


Abstract
Mutations in FUS cause amyotrophic lateral sclerosis (ALS), but the molecular pathways leading to neurodegeneration remain obscure. We previously found that U1 snRNP is the most abundant FUS interactor. Here, we report that components of the U1 snRNP core particle (Sm proteins and U1 snRNA), but not the mature U1 snRNP-specific proteins (U1-70K, U1A and U1C), co-mislocalize with FUS to the cytoplasm in ALS patient fibroblasts harboring mutations in the FUS nuclear localization signal (NLS). Similar results were obtained in HeLa cells expressing the ALS-causing FUS R495X NLS mutation, and mislocalization of Sm proteins is RRM-dependent. Moreover, as observed with FUS, knockdown of any of the U1 snRNP-specific proteins results in a dramatic loss of SMN-containing Gems. Significantly, knockdown of U1 snRNP in zebrafish results in motor axon truncations, a phenotype also observed with FUS, SMN and TDP-43 knockdowns. Our observations linking U1 snRNP to ALS patient cells with FUS mutations, SMN-containing Gems, and motor neurons indicate that U1 snRNP is a component of a molecular pathway associated with motor neuron disease. Linking an essential canonical splicing factor (U1 snRNP) to this pathway provides strong new evidence that splicing defects may be involved in pathogenesis and that this pathway is a potential therapeutic target.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron disease with no available treatment, and disease mechanisms are not understood (1,2). Although ∼90% of ALS cases are sporadic, mutations in numerous genes have been identified that cause familial ALS, and studies of these genes are leading to critical new insights into both forms of the disease (1–3). Several ALS-causing genes encode nuclear RNA/DNA binding proteins (4–7). These proteins are exemplified by FUS and TDP-43, and recently, Matrin3 and hnRNPA1 were added to the list (8–14). These proteins localize in the nucleus at steady state and have roles in RNA processing and other steps of gene expression (4–7,11).
The relevance of RNA/DNA-binding proteins to ALS is underscored by the observation that several other motor neuron diseases are caused by defects in these types of proteins. A well-known example is the childhood disease spinal muscular atrophy (SMA), which results from deficiency of the SMN protein (15), a component of the SMN complex. This complex localizes both diffusely in the cytoplasm and in nuclear Gems and is required for biogenesis of the spliceosomal snRNPs (16). We previously found that the ALS-causative protein FUS associates with the SMA-causative protein SMN, and both FUS and SMN are each required for Gem formation (17,18). TDP-43 also associates with both FUS and SMN and is required for Gem formation (19). Thus, these two motor neuron diseases are converging on the same molecular pathway, indicating its potential significance in pathogenesis. The ALS-causative proteins Matrin3 and hnRNPA1 interact with each other and also with TDP-43 (11,20), suggesting that they are also linked to this common pathway. Despite these associations among RNA/DNA binding proteins, it is not yet known how defects in these proteins or this pathway cause motor neuron disease. It is known that RNA/DNA binding proteins, such as TDP-43, FUS, and hnRNPA1, self-associate via low-complexity domains present in these proteins (5,7,21). This self-association is proposed to have a normal role in the cell, which is to trigger assembly of cellular bodies that concentrate factors with functions in the same pathway, thereby increasing the efficiency and fidelity of complex cellular pathways. Examples of such bodies include the nucleolus, Gems, nuclear speckle domains, and P-bodies (5,7,21). Pathogenesis may arise when these self assembly-prone proteins are mutated or altered in some manner and instead form cytoplasmic aggregates (5,7,22–23). The best-known example is observed with TDP-43, in which cytoplasmic aggregates are found in neuronal cells in the majority of ALS cases (24,25). FUS and hnRNPA1 aggregates have also been observed in some cases (5,10,21,26). It is not yet known whether the aggregates are pathogenic due to decreased function of these proteins in the nucleus and/or whether the aggregates themselves are toxic. A major challenge in the field is to sort these issues out and clearly define the pathways that are disrupted in motor neuron disease.
In light of our previous observations that FUS interacts directly with SMN and that both proteins function in the Gem pathway (17), we have now investigated the role of U1 snRNP in this pathway. Our interest in U1 snRNP stemmed from our observation that it is the most abundant factor that interacts with FUS in multiple assays in both HeLa and neuronal cells (17,27). These links between FUS and U1 snRNP, the SMN complex, and Gems were also corroborated in a new study in HeLa cells (28). In addition, as observed with FUS, the SMN complex is known to associate with U1 snRNP (29). However, the relationships between FUS, the SMN complex, and U1 snRNP, as well as the potential role of U1 snRNP in ALS are not yet understood. In this study, we carried out a series of assays to address these questions. We show that, as observed with FUS, the components of U1 snRNP are essential for Gem formation in HeLa cells. Moreover, ALS patient fibroblasts or HeLa cells bearing FUS mutations in the NLS show co-mislocalization of FUS with Sm core proteins to the cytoplasm. U1 snRNA is also mislocalized to the cytoplasm in these cells. To determine whether U1 snRNP has a cell-type specific role in motor neurons we knocked it down in zebrafish. This analysis revealed a striking truncation of motor axons. Together, our data link U1 snRNP to Gems, ALS patient cells with FUS mutations, and motor axons, raise the possibility that U1 snRNP is part of the pathway defective in motor neuron disease. The sequestration of Sm proteins and U1 snRNA by mutant FUS in ALS patient cells may also contribute to the toxicity of FUS bearing NLS mutations.
MATERIALS AND METHODS
Cell lines
HeLa cells and fibroblasts were cultured using standard conditions. Stable FUS and scrambled knockdown lines were reported (17). FUS R521C fibroblasts were from a 40-year-old female and FUS R514G were from a 39-year-old male. The control was from a 40-year-old female. The passage numbers for all experiments were between 15 and 18.
Plasmids
FLAG-tagged wild type FUS, FUS R495X, FUS R495X+RRM mut (changes in RRM: F305L, F341L, F359L and F368L) (30) and FUS 174-R495X were constructed by inserting fragments into the BamHI and XhoI sites of the vector pcDNA5/FRT/TO/i/FLAG, which contains the Ftz intron and 3x FLAG tags in the AflII and HindIII sites of pcDNA5/FRT/TO (Invitrogen), respectively.
