparison with previously published assignments from BMRB 18092 and confirmed with 15N-edited TOCSY and NOESY NMR spectra for the wild-type protein.
RESULTS
Yeast eRF1 mutants and associated phenotypes
We characterized the P1 pocket, by assessing the contribution of each of its residues to termination efficiency. We generated an alanine scanning mutant for each position in this region, by performing directed mutagenesis on the pHS8-SUP45 plasmid. All the positions mutated are represented in Figure 1, using human numbering. We will use human numbering to designate the mutations throughout this article, for the sake of clarity. The FS1ΔSUP45 S. cerevisiae strain was transformed with the mutant constructs and the plasmid bearing the wild-type SUP45 gene copy was shuffled out by 5-FOA counter selection. Two mutations, the T32A and N67A mutations, were incompatible with cell viability, as the corresponding strains did not grow on 5FOA medium. All the other mutant cells were viable at 30°C.
Quantification of the termination efficiency of yeast eRF1 mutants
Stop codon readthrough regulates gene expression in several organisms (30–33). These recoding motifs divert the ribosome to efficiently promote the incorporation of a near-cognate tRNA (34). They are very useful to quantify increase or decrease in translation termination efficiency (whereas only decreases in termination efficiency can be quantified using efficient stop codon contexts) (27). We used these sequences to assess the impact of the mutations on termination efficiency, by quantifying stop codon readthrough in the strains bearing mutant eRF1 plasmids, using the pAC dual reporter system with UAA, UAG or UGA (in the TMV readthrough context) at the lacZ-luc junction (29). Weaker termination efficiency corresponds to an increase in stop codon readthrough efficiency. Readthrough was quantified and compared with that obtained in the presence of the wild-type plasmid. We assessed the effects of nine mutations on translation termination efficiency at the three stop codons (Supplementary Figure S1 and Figure 2). We classified the mutations into four groups, according to their effects on termination efficiency. The first category included the R65A, K109A, L69A and V66A mutations, which increased readthrough for all three stop codons. The L69A mutant had a slightly stronger effect on UAG codons than on the other stop codons (Figure 2 and Supplementary Figure S1B), suggesting that the leucine residue in this position might be important for the recognition of the guanine residue in the third position of the codon. The replacement of the arginine and lysine residues in positions 65 and 109 with alanine residues resulted in the highest level of readthrough, with the R65A mutation causing at least a 6-fold increase in readthrough at UAA and UAG codons (Figure 2 and Supplementary Figure S1A and B), and the K109A mutation increasing readthrough at UAG codons by a factor of up to eight (Figure 2 and Supplementary Figure S1B). The second category included the T99A and E107A mutations, which decreased readthrough efficiency, by a factor of two, at all three stop codons. The N30A mutation, by contrast, had no significant impact on termination efficiency (Figure 2 and Supplementary Figure S1). The most interesting category was probably that containing the S33A and S70A mutations, which increased readthrough, specifically at the UGA stop codon (Figure 2 and Supplementary Figure S1C). Indeed, these two mutations resulted in a readthrough efficiency eight times higher than that of the wild-type protein at UGA stop codons, whereas termination at UAA and UAG codons was similar to that observed for the wild-type strain. The distance between the two serine residues is compatible with hydrogen bonding between them. A loss of interaction in the two alanine mutants may therefore account for their similar effects on translation termination (as both mutations would disrupt the same intramolecular interaction).
Figure 2.
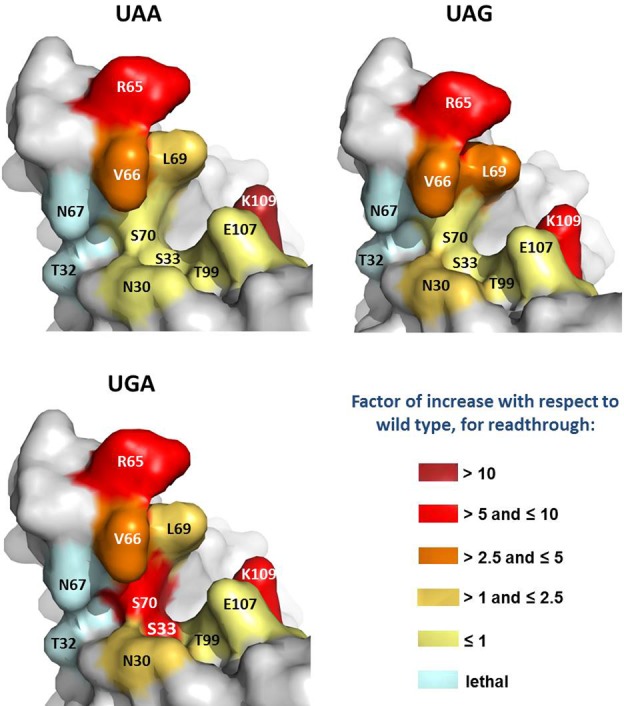
Impact of alanine mutations on readthrough efficiency. The fold-increase in readthrough induced by the alanine mutations with respect to the wild-type protein is shown for each stop codon; the impact on readthrough is represented by a gradient from yellow to red. Lethal mutations are shown in light blue.
