


Abstract
Ataxia-telangiectasia mutated (ATM) is needed for the initiation of the double-strand break (DSB) repair by homologous recombination (HR). ATM triggers DSB end resection by stimulating the nucleolytic activity of CtIP and MRE11 to generate 3′-ssDNA overhangs, followed by RPA loading and RAD51 nucleofilament formation. Here we show for the first time that ATM is also needed for later steps in HR after RAD51 nucleofilament formation. Inhibition of ATM after completion of end resection did not affect RAD51 nucleofilament formation, but resulted in HR deficiency as evidenced by (i) an increase in the number of residual RAD51/γH2AX foci in both S and G2 cells, (ii) the decrease in HR efficiency as detected by HR repair substrate (pGC), (iii) a reduced SCE rate and (iv) the radiosensitization of cells by PARP inhibition. This newly described role for ATM was found to be dispensable in heterochromatin-associated DSB repair, as KAP1-depletion did not alleviate the HR-deficiency when ATM was inhibited after end resection. Moreover, we demonstrated that ATR can partly compensate for the deficiency in early, but not in later, steps of HR upon ATM inhibition. Taken together, we describe here for the first time that ATM is needed not only for the initiation but also for the completion of HR.
INTRODUCTION
The phosphatidyl inositol 3-kinase-like kinase (PIKK) family member ataxia-telangiectasia mutated (ATM) is the master player of the DNA damage response (DDR), which coordinates a complex network of signaling cascades including cell cycle checkpoints and the repair of DNA double-strand breaks (DSB) in order to maintain genomic integrity (1). Upon induction of DSBs, ATM is activated and phosphorylates several DSB response proteins including the histone H2A variant H2AX over a large chromatin domain flanking the DSBs and initiates a series of downstream reactions including protein recruitment and post-translational protein modifications on this chromatin domain (2,3). Cells lacking ATM exhibit a severe DSB repair defect, checkpoint dysfunction, pronounced genomic instability and an extremely high radiosensitivity (4).
DSBs are mainly repaired by two repair pathways: non-homologous end joining (NHEJ) and homologous recombination (HR). NHEJ is a fast process and represents the major DSB repair pathway in mammalian cells, repairing DSBs in all cell cycle phases though predominately in G1 (5). HR is a rather slow and multiple repair process, which is restricted to the S/G2 phase, when an intact sister chromatin is available to allow error-free repair (5).
Several lines of evidence support a specific role for ATM in HR. In addition to H2AX, many HR factors such as BRCA1, BLM, NBS1, MRE11 and CtIP are ATM substrates (1). Small molecule inhibitors of ATM or siRNA-mediated ATM depletion reduce the phosphorylation and hence the activation of such substrates (6,7). Importantly, cells carrying homozygous ATM kinase-dead mutations show reduced HR and consequently increased sensitivity to Poly(ADP-ribose)-Polymerase (PARP) inhibition (8,9), mitomycin C (10) and topotecan (11).
Conceptually, HR is divided into three stages: presynapsis, synapsis and postsynapsis. In presynapsis, DSB ends are processed by nucleolytic enzymes to generate long stretches of single-stranded DNA (ssDNA)—a mechanism generally described as DNA end resection (12). In mammalian cells, the end resection step is initiated by the collaborative action of MRE11 and CtIP (13,14), with the generated ssDNA being subsequently coated with RPA (15). In a further step, both BRCA2 and RAD54 promote the exchange of RPA with RAD51, allowing RAD51 nucleofilament formation (12). In synapsis, the nucleofilament mediates the homology search and strand invasion to form the D-loop (16). In postsynapsis, RAD51 is assumed to dissociate from the ends to allow for further steps such as DNA synthesis (17,18). Thus far, ATM is only known to be engaged in presynapsis by stimulating DSB end resection through the phosphorylation and activation of nuclease enzymes such as CtIP, MRE11, EXO1 and BLM (1). Consequently, ATM-deficient or inhibited cells exhibit impaired DSB end resection as indicated by the smaller number of RPA foci observed at DSBs (19,20). Whether or not ATM is involved in the other two stages of HR is unclear.
Here, we present strong evidences that ATM is also involved in HR after completion of the presynapsis stage. We show that ATM inhibition after DSB end resection did not affect RAD51 nucleofilament formation, but did result in a reduced HR efficiency with an enhanced number of residual RAD51 and yH2AX foci in both S and G2 cells. This effect is not related to the role of ATM in DSB repair in heterochromatin (HC), as the knockdown of KAP1 did not alleviate the HR deficiency driven by ATM inhibition after end resection. Moreover, we demonstrate that ATR can partially reverse the effect of ATM inhibition on HR in the presynapsis stage, but not after its completion.
MATERIALS AND METHODS
Cell culture, X-irradiation and inhibitors
The human cervical carcinoma cell lines HeLa, HeLa-pGC (containing the gene conversion substrate) and the human lung carcinoma cell line A549 were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco-Invitrogen) supplemented with 10% Fetal Calf Serum (FCS). Irradiation was performed as previously described (200 keV, 15 mA, additional 0.5-mm Cu filter at a dose rate of 0.8 Gy/min) (21). To inhibit the kinase activity of ATM and ATR, 10-μM KU55933 and 0.02-μM VE-821 were used, respectively, and 25-μM Mirin was applied to inhibit the nuclease activity of MRE11. To inhibit PARP activity, we used 1-μM Olaparib.