Antibodies
Antibodies to Gemin8 (IF8) and Sm (Y12) were from Abcam. SMN (Mansma1) was from the Developmental Studies Hybridoma Bank. Coilin (F-7), FLAG, Gemin3 (C-5), Gemin4 (E-8), Gemin5 (10G11), Gemin6 (20H8), Lamin A/C, Snurportin (B-12), U1A (BJ-7) and U1C (5C9) were from Santa Cruz. TMG (K121) and U1-70K (9C4.1) from Millipore. Tubulin (DM1A) and SFRS2 were from Sigma. The FUS antibody was described (17).
Immunofluorescence (IF)
IF was carried out using antibodies against FUS (1:2000), SMN (1:400), U1-70K (1:2000), U1A (1:1000), U1C (1:1000), U2B” (1:400), FLAG (1:1000), Y12 (1:1000), Coilin (1:1000) and SRSF2 (1:1000). HeLa cells were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) for 15 min. Cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min. For IF, cells were incubated in the primary antibody overnight at 4°C. After three washes in PBS, the secondary antibody was added for 1 h at room temperature, followed by three washes in PBS. Primary antibodies were diluted in 10% calf serum in PBS. The secondary antibodies were mouse Alexa-488 and rabbit Alexa-647 diluted 1:1000 in 10% calf serum in PBS. Images were captured with a Nikon TE2000U inverted microscope, with a PerkinElmer ultraview spinning disk confocal, and a 20× objective using Metamorph software (Molecular Devices, Sunnyvale, CA, USA).
Fluorescence in situ hybridization (FISH)
FISH for U1 snRNA was performed using a biotin-labeled LNA probe (5′-GGTATCTCCCCTGCCAGGTAAGTAT-3′) (31), obtained from Exiqon. Hybridization was performed in 50% formamide, 2× SSC, 50 mM sodium phosphate (pH 7.0), and 10% dextran sulfate, containing 10 nM LNA probe, at 37°C overnight. After hybridization, cells were washed in 2× SSC + 0.1% Triton X-100, followed by detection with a fluorescent Alexa Fluor 594 streptavidin conjugate. Cells were washed three times for 5 min each at 37°C in 4× SSC + 0.1% Triton X-100, followed by washes in 2× and 1× SSC, with a final wash in PBS at 25°C.
Quantitative RT-PCR
Total RNA was extracted with Trizol and treated with DNase I for 15 min at 37°C. DNase I stop solution was added and the mixture was incubated at 65°C to inactivate DNase. After phenol extraction and ethanol precipitation, RNA was dissolved in H2O. One microgram of RNA was used for each RT reaction. RNA was heated at 75°C for 10 min in the presence of first strand transcription buffer and gene specific primer (U1 snRNA Forward: 5′-GATACCATGATCACGAAGGTGGTT-3′, Reverse: 5′-CACAAATTATGCAGTCGAGTTTCC-3′, 5S RNA Forward: 5′-CGGCCATACCACCCTGAAC-3′, Reverse: 5′-GCGGTCTCCCATCCAAGTAC-3′). The mixture was then immediately chilled on ice for 5 min. deoxynucleotide triphosphate (dNTP) mixture, dithiothreitol (DTT), and RNase inhibitor were add and incubated at 37°C for 2 min. M-MLV RTase was then added and the RT reaction was carried out. qPCR was performed using SYBR® Green PCR Master Mix (Invitrogen) using an ABI 7300 real-time PCR system following the manufacturer's instructions. Data analysis was performed with SDS 2.3 software (Applied Biosystems). Values were normalized to Actin. Reactions were carried out in triplicate and data were analyzed using the comparative (ΔΔC) method.
Microinjection of zebrafish embryos
The spawning of transgenic Tg(mnx1:GFP) zebrafish that express GFP in ventrally projecting motor axons and the care and husbandry of the embryos were carried out according to standard protocols (32). The embryos were injected at the one- to two-cell stage with the following morpholino oligonucleotides (MO): U1-70K (5′-TGTTCACCTCAGTTTTTCATTCGGC-3′), U1 AMO (5′-GGTATCTCCCCTGCCAGGTAAGTAT-3′) (31), and standard control MO (5′-CCTCTTACCTCAGTTACAATTTATA-3′) (GeneTools Eugene, OR, USA). For the rescue of U1-70K MO injection, the mutated U1-70K open reading frame (to abolish the MO binding site) was amplified with forward primer 5′- GAATTCGGATCCATGACGCAATTCTTGCCGCCGAACCTGTTGG-3′ and reverse primer 5′-CTCGAGTCTAGATTAGTACTCATCACCCTGGGC-3′ and sub-cloned into the pCS2+ vector using BamHI and XbaI sites. Plasmid DNA was linearized with NotI and capped RNA was generated using the mMESSAGE mMACHINE Sp6 kit (Ambion, Austin, TX, USA) following the manufacturer's protocol. For the rescue experiment, 50 pg of synthesized U1-70K mRNA was co-injected with 0.7 ng of U1-70K MO into one- to two-cell stage Tg(mnx1:GFP) embryos.
Zebrafish motor axon truncation
To visualize motor axons in GFP transgenic animals, Tg(mnx1:GFP) zebrafish were fixed with 4% paraformaldehyde in PBS overnight at 4°C, washed with PBST for 10 min, three times and mounted on slides for observation using a Zeiss Radiance 2100 laser scanning system together with Laser-Sharp and LSM imaging software (Carl Zeiss MicroImaging, Inc.). Ten motor axons from each side of the embryo were scored (total of 20 per embryo) at 28 h post-fertilization and used to classify the embryo as one truncation, two truncations, three truncations or no defect. Three sets of injections of embryos spawned the same day from different parents were performed per condition and 200 embryos (4000 motor axons) scored in each experiment. Statistical analysis was carried out using Prism 6 (GraphPad) software. The distribution of larval classifications was analyzed by comparing median values of each group with a two-tailed student's t test. Data are represented as mean and standard error of the mean (SEM) from independent experiments and P values are indicated as follows: *P < 0.05; **P < 0.01; ***P < 0.001.