The nucleotide context of the stop codon can greatly affect termination efficiency. We therefore investigated the influence of the stop codon environment, using the alanine mutant proteins with the strongest effects on stop codon readthrough. We used the three stop codons in an efficient termination environment, to quantify readthrough efficiency for these mutants. In this new nucleotide context, we again observed an increase in readthrough efficiency for the R65A and K109A mutant proteins, for all three stop codons (Supplementary Figure S2), with readthrough levels at least six times greater than wild-type levels for R65A at UGA codons (Supplementary Figure S2C) and four times wild-type levels for K109A at UAG codons (Supplementary Figure S2B). The S33A and S70A mutant proteins displayed high levels of readthrough, specifically at the UGA codon (Supplementary Figure S2C). Thus, the nucleotide context of the stop codon has no impact on the readthrough phenotype of eRF1 mutants.
Genetic analysis of the contributions of the serine 33 and serine 70 residues to termination efficiency
The S33A and S70A mutants had identical termination efficiency phenotypes. We analysed these mutations further, by investigating the possibility that they might act in synergy. We constructed the S33A-S70A double mutant and measured readthrough at the three leaky stop codons. The termination profile for the double mutant was similar to that of the two single mutants, S33A and S70A (Figure 3A). Indeed, the S33A-S70A mutant displayed an increase in readthrough specifically with the UGA codon (34%), although this increase was slightly smaller than that observed for each single mutant, at only five times the wild-type readthrough level (7%). As for the single mutants, termination efficiency at the UAA and UAG stop codons was not affected by the presence of the double alanine mutation.
Figure 3.
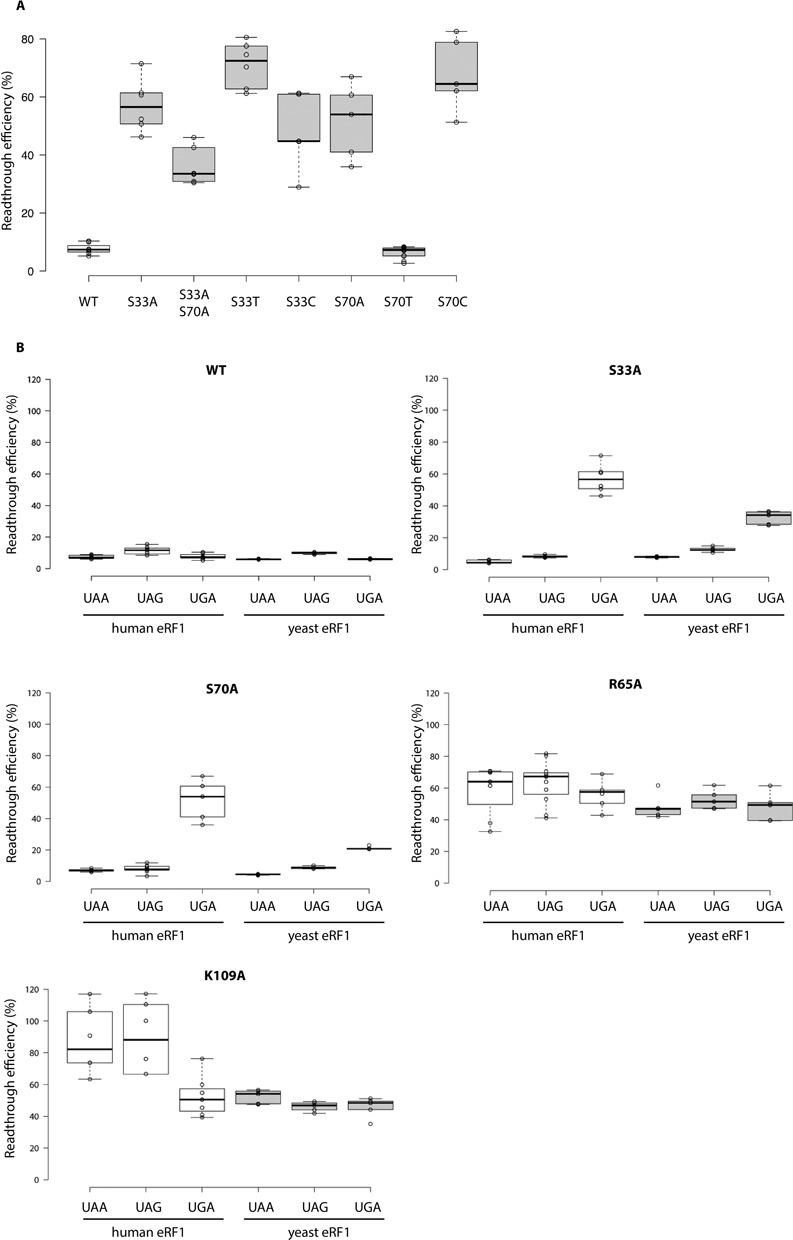
(A) Readthrough efficiency in the TMV context for alanine, threonine and cysteine mutants at positions 33 and 70, for the UGA stop codon. The lines in the centres of the boxes indicate the median values; the box limits indicate the 25th and 75th percentiles calculated with R software; the whiskers extend to the minimum and maximum values; data points are plotted as open circles. n ≥ 5 sample points. Wild-type values are indicated in white boxes; mutant protein values are indicated in grey boxes. Readthrough efficiency is shown for the UGA stop codon. Wild-type, S33A and S70A values were identical to those shown in Supplementary Figure S1. (B) Readthrough efficiency in the TMV context, for alanine mutants of the yeast and human eRF1 P1 pocket. The lines in the centres of the boxes indicate the median values; the box limits indicate the 25th and 75th percentiles calculated with R software; the whiskers extend to the minimum and maximum values; data points are plotted as open circles. n ≥ 5 sample points. The values obtained with human eRF1 are indicated in white boxes. The values obtained with the yeast eRF1 (Sup45p) are indicated in grey boxes.