RESULTS/DISCUSSION
U1 snRNP-specific proteins are essential for Gem formation in HeLa cells
Previous studies revealed that FUS is required for the presence of Gems in HeLa cells, motor neurons, fibroblasts and other cell types (17,18). As U1 snRNP is such an abundant FUS interactor, we first asked whether knock down of U1 snRNP components affects Gem levels in HeLa cells, using FUS and scrambled knockdowns for controls. The FUS and U1 snRNP-specific protein knockdowns were efficient as revealed by Western analysis (Figure 1A), and no significant differences were observed in the levels of factors that associate with U1 snRNP, including Sm proteins, SMN complex components, and snurportin (Figure 1B). Consistent with previous work, IF staining revealed that Gems are present in the control knockdown cells but lost in the FUS knockdown cells (Figure 1C, panels i and ii). Significantly, near complete Gem loss was observed in the three U1 snRNP protein-specific knockdown cells whereas control cells had normal Gem levels (Figure 1C, panels iii–vi). These data indicate that U1-70K, U1A, and U1C are each required for the presence of Gems in HeLa cells. Our data support the recent report that U1-70K knockdown results in Gem loss in HeLa cells (33). Knockdowns of U1A and U1C were not examined in their study (33). In a number of cell types, including HeLa, Gems are found in association with Cajal bodies, which are thought to function in snRNP biogenesis and/or recycling (29). Consistent with previous work, the Cajal body marker coilin, co-localizes with SMN in control knockdown cells (Supplementary Figure S1A). However, in the FUS or U1 snRNP-specific knockdown cells, coilin-stained foci are still detected whereas SMN staining of these foci is lost (Supplementary Figure S1A). These data indicate that FUS and each of the U1 snRNP-specific proteins is required for the presence of Gems but not Cajal bodies in HeLa cells. We also observed no apparent effect of FUS or U1 snRNP-specific protein knockdowns on nuclear speckle domains, which contain snRNPs and other splicing factors (Supplementary Figure S1B). The observation that U1 snRNP, as found for FUS, TDP-43, and SMN, is required for normal Gem levels indicates that U1 snRNP is another key component of a molecular pathway that may be disrupted in motor neuron disease.
Figure 1.
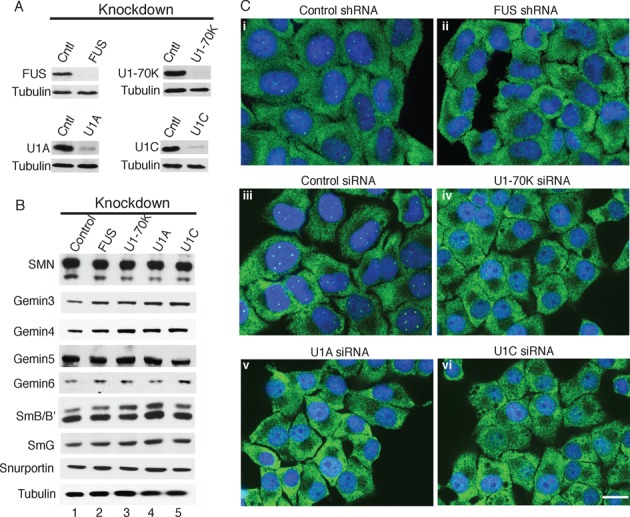
U1 snRNP-specific proteins are required for Gem formation. (A and B) HeLa cells stably expressing shRNA against FUS or transfected with siRNAs against U1-70K, U1A, or U1C were used to knock down these proteins. Scrambled shRNA or siRNA was used as a negative control. The knockdowns were analyzed by Western using the indicated antibodies. Tubulin was used as a loading control. (C) IF of the indicated knockdown cells with SMN antibodies (green) was used to detect Gems. DAPI (blue) was used to mark the nucleus. The merged images are shown. Scale bar: 20 μm.
SmB snRNP core proteins are mislocalized to the cytoplasm in ALS patient fibroblasts bearing NLS mutations in FUS
FUS is known to be mislocalized to the cytoplasm in ALS patient fibroblasts bearing mutations in the NLS, which is located between amino acids 495–526 (34). The mislocalization is only partial because the cells contain one normal and one mutant copy of FUS. Consistent with previous studies, confocal images of IF staining showed that FUS is properly localized to the nucleus in fibroblasts from unaffected controls and from patients with Alzheimer's disease (AD) or Parkinson's disease (PD) (Figure 2A and B, panels i, and Supplementary Figure S2A, i–v). In contrast, FUS is mislocalized to the cytoplasm in ALS patient fibroblasts bearing an R514G (Figure 2A, panel vi) or R521C (Figure 2B, panel vi) mutation. This mislocalization of FUS in the ALS versus control fibroblasts can also be seen in lower magnification widefield microscopy images of cell fields (Supplementary Figure S2B and C, panels i–ii).
Figure 2.
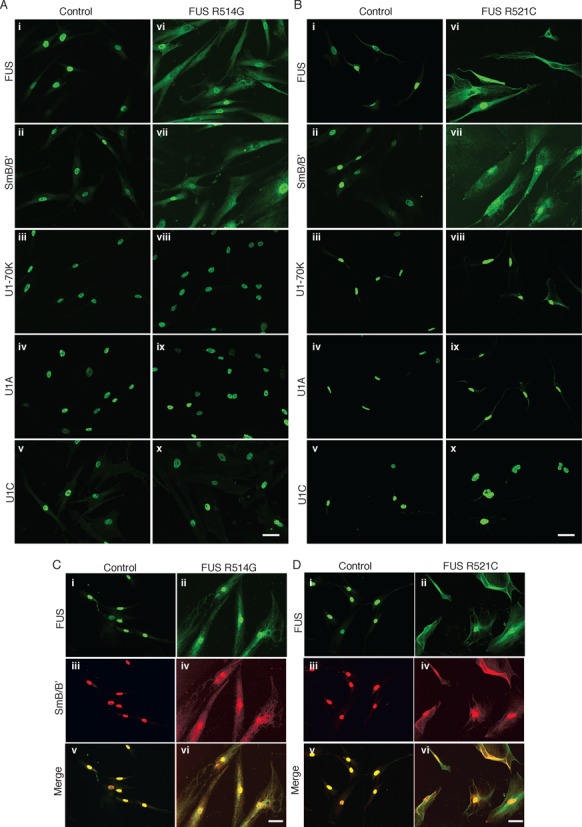
FUS and SmB are mislocalized to the cytoplasm in ALS patient fibroblasts bearing FUS mutations in the NLS. (A and B) IF with antibodies against FUS, SmB, U1-70K, U1A, and U1C was carried out with ALS patient cells carrying mutations in FUS R514G (A) or R521C (B). Control fibroblasts from an unaffected individual (#34) were used as a control (and see Supplementary Figure S2 for five additional control fibroblasts). Scale bar: 50 μm. (C and D) The indicated patient fibroblasts were co-stained with FUS and SmB antibodies. The merged images are shown. Scale bar: 50 μm.
U1 snRNP contains the Sm snRNP core proteins (B/B’, D1, D2, D3, E, F, G), the U1 snRNP-specific proteins (U1-70K, U1A and U1C) and U1 snRNA. In light of our previous observation that all of these components of U1 snRNP abundantly associate with FUS (17), we next examined the distribution of U1 snRNP components in the ALS patient and control fibroblasts. IF staining of control cells revealed that SmB/B’ (hereafter referred to in the text as SmB) as well as U1-70K, U1A, and U1C localized in the nucleus (Figure 2A and B, panels ii–v, Supplementary Figure S2A panels vi–x, Figure S2B, and Figure S2C panel iii). A weak signal for SmB was also detected in the cytoplasm of control cells. This low level of cytoplasmic SmB signal is expected because the early steps of snRNP biogenesis take place in the cytoplasm, including loading of the Sm core proteins onto the snRNA via the SMN complex (35). The snRNP-specific proteins (U1-70K, U1A, U1C) are subsequently loaded onto the snRNA in the nucleus (36). In striking contrast to the control cells, in the ALS patient fibroblasts bearing mutations in the FUS NLS, IF staining of SmB was not only detected in the nucleus, but also was abundantly detected in the cytoplasm (Figure 2A and B, panel vii). The mislocalization of SmB in the ALS patient fibroblasts versus control can readily be seen in lower magnification images of the ALS patient fibroblasts (Supplementary Figures S2B and S2C). Mislocalization is not due to general leakiness of the nuclei as evidenced by the observation that the three U1 snRNP-specific proteins (U1-70K, U1A, U1C) were properly localized to the nucleus in the ALS patient fibroblasts (Figure 2A and B, panels viii–x). Analysis of the fibroblasts by Western revealed that the overall levels of FUS and SmB were similar (Supplementary Figure S2). In support of the IF data, the Westerns showed increased levels of both FUS and SmB in the cytoplasm in the ALS patient (R514G) versus control fibroblasts whereas the U1 snRNP-specific proteins were only detected in the nucleus in both types of fibroblasts (Supplementary Figure S2D). We did not observe an obvious decrease in the nuclear levels of FUS and SmB in the patient cells versus control. This may be because some of the ALS patient cells show complete absence of FUS from the nucleus whereas other cells in the population have higher levels of nuclear FUS. Therefore, the net amount of nuclear FUS may not differ in the total cell population. We note that the FUS patient cell lines grow extremely poorly compared to the control, AD, and PD lines used in this study. This poor growth was observed with different aliquots of the cells, and FUS R521C grows so poorly that we were unable to obtain sufficient quantities of cells for Westerns or other biochemical studies. Together, our data indicate that FUS and SmB, but not the U1 snRNP-specific proteins, are mislocalized to the cytoplasm in the ALS patient fibroblasts bearing mutations in the FUS NLS. We attempted to analyze additional Sm core proteins by both IF and Western but were not able to obtain antibodies that worked in these assays. However, it is well known that the Sm core proteins associate tightly with one another and co-IP in a stoichiometric complex (37). Thus, although it remains to be shown directly, the mislocalization of SmB may serve as a proxy for mislocalization of the entire Sm core. Our observation that SmB mislocalizes to the cytoplasm when mutant FUS mislocalizes is consistent with previous work in which transfected NSC-34 cells expressing FUS with NLS mutations showed a similar mislocalization of SmB (38).
SmB co-mislocalizes with FUS in the cytoplasm
Our previous biochemical data showed that SmB associates with FUS in an RRM-dependent, but NLS-independent manner (17). Thus, we sought to determine whether mislocalization of SmB could be explained by its association with cytoplasmic FUS in the ALS patient cells. As shown in Figure 2C and D (panel ii), the extent of FUS mislocalization varies between mostly nuclear to mostly cytoplasmic in the ALS fibroblast populations. Significantly, co-staining revealed that SmB and FUS are mislocalized to similar extents in each of the cells (Figure 2C and D, panels ii and iv). These data support the conclusion that mislocalization of SmB occurs because it is sequestered in the cytoplasm by mislocalized mutant FUS. As FUS is known to form intermolecular interactions with itself (17,39), it is possible that the differences in levels of SmB and FUS mislocalization are due to mutant FUS sequestering variable levels of wild type FUS, which, in turn, sequesters variable levels of SmB.
U1 snRNA mislocalizes to the cytoplasm in ALS patient fibroblasts with FUS NLS mutations
It is known that U1 snRNA associates with the Sm core proteins in the cytoplasm during snRNP biogenesis. Accordingly, we next investigated the localization of U1 snRNA in the ALS patient fibroblasts using FISH. This analysis revealed that U1 snRNA was mislocalized to the cytoplasm in both the FUS R514G (Figure 3A, panel ii) and R521C (Figure 3A, panel iv) patient fibroblasts whereas U1 snRNA was only nuclear in the control fibroblasts (Figure 3A, panels i and iii). However, we also observed that the overall FISH signal for U1 snRNA was stronger in both of the ALS patient cell lines compared to the control (Figure 3A). This increased level of U1 snRNA was validated by qPCR, which revealed ∼1.5 fold more U1 snRNA in the ALS versus control fibroblasts (Figure 3B). It is possible that this upregulation occurs because the nuclear levels of Sm proteins have decreased due to their mislocalization to the cytoplasm by mutant FUS. A similar upregulation of snRNAs was observed previously in an analysis of sporadic ALS patient neuronal cells containing TDP-43 aggregates (18). Thus, upregulation may be a common feature that occurs with mislocalized or lower levels of snRNP proteins and/or their associated proteins. The observation the SmB and U1 snRNA are mislocalized to the cytoplasm (this study and (38)) could explain the decreased levels of Gems observed in ALS patient fibroblasts and in cells expressing FUS with NLS mutations (17,28,38). It has also been reported that mutant FUS forms nuclear aggregates in ALS patient fibroblasts, and the decreased Gem levels may also be due to loss of function of this mutant nuclear FUS (34,40).
Figure 3.