These phenotypes were compatible with the hypothetical presence of a hydrogen bond between the two serine residues. We therefore replaced these residues with threonine residues, which are chemically similar in that they can form hydrogen bonds via their alcohol groups. We reasoned that if this potential interaction between the two amino acids was involved in stop codon recognition, then the replacement of serine residues with threonine residues would have no effect on termination efficiency. We therefore constructed the S33T and S70T mutants and quantified readthrough efficiency at the three stop codons. Unexpectedly, we found that the two mutants behaved differently. The S33T mutant yielded higher levels of readthrough than the alanine mutant, with 72% at the UGA codon, whereas the S70T mutation almost completely restored termination efficiency to wild-type levels at the UGA stop codon (7%) (Figure 3A). Thus, an ability to form a hydrogen bond is the key determinant feature of the S70 residue. We decided to investigate whether the chemical structure of these residues was also important. We replaced the serine residues with cysteine residues, which are structurally homologous, reasoning that if chemical structure was the key determinant, then this change would have no impact on translation termination efficiency. The readthrough quantification results revealed that the S33C and S70C mutants had phenotypes similar to that of the alanine mutants with the UGA codon. Readthrough levels of 45 and 65% were measured for the S33C and S70C mutants, respectively, corresponding to levels 7 and 10 times those recorded for the wild-type protein for the UGA codon (Figure 3A). Thus, at position 33, both the structural and chemical properties of serine are important for efficient termination whereas, at position 70, the key feature seems to be an ability to hydrogen bond.
Conformational analysis of the S33A, S70A, R65A and K109A mutant proteins by NMR on the N-terminal domain of human eRF1
We investigated the effect of eRF1 mutations on termination efficiency in more detail, by performing NMR analysis on some of the most interesting mutants. We analysed the S33A and S70A mutants, which were considered particularly interesting because of their UGA specificity, and the R65A and K109A mutants, which displayed very high levels of readthrough for all three stop codons. These experiments were performed with the human protein, so we first checked that these mutations had similar impacts on termination efficiency for human eRF1 and yeast eRF1 (Figure 3B).
We produced and purified the 15N-labelled eRF1 N-terminal domain of the wild-type protein and the four selected mutant proteins, and performed 1H-15N HSQC experiments with the purified proteins, to detect the chemical shifts of each residue (Figure 4A and Supplementary Figure S3). The chemical shifts of the backbone amides of the wild-type and mutant N domains were compared by calculating minimal map values on the basis of peaks displaying a shift between wild-type and mutant 1H-15N HSQCs. The most strongly affected residues are shown in red on the 3D structure of heRF1 (PDB 1DT9) for each mutant (Figure 4B).