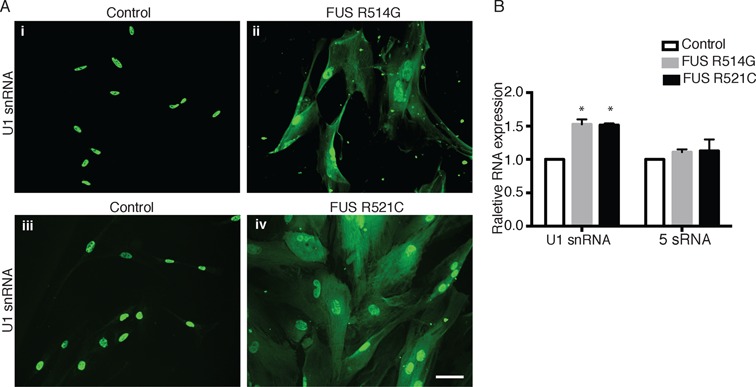
U1 snRNA is mislocalized to the cytoplasm in ALS patient fibroblasts bearing NLS mutations in FUS. (A) FISH was carried out to detect U1 snRNA in ALS patient fibroblasts harboring FUS mutations (R514G or R521C) and in control fibroblasts. Scale bar: 50 μM. (B) U1 snRNA and 5S RNA levels in ALS patient and control fibroblasts were determined by RT-qPCR in three separate experiments that were conducted in triplicate. Values are expressed as mean ± SD. *P < 0.01, two-tailed Student's t test.
SmB co-mislocalizes with FUS R495X to the cytoplasm in an RRM-dependent manner
To further investigate the cytoplasmic sequestration of SmB by FUS containing NLS mutations, we expressed the FUS mutant, R495X, in HeLa cells. This mutation truncates the NLS, which causes a severe and early onset form of ALS (34,41–42). In addition, FUS R495X is known to be strongly mislocalized to the cytoplasm (34). When we expressed wild type FLAG-tagged FUS in HeLa cells, IF staining with a FLAG antibody revealed that FUS was nuclear (Figure 4A, panel i). In contrast, FLAG-tagged FUS R495X was mainly detected diffusely in the cytoplasm (Figure 4A, panel iv). Analysis of the localization of U1 snRNP components revealed that in all of the cells in which FUS R495X was detected diffusely in the cytoplasm, a low but reproducible level of SmB was also diffuse in the cytoplasm (Figure 4A, compare panels iv and v). In contrast, the U1-snRNP specific proteins (U1-70K and U1A) were only detected in the nucleus in both the wild type and R495X transfected cells (Supplementary Figure S3A and S3B). We previously found that SmB associates with FUS in an RNA-dependent manner and requires the FUS RRM (17). Accordingly, we next mutated the RRM in FUS R495X and examined SmB localization. This analysis revealed that although FUS was mislocalized, SmB remained nuclear (Figure 4A, panels vii and viii). We conclude that SmB is sequestered in the cytoplasm by mutant FUS, and the RRM is required for this sequestration. The observation that the U1 snRNP-specific proteins are properly localized in the nucleus indicates that detection of SmB in the cytoplasm is not due to a general leakiness of the nuclei in cells containing FUS R495X. A likely reason that the Sm proteins and U1 snRNA are mislocalized by mutant FUS is because they are present in the cytoplasm during the SMN complex-dependent assembly of the U1 snRNP core particle. Thus, mutant FUS in the cytoplasm has the opportunity to interact with the Sm proteins and U1 snRNA and sequester these factors in the cytoplasm. Consistent with this conclusion, previous work showed that FUS specifically associates with U1 snRNP core particles (43). In the case of the U1 snRNP-specific proteins (U1-70K, U1A and U1C), they are known to be imported independently of the U1 snRNP core particle (44). Moreover, the U1 snRNP-specific proteins are known to first associate with the U1 snRNP core particle in the nucleus during assembly of the mature U1 snRNP (44). Together, these data may explain why mutant FUS causes mislocalization of the Sm proteins and U1 snRNA while the U1 snRNP-specific proteins are properly localized. We note that the number of cells expressing mutant or wild type FUS was low in our system and it was therefore not possible to carry out biochemical studies using these cells.
Figure 4.
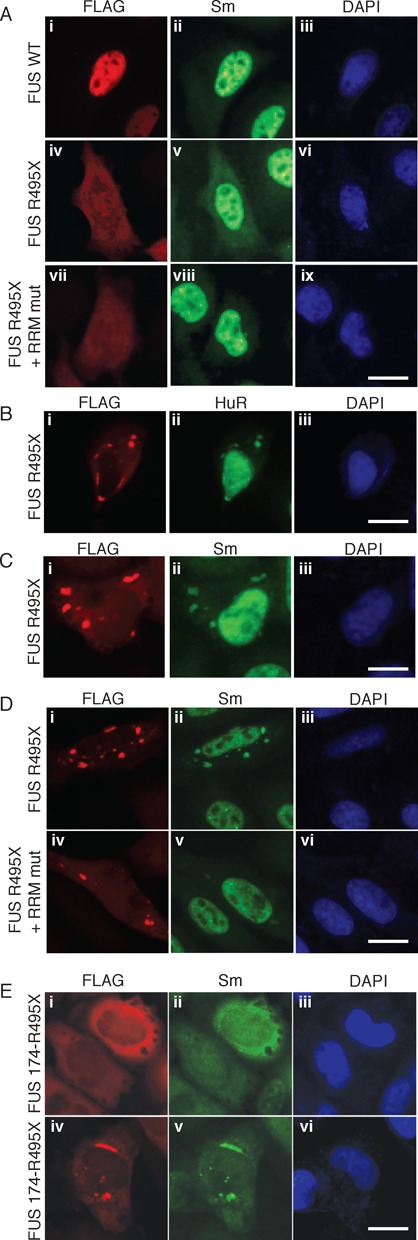
SmB is mislocalized to the cytoplasm in an RRM-dependent manner in HeLa cells expressing FUS bearing the ALS-causative R495X NLS mutation. (A) HeLa cells were transfected with FLAG-tagged WT FUS (panels i–iii), FUS R495X (panels iv–vi), or FUS R495X + RRMmut (panels vii–ix) followed by IF with FLAG or SmB antibodies. (B and C) HeLa cells were transfected with FLAG-tagged FUS R495X, and IF was carried out with antibodies against FLAG (B, C), HuR (B) or Sm (C). (D) HeLa cells were transfected with FLAG-tagged FUS R495X (panels i–iii), or FUS R495X + RRMmut (panels iv–vi) followed by IF with FLAG or SmB antibodies. (E) HeLa cells were transfected with Flag-tagged FUS 174-495X and IF was carried out with FLAG or SmB antibodies. (A–E) DAPI was used to detect the nucleus. Scale bar: 20 μm.
FUS and Sm proteins co-aggregate in stress granules in HeLa cells expressing FUS R495X
During our analyses of the transfected HeLa cells, we observed that FUS R495X, but not wild type FUS, formed cytoplasmic aggregates in a subset (∼5%) of the cells (Figure 4B, panel i). Moreover, these aggregates co-localized with the stress granule marker, HuR (Figure 4B, panel ii). Strikingly, we found that SmB also co-aggregates with FUS R495X (Figure 4C, panels i and ii) whereas the U1 snRNP-specific proteins were properly localized to the nucleus (Supplementary Figure S3C, panels ii and vi). Moreover, SmB does not aggregate with FUS R495X when the RRM is mutated (Figure 4D, panel v). We conclude that SmB, but not the U1 snRNP-specific proteins, co-aggregates with FUS R495X in cytoplasmic stress granules in an RRM-dependent manner. It is known that proteins such as FUS and TDP-43 form cytoplasmic aggregates in neuronal cells (21,24–25). Thus, our observation that FUS can form aggregates when expressed in HeLa cells may be related to aggregate formation in neuronal cells. In previous studies in yeast and drosophila, the RNA binding domains of FUS and TDP-43 were found to be essential for mislocalization of the respective proteins to stress granules as well as for their toxic effects (25,45–46,30). Together, these observations support the view that the RNA binding capability of ALS causing proteins, such as FUS and TDP-43, plays a role in their pathogenesis.
Role of the FUS prion-like domain in localization of SmB
The N-terminus of FUS contains a loosely structured, prion-like domain that participates in FUS aggregation and is thought to play a critical role in ALS pathogenesis (1,4–5,7,21,47–49). To determine whether this N-terminal domain of FUS affects SmB localization, we constructed FUS 174-R495X, in which both the N-terminal region and the NLS are truncated. This analysis revealed a dramatic diffuse mislocalization of both FUS and SmB to the cytoplasm (Figure 4E, panels i and ii). In addition, SmB was observed in co-aggregates with FUS 174-R495X in the cytoplasm, accompanied by a depletion of SmB staining from the nucleus (Figure 4E, panels iv and v). In contrast, to SmB, U1-70K is detected in the nucleus of cells expressing FUS 174-R495X regardless of whether FUS is diffusely mislocalized to the cytoplasm or present in cytoplasmic aggregates (Supplementary Figure 3D, compare panels i–ii and iv–v). It is known that FUS forms an intramolecular interaction (39) between the N and C termini of the protein. Thus, it is possible that the dramatic mislocalization of SmB is due to exposure of the FUS RRM when the N-terminus is truncated and cannot form this intramolecular interaction, as it may increase the levels of RRM-dependent SmB binding to FUS.
Knockdown of U1 snRNP in zebrafish causes motor neuron defects
Previously, morpholino oligonucleotides (MOs) were used to decrease the levels of SMN during zebrafish development, revealing that SMN functions in motor axon outgrowth and branching (26). Similar phenotypes were observed when FUS or TDP-43 was knocked down in zebrafish (50,51). To investigate a role for U1 snRNP in motor neurons, we knocked down U1 snRNP in zebrafish by injecting U1-70K MO into transgenic Tg(mnx1:GFP) embryos at the one- to two-cell stage. When 1.4 ng of MOs was injected, 75% (n = 600) of embryos injected with the U1-70K MO survived to 28 h post-fertilization (hpf) compared to 94% (n = 600) injected with the control MO. Significantly, truncations of motor axons were observed in the surviving 28 hpf embryos (Supplementary Figure S4A, panel ii). However, these embryos showed abnormal morphology, such as shortened body axis and kinked tail (Supplementary Figure S4B, panel iii). When only 0.7 ng U1-70K MO was injected, no abnormal morphology was observed (Supplementary Figure S4B, panel ii). However, we observed striking truncations of motor axons in these 28 hpf embryos (Figure 5A, panel ii, arrows), which is quantitated in Figure 5B. Moreover, the motor axon defect was rescued when U1-70K MO was co-injected with U1-70K mRNA (Figure 5B, panel iii), indicating that the axon defect is specifically caused by U1-70K knockdown.
Figure 5.
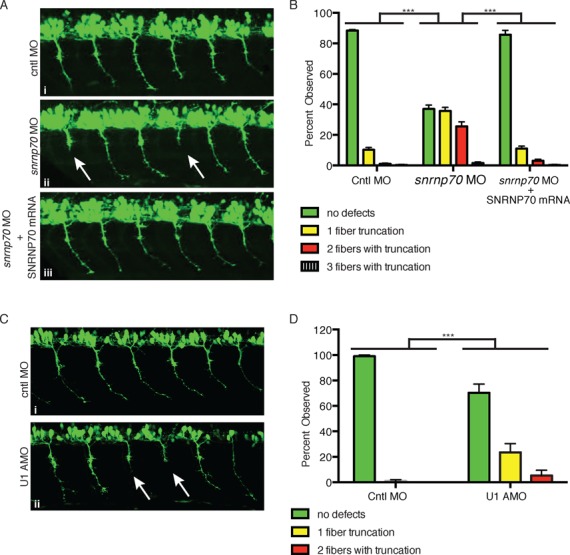
Knock down of U1 snRNP causes motor neuron defects in zebrafish. (A) Representative lateral views of motor axons in 28 hpf Tg(mnx1:GFP) zebrafish embryos injected with a negative control MO (0.7 ng, panel i), an MO targeting U1-70K (0.7 ng, panel ii) or a combination of U1-70K MO and U1-70K mRNA (panel iii). Arrows indicate truncated axons. (B) Quantitation of data shown in (A). Embryos were classified based on the number (0, 1, 2 or 3) of motor axon truncations and the percentage of each group is shown. (C) Representative lateral views of motor axons from 28 hpf zebrafish embryos that were injected with control MO (i) or U1 AMO (ii) are shown. Arrows indicate truncated axons. (D) Quantitation of data shown in (C). Embryos were classified based on the number (0, 1 or 2) of motor axon truncations and the percentage of each group is shown. Data in all graphs are represented as mean and standard error of the mean.