Figure 4.
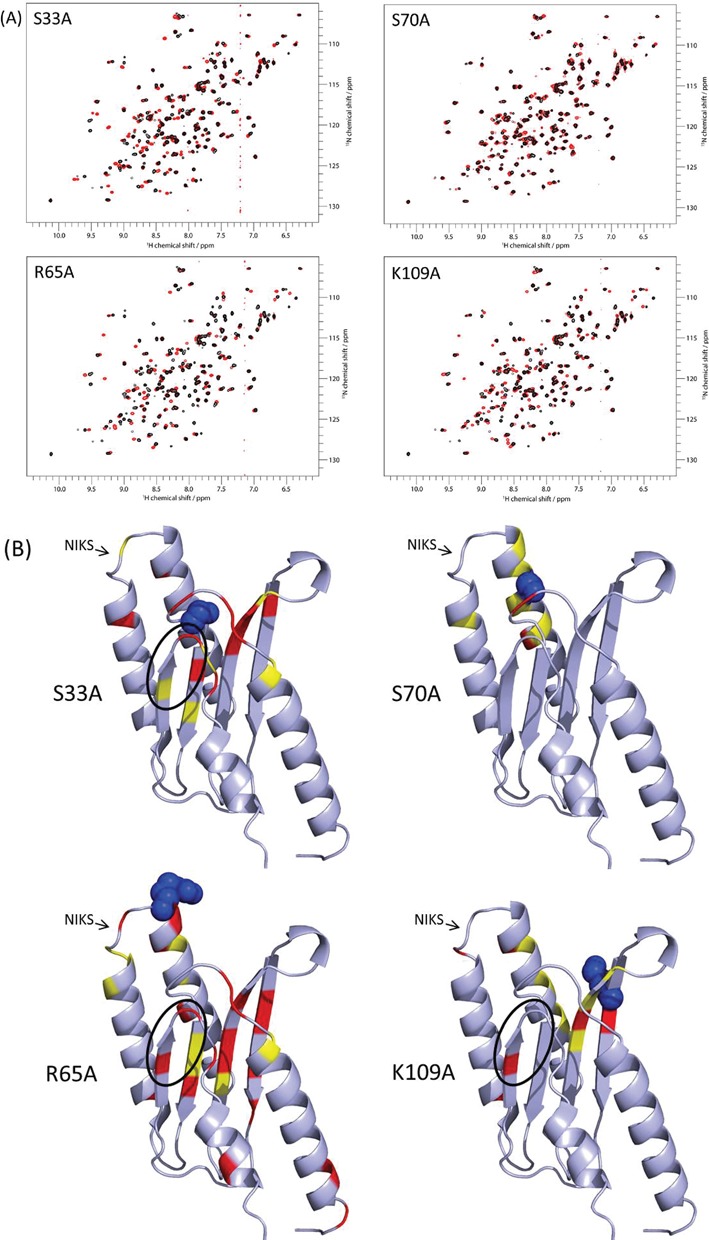
Impact of alanine mutations on the structural conformation of the eRF1 N domain. (A) NMR spectra of the N domains of heRF1 mutants. Proteins are labelled with 15N. Proton chemical shifts are represented on the horizontal axis, with nitrogen on the vertical axis. Minimal map values were calculated from peaks displaying a shift between the wild-type (in black) and mutant (in red) HSQC spectra. (B) Location of the observed chemical shifts on the structure of the eRF1 N domain. The most significant shifts are highlighted in red, and the least significant shifts are shown in yellow. The YxCxxxF (black circle) and NIKS motifs are indicated.
NMR showed chemical shifts between the S70A mutant and wild-type proteins to be minimal (Figure 4A and B), indicating that the structural conformation of this mutant differed little, if at all, from that of the wild-type protein. By contrast, several significant shifts were common to the three other mutants: S33A, R65A and K109A (Figure 4A and B). The residues that are most strongly affected by the mutations are well conserved between yeast and human. They included, in particular, the universally conserved YxCxxxF (125–131) motif, which is known to be involved in the decoding of the purines in the second and third positions of the stop codon. The leucine 126 residue was found to have shifted in the S33A, R65A and K109A mutants, and the asparagine 129 and phenylalanine 131 residues displayed shifts in the S33A and R65A mutants. The residues of the conserved NIKS motif were affected in both the R65A and K109A mutants (K63 and N61, respectively). In the S33A and R65A mutants, we also detected a different conformation of the R28 and G29 residues, which are located close to the GTS motif (31–33). The shifted amino acids detected in both the R65A and K109A mutant proteins also included S46, R68 and V110. With the exceptions of A8, V12 and A138, all the shifted residues were located close to the mutated positions, surrounding the P1 pocket. Thus, the impact of the S33A, S70A, R65A and K109A mutations on the conformation of the N domain appears to be local, rather than remote.
DISCUSSION
Many studies involving mutagenesis or cross-linking experiments have investigated the mechanism of stop codon recognition by eukaryotic termination factor eRF1 (14,15,17). Some of these studies used in vitro reconstituted translation systems to assay the activity of eRF1 mutants and to identify the residues critical for stop codon recognition (22,26). Others used organisms with a variant genetic code to identify the amino acids of eRF1 responsible for unipotency or bipotency (18,21–23). However, the way in which eRF1 recognizes the stop codon remains unclear. We used a combination of genetic approaches and NMR to characterize a region of the eRF1 N domain previously implicated in translation termination efficiency, and potentially responsible for interactions with mRNA.