As another approach for investigating the role of U1 snRNP in axons, we injected zebrafish embryos with an antisense morpholino (U1 AMO) that base pairs to U1 snRNA and prevents its base pairing to 5′ splice sites (31). When we injected low levels (1.5 ng) of the U1 AMO or control MO into transgenic Tg(mnx1:GFP) embryos, morphological abnormalities were not observed. Significantly, however, motor axon truncation was observed when 1.5 ng of the AMO was injected, and no defects were observed when the control MO was injected (Figure 5C, panels i and ii; quantitated in Figure 5D). Together, these data indicate that knockdown of U1 snRNP specifically causes motor neuron defects in developing zebrafish.
In this study, we have reported that the essential canonical splicing factor, U1 snRNP, which is also an abundant FUS and SMN interactor, is not properly localized to the nucleus in ALS patient fibroblasts, is necessary for Gem formation, and plays a role in motor axon outgrowth in zebrafish. These observations indicate that there is a growing list of proteins, cellular machineries, and RNP bodies that function in the pre-mRNA splicing pathway and also have a particularly important role in motor neurons. So far, this list includes SMN and the SMN complex, FUS, TDP-43, hnRNPA1, MATRIN3 and now U1 snRNP. Although all of these factors have functions in processes other than splicing, the addition of U1 snRNP to the list increases the likelihood that defective splicing may be one of the key processes that is pathogenic in motor neuron disease. Consistent with this view, ALS-causing mutations such as FUS R521C as well as knockdown of FUS, TDP-43 and SMN all result in splicing defects (52–54).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank C.E. Beattie (Ohio State University) for Tg(mnx1:GFP) zebrafish. HeLa cells were obtained from the National Cell Culture Center Biovest International (Minneapolis, MN), and we thank the Nikon imaging center at Harvard Medical School for assistance with microscopy. The authors declare no conflict of interest.
Footnotes
Present addresses:
Tomohiro Yamazaki, Institute for Genetic Medicine, Hokkaido University, Sapporo 060-0815, Japan.
Yoan Eliasse, Department of Biology, Polytech Nice-Sophia Engineering school, 930 Route des Colles, 06903 Sophia Antipolis, France.
FUNDING
National Institutes of Health [GM043375 to R.R.]; Amyotrophic Lateral Sclerosis (ALS) Therapy Alliance [2013-S-006 to R.R.]. Funding for open access charge: National Institutes of Health [GM043375 to R.R.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Robberecht W., Philips T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 2.Renton A.E., Chio A., Traynor B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leblond C.S., Kaneb H.M., Dion P.A., Rouleau G.A. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp. Neurol. 2014;262:91–101. doi: 10.1016/j.expneurol.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Ling S.C., Polymenidou M., Cleveland D.W. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y.R., King O.D., Shorter J., Gitler A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013;201:361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemmens R., Moore M.J., Al-Chalabi A., Brown R.H. Jr, Robberecht W. RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci. 2010;33:249–258. doi: 10.1016/j.tins.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Ramaswami M., Taylor J.P., Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vance C., Rogelj B., Hortobagyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sreedharan J., Blair I.P., Tripathi V.B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J.C., Williams K.L., Buratti E., et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim H.J., Kim N.C., Wang Y.D., Scarborough E.A., Moore J., Diaz Z., MacLea K.S., Freibaum B., Li S., Molliex A., et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson J.O., Pioro E.P., Boehringer A., Chia R., Feit H., Renton A.E., Pliner H.A., Abramzon Y., Marangi G., Winborn B.J., et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014;17:664–666. doi: 10.1038/nn.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwiatkowski T.J. Jr, Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T., et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 13.Van Deerlin V.M., Leverenz J.B., Bekris L.M., Bird T.D., Yuan W., Elman L.B., Clay D., Wood E.M., Chen-Plotkin A.S., Martinez-Lage M., et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rutherford N.J., Zhang Y.J., Baker M., Gass J.M., Finch N.A., Xu Y.F., Stewart H., Kelley B.J., Kuntz K., Crook R.J., et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G., Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 16.Battle D.J., Kasim M., Yong J., Lotti F., Lau C.K., Mouaikel J., Zhang Z., Han K., Wan L., Dreyfuss G. The SMN complex: an assembly machine for RNPs. Cold Spring Harb. Symp. Quant. Biol. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- 17.Yamazaki T., Chen S., Yu Y., Yan B., Haertlein T.C., Carrasco M.A., Tapia J.C., Zhai B., Das R., Lalancette-Hebert M., et al. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012;2:799–806. doi: 10.1016/j.celrep.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuiji H., Iguchi Y., Furuya A., Kataoka A., Hatsuta H., Atsuta N., Tanaka F., Hashizume Y., Akatsu H., Murayama S., et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol. Med. 2013;5:221–234. doi: 10.1002/emmm.201202303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shan X., Chiang P.M., Price D.L., Wong P.C. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 2010;107:16325–16330. doi: 10.1073/pnas.1003459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buratti E., Brindisi A., Giombi M., Tisminetzky S., Ayala Y.M., Baralle F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005;280:37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- 21.Thomas M., Alegre-Abarrategui J., Wade-Martins R. RNA dysfunction and aggrephagy at the centre of an amyotrophic lateral sclerosis/frontotemporal dementia disease continuum. Brain. 2013;136:1345–1360. doi: 10.1093/brain/awt030. [DOI] [PubMed] [Google Scholar]
- 22.Dewey C.M., Cenik B., Sephton C.F., Johnson B.A., Herz J., Yu G. TDP-43 aggregation in neurodegeneration: are stress granules the key. Brain Res. 2012;1462:16–25. doi: 10.1016/j.brainres.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bentmann E., Haass C., Dormann D. Stress granules in neurodegeneration–lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J. 2013;280:4348–4370. doi: 10.1111/febs.12287. [DOI] [PubMed] [Google Scholar]
- 24.Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 25.Nonaka T., Masuda-Suzukake M., Arai T., Hasegawa Y., Akatsu H., Obi T., Yoshida M., Murayama S., Mann D.M., Akiyama H., et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4:124–134. doi: 10.1016/j.celrep.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Lee E.B., Lee V.M., Trojanowski J.Q. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groen E.J., Fumoto K., Blokhuis A.M., Engelen-Lee J., Zhou Y., van den Heuvel D.M., Koppers M., van Diggelen F., van Heest J., Demmers J.A., et al. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum. Mol. Genet. 2013;22:3690–3704. doi: 10.1093/hmg/ddt222. [DOI] [PubMed] [Google Scholar]
- 28.Sun S., Ling S.C., Qiu J., Albuquerque C.P., Zhou Y., Tokunaga S., Li H., Qiu H., Bui A., Yeo G.W., et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 2015;6:6171. doi: 10.1038/ncomms7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q., Fischer U., Wang F., Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–1021. doi: 10.1016/s0092-8674(00)80367-0. [DOI] [PubMed] [Google Scholar]
- 30.Sun Z., Diaz Z., Fang X., Hart M.P., Chesi A., Shorter J., Gitler A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9:e1000614. doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaida D., Berg M.G., Younis I., Kasim M., Singh L.N., Wan L., Dreyfuss G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cottenie E., Kochanski A., Jordanova A., Bansagi B., Zimon M., Horga A., Jaunmuktane Z., Saveri P., Rasic V.M., Baets J., et al. Truncating and missense mutations in IGHMBP2 cause Charcot-Marie tooth disease type 2. Am. J. Hum. Genet. 2014;95:590–601. doi: 10.1016/j.ajhg.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stejskalova E., Stanek D. The splicing factor U1-70K interacts with the SMN complex and is required for nuclear gem integrity. J. Cell Sci. 2014;127:3909–3915. doi: 10.1242/jcs.155838. [DOI] [PubMed] [Google Scholar]
- 34.Bosco D.A., Lemay N., Ko H.K., Zhou H., Burke C., Kwiatkowski T.J. Jr, Sapp P., McKenna-Yasek D., Brown R.H. Jr, Hayward L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010;19:4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pellizzoni L., Yong J., Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 36.Yong J., Kasim M., Bachorik J.L., Wan L., Dreyfuss G. Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol. Cell. 2010;38:551–562. doi: 10.1016/j.molcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urlaub H., Raker V.A., Kostka S., Luhrmann R. Sm protein-Sm site RNA interactions within the inner ring of the spliceosomal snRNP core structure. EMBO J. 2001;20:187–196. doi: 10.1093/emboj/20.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerbino V., Carri M.T., Cozzolino M., Achsel T. Mislocalised FUS mutants stall spliceosomal snRNPs in the cytoplasm. Neurobiol. Dis. 2013;55:120–128. doi: 10.1016/j.nbd.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Wang X., Arai S., Song X., Reichart D., Du K., Pascual G., Tempst P., Rosenfeld M.G., Glass C.K., Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–130. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz J.C., Podell E.R., Han S.S., Berry J.D., Eggan K.C., Cech T.R. FUS is sequestered in nuclear aggregates in ALS patient fibroblasts. Mol. Biol. Cell. 2014;25:2571–2578. doi: 10.1091/mbc.E14-05-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waibel S., Neumann M., Rabe M., Meyer T., Ludolph A.C. Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology. 2010;75:815–817. doi: 10.1212/WNL.0b013e3181f07e26. [DOI] [PubMed] [Google Scholar]
- 42.Waibel S., Neumann M., Rosenbohm A., Birve A., Volk A.E., Weishaupt J.H., Meyer T., Muller U., Andersen P.M., Ludolph A.C. Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: a clinico-genetic study in Germany. Eur. J. Neurol. 2013;20:540–546. doi: 10.1111/ene.12031. [DOI] [PubMed] [Google Scholar]
- 43.Hackl W., Luhrmann R. Molecular cloning and subcellular localisation of the snRNP-associated protein 69KD, a structural homologue of the proto-oncoproteins TLS and EWS with RNA and DNA-binding properties. J. Mol. Biol. 1996;264:843–851. doi: 10.1006/jmbi.1996.0681. [DOI] [PubMed] [Google Scholar]
- 44.Romac J.M., Graff D.H., Keene J.D. The U1 small nuclear ribonucleoprotein (snRNP) 70K protein is transported independently of U1 snRNP particles via a nuclear localization signal in the RNA-binding domain. Mol. Cell. Biol. 1994;14:4662–4670. doi: 10.1128/mcb.14.7.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson B.S., Snead D., Lee J.J., McCaffery J.M., Shorter J., Gitler A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009;284:20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voigt A., Herholz D., Fiesel F.C., Kaur K., Muller D., Karsten P., Weber S.S., Kahle P.J., Marquardt T., Schulz J.B. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS One. 2010;5:e12247. doi: 10.1371/journal.pone.0012247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gitler A.D., Shorter J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion. 2011;5:179–187. doi: 10.4161/pri.5.3.17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwon I., Kato M., Xiang S., Wu L., Theodoropoulos P., Mirzaei H., Han T., Xie S., Corden J.L., McKnight S.L. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155:1049–1060. doi: 10.1016/j.cell.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwartz J.C., Wang X., Podell E.R., Cech T.R. RNA seeds higher-order assembly of FUS protein. Cell Rep. 2013;5:918–925. doi: 10.1016/j.celrep.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kabashi E., Bercier V., Lissouba A., Liao M., Brustein E., Rouleau G.A., Drapeau P. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 2011;7:e1002214. doi: 10.1371/journal.pgen.1002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kabashi E., Lin L., Tradewell M.L., Dion P.A., Bercier V., Bourgouin P., Rochefort D., Bel Hadj S., Durham H.D., Vande Velde C., et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2010;19:671–683. doi: 10.1093/hmg/ddp534. [DOI] [PubMed] [Google Scholar]
- 52.Qiu H., Lee S., Shang Y., Wang W.Y., Au K.F., Kamiya S., Barmada S.J., Finkbeiner S., Lui H., Carlton C.E., et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 2014;124:981–999. doi: 10.1172/JCI72723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou Y., Liu S., Liu G., Ozturk A., Hicks G.G. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013;9:e1003895. doi: 10.1371/journal.pgen.1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoell J.I., Larsson E., Runge S., Nusbaum J.D., Duggimpudi S., Farazi T.A., Hafner M., Borkhardt A., Sander C., Tuschl T. RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 2011;18:1428–1431. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.