We performed alanine scanning mutagenesis over the P1 pocket region and measured the readthrough efficiency in yeast for each resulting mutant. Two mutations, T32A and N67A, were not compatible with viability. It is possible that their location at the entrance of the pocket renders these two residues critical for the correct conformation of this region and that their replacement with alanine residues prevents translation termination. We classified the alanine mutations according to their impact on termination efficiency. L69A was one of the mutations that increased readthrough levels at all three stop codons. However, its slightly stronger effect on UAG suggests that this residue may play an important role in recognition of the guanine in the third position. The S33A and S70A mutations constituted an interesting category of mutations, with effects on readthrough specific to the UGA stop codon. This phenotype has already been reported for positions 33 and 70, but only in vitro (22,26). Our findings provide the first demonstration of the importance of these residues in vivo. There are two possible explanations for the role of the serine 33 and 70 residues in UGA decoding. First, both these residues may interact with the stop codon, probably with the guanine residue in the second position. Second, there may be a direct interaction between the two serine residues which is critical for the recognition of the guanine in the second position. The phenotype of the S33A-S70A mutant, which was similar to that of the single mutants, is more consistent with this second hypothesis (Figure 3A). Indeed, if both these residues interacted with the stop codon, we would expect the double mutant to have a stronger phenotype than the single mutants. We investigated the possible presence of a hydrogen bond between S33 and S70, by replacing these residues individually with threonine residues, a chemical homologue of serine. Quantification of the level of readthrough induced by the S33T and S70T mutations revealed that the replacement of the serine residue with a threonine residue at position 70 had almost no effect, whereas a similar substitution at position 33 resulted in high levels of readthrough at UGA codons (Figure 3A). Surprisingly S30T has been previously reported to decrease UGA recognition in vitro (26). This discrepancy probably reflects either differences in termination stringency in vivo and in vitro or differences in the termination process itself as the in vitro data have been obtained with a stop codon very close to the initiation codon (as a premature termination codon). We believe that our results better represent the termination process that occurs at the end of the coding sequences. Thus, efficient UGA recognition requires the serine at position 70 to be able to form a hydrogen bond. However, this is clearly not sufficient at position 33, as the S33T mutation does not restore the wild-type phenotype. We then replaced the serine residues with cysteine residues, which are similar in structure to serine residues but cannot form hydrogen bond. The results obtained with these mutants confirmed our hypothesis. The introduction of a cysteine residue to replace the serine in position 70 did not restore wild-type termination, highlighting the importance of hydrogen bond formation. Similar results were obtained for position 33, demonstrating the importance of the shape and chemical groups of serine at this position.
We then carried out an NMR analysis on the S33A and S70A mutant proteins, to add to the genetic data and build a model explaining the phenotypes observed. A comparison of the spectra of the wild-type and mutant proteins revealed that the S70A mutation had no significant impact on the local conformation of the eRF1 N domain (Figure 4A and B). This is consistent with the observed levels of readthrough and our hypothesis that the codon specificity of the S70A mutant reflects the loss of a potential interaction, rather than a change in local structure. By contrast, NMR for the S33A mutant showed that the presence of an alanine residue in this position led to local conformational changes in the eRF1 N domain (Figure 4A and B). It also confirmed that the shape of the serine residue at position 33 was important. A comparison of the wild-type and S33A mutant proteins spectra highlighted the shifts of the N129 and F131 residues from the universally conserved YxCxxxF motif, which has already been implicated in recognition of the second and third bases (19,20). The serine 33 residue is part of the conserved GTS loop, the conformation of which is modified by mutations in the YxCxxxF motif or surrounding sequences (25). Mutations affecting the GTS loop would thus be expected to induce changes in the conformation of the YxCxxxF motif. The shifted residues in the S33A mutant also included R28, G29, G31 and I35, which are located close to S33 in the secondary sequence of the protein, but are probably too far away from this residue in the 3D structure to interact directly with it. As for the YxCxxxF motif residues, the impact of mutations affecting these amino acids probably reflects local changes in conformation triggered by the presence of the alanine residue. We suggest that a direct interaction between S33 and S70 is required for the decoding of the guanine residue in the second position. This interaction would imply a particular conformation of the GTS loop, dependent on the YxCxxxF motif.
We characterized the P1 pocket in more detail, by studying the R65A and K109A mutants by NMR. These mutants displayed very high levels of readthrough at all three stop codons (Figure 2 and Supplementary Figure S1). We checked that this phenotype was not due to destabilization of the mutant proteins, by western blotting to assess their production and stability (Supplementary Figure S4). It is possible that R65 and K109 are directly involved in the decoding of the uracil residue in first position and that the mutation of these residues prevents stop codon recognition. However, this hypothesis is not consistent with previous findings of a direct interaction between this nucleotide and the K63 residue of the NIKS motif (15). It appears more plausible that R65 and K109 are important for the structure of the eRF1 N domain and, more specifically, for that of the P1 pocket. As these two residues are located at the edge of the cavity, we presume that they are critical for the correct conformation of the pocket. NMR revealed shifts of many residues with respect to the wild-type protein, in these two mutant proteins. The K109A mutant displayed a less marked change in conformation, but many of the chemical shifts observed were similar to those for the R65A mutant. The key observation accounting for the effect of these mutations on termination efficiency at the three stop codons is probably their impact on the residues of the NIKS motif in both the R65A and K109A mutants. Lysine and arginine are both positively charged amino acids. This suggests the possibility that they interact with RNA through the phosphate groups, which are negatively charged. Such an interaction might involve rRNA, but the orientation of the lateral chain of the arginine 65 and lysine 109 residues is more consistent with contact with the sugar-phosphate backbone of the mRNA. The role of these two residues may be to position the mRNA correctly in the pocket. As observed for the S33A mutant, some residues of the YxCxxxF motif are strongly affected by the R65A and K109A mutations. We suggest that, as for the GTS loop, the conformation of the YxCxxxF motif is probably closely related to that of the P1 pocket. Thus, a mutation affecting one of these regions is likely to result in changes in the conformation of the other.
Our results demonstrate the crucial role of the P1 pocket in stop codon recognition. By combining genetic and structural data, we developed a model in which the stop codon initially interacts with eRF1 via the NIKS motif and then with the P1 cavity (Figure 5A and B). We showed that some of the residues of the P1 pocket, such as R65 and K109, were essential for the correct conformation of the cavity. These residues potentially act together with T32 and N67 ensuring the correct positioning of the mRNA. Others, such as L69, may recognize the stop codon through direct interaction. A thorough analysis of the roles of the serine 33 and serine 70 residues implicated both in the recognition of the guanine residue in the second position, probably via interactions between these two residues. Structural analyses of the mutant proteins with alanine residues in these positions suggested that the direct interaction of these residues was governed by the conformation of the YxCxxxF motif, which seems to play a key role in determining the conformation of the GTS loop and, more generally, of the whole cavity. The role of the YxCxxxF motif in stop codon recognition may therefore be indirect, mediated through its impact on the P1 pocket. Our model is consistent with the recently determined cryo-EM structure of the pretermination complex in the mammalian ribosome (13). Indeed, the interaction of the stop codon with P1 would necessitate large conformational changes within N domain (Figure 5C), potentially corresponding to those observed on cryo-EM. Our model supports a two-step process of stop codon recognition, as suggested by other groups (26,35). We suggest that the stop codon is recognized through the NIKS motif when eRF1 enters the ribosome in association with eRF3. Following the release of eRF3, the binding of ABCE1/Rli1 induces changes in the conformation of the eRF1 N domain allowing the stop codon to reach the P1 pocket, where the second and third nucleotides are decoded (Figure 6). This new model is entirely consistent with the latest cryo-EM data and published genetic and biochemical findings. Given the importance of P1 in stop codon recognition, it would be interesting to search for drugs able to bind to this cavity and, thus, to inhibit translation termination. This would induce a strong suppressor phenotype that might help to restore the expression of proteins inactivated by a premature termination codon in genetic diseases.
Figure 5.
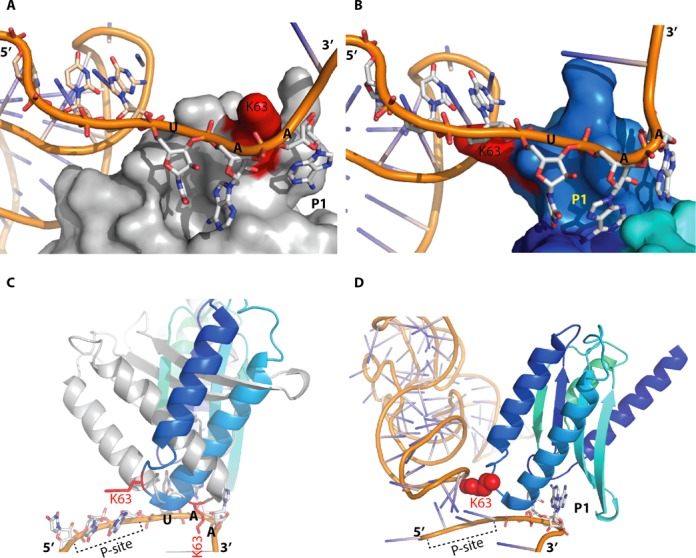
Interactions between the stop codon and the eRF1 N-terminal domain. (A) The first step in the recognition process, with the stop codon located near the NIKS motif (K63 in red) and the P-site tRNA shown in orange on the left. (B) Second step in the recognition process, involving the interaction of the stop codon with the P1 pocket. The P-site tRNA is shown in orange on the left. (C) The interaction between the stop codon and the P1 pocket requires changes in the conformation of the N domain. eRF1 is shown in grey for the conformation adopted during the first step, as in panel (A), and in blue for the conformation adopted during the second step. The P-site tRNA is not shown, for the sake of clarity. (D) As in panel (B), eRF1 is in the conformation required for the second step of stop codon recognition, with K63 indicated in red.
Figure 6.
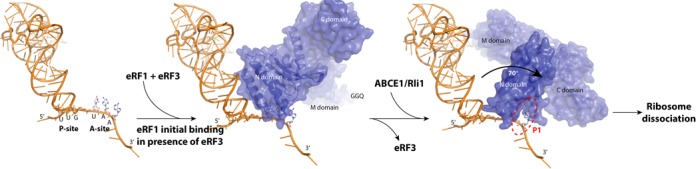
Proposed model of translation termination by eRF1. The stop codon is recognized by eRF1 in complex with eRF3. Following GTP hydrolysis, eRF3 is released and replaced by ABCE1, inducing changes in the conformation of the eRF1 N domain. These structural changes allow the stop codon to interact with the P1 pocket and the GGQ motif to be in close proximity to the CCA end of the P-site tRNA for the induction of peptide release before ribosome dissociation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Alex Edelman & Associates for correcting English usage. We would like to thank Valerie Heurgué-Hamard for kindly providing the eRF1 antibody and the human eRF1 cDNA.
FUNDING
The French Foundation ARC [SFI20101201647, PJA 20131200234]; the French Association ‘Ligue Nationale contre le cancer’ [3FI10167LVCY]; the ANR [ANR-2011-BSV6-011, ANR-13-BSV8-0012-02 to O.N.]; the ARC [to S.B.]; Wellcome Trust Equipment Award [091163/Z/10/Z to M.J.H.]. Funding for open access charge: the French Foundation ARC [20131200234]; the French Association ‘Ligue Nationale contre le cancer’ [3FI10167LVCY]; the ANR [ANR-2011-BSV6-011, ANR-13-BSV8-0012-02].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kisselev L., Ehrenberg M., Frolova L. Termination of translation: interplay of mRNA, rRNAs and release factors. EMBO J. 2003;22:175–182. doi: 10.1093/emboj/cdg017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shoemaker C.J., Green R. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc. Natl. Acad. Sci. U.S.A. 2011;108:E1392–E1398. doi: 10.1073/pnas.1113956108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frolova L.Y., Merkulova T.I., Kisselev L.L. Translation termination in eukaryotes: polypeptide release factor eRF1 is composed of functionally and structurally distinct domains. RNA. 2000;6:381–390. doi: 10.1017/s135583820099143x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song H., Mugnier P., Das A.K., Webb H.M., Evans D.R., Tuite M.F., Hemmings B.A., Barford D. The crystal structure of human eukaryotic release factor eRF1–mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- 5.Bertram G., Bell H.A., Ritchie D.W., Fullerton G., Stansfield I. Terminating eukaryote translation: domain 1 of release factor eRF1 functions in stop codon recognition. RNA. 2000;6:1236–1247. doi: 10.1017/s1355838200000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seit-Nebi A., Frolova L., Justesen J., Kisselev L. Class-1 translation termination factors: invariant GGQ minidomain is essential for release activity and ribosome binding but not for stop codon recognition. Nucleic Acids Res. 2001;29:3982–3987. doi: 10.1093/nar/29.19.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frolova L.Y., Tsivkovskii R.Y., Sivolobova G.F., Oparina N.Y., Serpinsky O.I., Blinov V.M., Tatkov S.I., Kisselev L.L. Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA. 1999;5:1014–1020. doi: 10.1017/s135583829999043x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heurgue-Hamard V., Champ S., Mora L., Merkulova-Rainon T., Kisselev L.L., Buckingham R.H. The glutamine residue of the conserved GGQ motif in Saccharomyces cerevisiae release factor eRF1 is methylated by the product of the YDR140w gene. J. Biol. Chem. 2005;280:2439–2445. doi: 10.1074/jbc.M407252200. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Z., Saito K., Pisarev A.V., Wada M., Pisareva V.P., Pestova T.V., Gajda M., Round A., Kong C., Lim M., et al. Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev. 2009;23:1106–1118. doi: 10.1101/gad.1770109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merkulova T.I., Frolova L.Y., Lazar M., Camonis J., Kisselev L.L. C-terminal domains of human translation termination factors eRF1 and eRF3 mediate their in vivo interaction. FEBS Lett. 1999;443:41–47. doi: 10.1016/s0014-5793(98)01669-x. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J., Korostelev A., Lancaster L., Noller H.F. Crystal structures of 70S ribosomes bound to release factors RF1, RF2 and RF3. Curr. Opin. Struct. Biol. 2012;22:733–742. doi: 10.1016/j.sbi.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.des Georges A., Hashem Y., Unbehaun A., Grassucci R.A., Taylor D., Hellen C.U., Pestova T.V., Frank J. Structure of the mammalian ribosomal pre-termination complex associated with eRF1.eRF3.GDPNP. Nucleic Acids Res. 2014;42:3409–3418. doi: 10.1093/nar/gkt1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Preis A., Heuer A., Barrio-Garcia C., Hauser A., Eyler D.E., Berninghausen O., Green R., Becker T., Beckmann R. Cryoelectron microscopic structures of eukaryotic translation termination complexes containing eRF1-eRF3 or eRF1-ABCE1. Cell Rep. 2014;8:59–65. doi: 10.1016/j.celrep.2014.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bulygin K.N., Khairulina Y.S., Kolosov P.M., Ven'yaminova A.G., Graifer D.M., Vorobjev Y.N., Frolova L.Y., Kisselev L.L., Karpova G.G. Three distinct peptides from the N domain of translation termination factor eRF1 surround stop codon in the ribosome. RNA. 2010;16:1902–1914. doi: 10.1261/rna.2066910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavatte L., Seit-Nebi A., Dubovaya V., Favre A. The invariant uridine of stop codons contacts the conserved NIKSR loop of human eRF1 in the ribosome. EMBO J. 2002;21:5302–5311. doi: 10.1093/emboj/cdf484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merritt G.H., Naemi W.R., Mugnier P., Webb H.M., Tuite M.F., von der Haar T. Decoding accuracy in eRF1 mutants and its correlation with pleiotropic quantitative traits in yeast. Nucleic Acids Res. 2010;38:5479–5492. doi: 10.1093/nar/gkq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frolova L., Seit-Nebi A., Kisselev L. Highly conserved NIKS tetrapeptide is functionally essential in eukaryotic translation termination factor eRF1. RNA. 2002;8:129–136. doi: 10.1017/s1355838202013262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito K., Frolova L., Seit-Nebi A., Karamyshev A., Kisselev L., Nakamura Y. Omnipotent decoding potential resides in eukaryotic translation termination factor eRF1 of variant-code organisms and is modulated by the interactions of amino acid sequences within domain 1. Proc. Natl. Acad. Sci. U.S.A. 2002;99:8494–8499. doi: 10.1073/pnas.142690099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conard S.E., Buckley J., Dang M., Bedwell G.J., Carter R.L., Khass M., Bedwell D.M. Identification of eRF1 residues that play critical and complementary roles in stop codon recognition. RNA. 2012;18:1210–1221. doi: 10.1261/rna.031997.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolosov P., Frolova L., Seit-Nebi A., Dubovaya V., Kononenko A., Oparina N., Justesen J., Efimov A., Kisselev L. Invariant amino acids essential for decoding function of polypeptide release factor eRF1. Nucleic Acids Res. 2005;33:6418–6425. doi: 10.1093/nar/gki927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salas-Marco J., Fan-Minogue H., Kallmeyer A.K., Klobutcher L.A., Farabaugh P.J., Bedwell D.M. Distinct paths to stop codon reassignment by the variant-code organisms Tetrahymena and Euplotes. Mol. Cell. Biol. 2006;26:438–447. doi: 10.1128/MCB.26.2.438-447.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eliseev B., Kryuchkova P., Alkalaeva E., Frolova L. A single amino acid change of translation termination factor eRF1 switches between bipotent and omnipotent stop-codon specificity. Nucleic Acids Res. 2011;39:599–608. doi: 10.1093/nar/gkq759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lekomtsev S., Kolosov P., Bidou L., Frolova L., Rousset J.P., Kisselev L. Different modes of stop codon restriction by the Stylonychia and Paramecium eRF1 translation termination factors. Proc. Natl. Acad. Sci. U.S.A. 2007;104:10824–10829. doi: 10.1073/pnas.0703887104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chavatte L., Kervestin S., Favre A., Jean-Jean O. Stop codon selection in eukaryotic translation termination: comparison of the discriminating potential between human and ciliate eRF1s. EMBO J. 2003;22:1644–1653. doi: 10.1093/emboj/cdg146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong L.E., Li Y., Pillay S., Frolova L., Pervushin K. Selectivity of stop codon recognition in translation termination is modulated by multiple conformations of GTS loop in eRF1. Nucleic Acids Res. 2012;40:5751–5765. doi: 10.1093/nar/gks192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kryuchkova P., Grishin A., Eliseev B., Karyagina A., Frolova L., Alkalaeva E. Two-step model of stop codon recognition by eukaryotic release factor eRF1. Nucleic Acids Res. 2013;41:4573–4586. doi: 10.1093/nar/gkt113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatin I., Fabret C., Rousset J.P., Namy O. Molecular dissection of translation termination mechanism identifies two new critical regions in eRF1. Nucleic Acids Res. 2009;37:1789–1798. doi: 10.1093/nar/gkp012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Namy O., Hatin I., Stahl G., Liu H., Barnay S., Bidou L., Rousset J.P. Gene overexpression as a tool for identifying new trans-acting factors involved in translation termination in Saccharomyces cerevisiae. Genetics. 2002;161:585–594. doi: 10.1093/genetics/161.2.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stahl G., Bidou L., Rousset J.P., Cassan M. Versatile vectors to study recoding: conservation of rules between yeast and mammalian cells. Nucleic Acids Res. 1995;23:1557–1560. doi: 10.1093/nar/23.9.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schueren F., Lingner T., George R., Hofhuis J., Dickel C., Gartner J., Thoms S. Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. eLife. 2014;3:e03640. doi: 10.7554/eLife.03640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Namy O., Duchateau-Nguyen G., Hatin I., Hermann-Le Denmat S., Termier M., Rousset J.P. Identification of stop codon readthrough genes in Saccharomyces cerevisiae. Nucleic Acids Res. 2003;31:2289–2296. doi: 10.1093/nar/gkg330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skuzeski J.M., Nichols L.M., Gesteland R.F., Atkins J.F. The signal for a leaky UAG stop codon in several plant viruses includes the two downstream codons. J. Mol. Biol. 1991;218:365–373. doi: 10.1016/0022-2836(91)90718-l. [DOI] [PubMed] [Google Scholar]
- 33.Loughran G., Chou M.Y., Ivanov I.P., Jungreis I., Kellis M., Kiran A.M., Baranov P.V., Atkins J.F. Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 2014;42:8928–8938. doi: 10.1093/nar/gku608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blanchet S., Cornu D., Argentini M., Namy O. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42:10061–10072. doi: 10.1093/nar/gku663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bulygin K.N., Khairulina Y.S., Kolosov P.M., Ven'yaminova A.G., Graifer D.M., Vorobjev Y.N., Frolova L.Y., Karpova G.G. Adenine and guanine recognition of stop codon is mediated by different N domain conformations of translation termination factor eRF1. Nucleic Acids Res. 2011;39:7134–7146. doi: 10.1093/nar/gkr376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.