Colony formation assay
For colony formation, cells were seeded and allowed to adhere before drug treatment or irradiation. A specific ATM inhibitor (10-mM KU55933), MRE11 inhibitor (25-μM Mirin) or PARP-1 inhibitor (1-μM Olaparib) was added 1 h prior to irradiation or 2 h post-IR and was kept in the medium for 24 h. Cells were subsequently incubated in drug-free medium for colony formation for 2–3 weeks and thereafter stained with crystal violet. Colonies of 50 cells or more were counted manually and survival curves were derived from triplicates of at least three independent experiments.
DSB repair reporter assay for HR
To induce DSBs, HeLa cells containing the stably integrated reporter construct for gene conversion pGC were transfected with the I-SceI expression vector pCMV3xnls-I-SceI (1 μg) using Fugene HD (Promega) as a transfection reagent. Forty-eight hours after transfection, the cells were assessed for green fluorescence by flow cytometry (FACScan, BD Bioscience).
Immunofluorescence
Cells grown on cover slips were washed once with cold phosphate buffered saline (PBS) and fixed with 4% para-formaldehyde/PBS for 10 min. Fixed cells were permeabilized with 0.2% Triton X-100/PBS on ice for 5 min. The cells were incubated overnight with primary antibodies: mouse monoclonal anti-phospho-S139-H2AX antibody (Millipore) at a dilution of 1:300, mouse monoclonal anti-RAD51 antibody (Abcam 14B4) at a dilution of 1:1000, mouse monoclonal anti-RPA antibody (Santa Cruz Biotechnology) at a dilution of 1:600 and rabbit monoclonal anti-CenpF antibody (Lifespan Biosciences) at a dilution of 1:750. After being washed three times with cold PBS, the cells were incubated for 1 h with secondary anti-mouse Alexa-fluor594 (Invitrogen) at a dilution of 1:500 or anti-rabbit Alexa-fluor488 (Invitrogen) at a dilution of 1:600. The nuclei were counterstained with 4′-6-diamidino-2-phenylindole (DAPI, 10 ng/ml). Slides were mounted in Vectashield mounting medium (Vector Laboratories). Immunofluorescence was observed with the Zeiss AxioObserver.Z1 microscope (objectives: ECPlnN 40x/0.75 DICII, resolution 0.44 μm; Pln Apo 63x/1.4Oil DICII, resolution 0.24 μm; EC PlnN 100x/1.3 Oil DICII, resolution 0.26 μm and filters: Zeiss 43, Zeiss 38, Zeiss 49). Semi-confocal images were obtained using the Zeiss Apotome, Zeiss AxioCamMRm and Zeiss AxioVision Software.
RNA interference
ATM and MRE11 siRNAs employed in this study were SMARTpools (Thermo Fisher). RNAi transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. KAP1 siRNA transfection was performed using Lipofectamine 2000 (Invitrogen, Germany) according the manufacturer's protocol. The KAP1 siRNA oligonucleotide (5′-CAGTGCTGCACTAGCTGTGAGGATA-3′) was ordered from Eurofins Genomics, Germany.
Sister chromatid exchange assay
Analysis of sister chromatid exchange (SCE) was performed as previously described (22). Briefly, A549 cells were grown for 48 h (about two replication rounds) in medium containing 25-μM BrdU before irradiation with 2 Gy. Two hundred nanograms per milliliter colcemid were added for 18 h to collect cells in metaphase. Metaphases were spread on slides and fixed with 4% para-formaldehyde for 10 min. Slides were then incubated for 1 h with Anti-rat anti-BrdU (AbD Serotec, ICR1) at a dilution of 1:1000. After being washed three times with cold PBS, metaphases were incubated for 1.5 h with anti-rat Alexa Fluor 555 (Molecular Probes) at a dilution of 1:1000. DAPI was used for counterstaining and immunofluorescence was observed with the Zeiss AxioObserver.Z1 microscope as described above.
Caspase activity
Detection of caspase activity was performed utilizing the FAMFLICA ™ Poly Caspases Assay Kit (Immunochemistry Technologies) according to the manufacturer's instructions. Data acquisition was performed on FACS Canto flow cytometer (BD Biosciences) using FACS Diva software (Becton Dickinson).
Graphs and statistics
Unless stated otherwise, experiments were independently repeated at least three times. Data points represent the mean ± SEM of all individual experiments. Statistical analysis, data fitting and graphics were performed with the GraphPad Prism 5.0 program (GraphPad Software).
RESULTS
Simultaneous inactivation of ATM and MRE11 shows a synergistic inhibitory effect on HR
ATM is known to be important for HR and—as a consequence—the down-regulation of ATM strongly reduces HR (6,23). To confirm this, we generated stable human HeLa cells carrying a single integrated copy of the HR substrate pGC (24) (Figure 1A, upper panel). Cells were treated with 10-μM ATM inhibitor (Ku55933) immediately prior to the induction of DSBs via transfection with the I-SceI-expression vector (pCMV-ISceI-3NLS). After 48 h, the percent of GFP-positive cells (GFP+ cells) as an indication for HR events was monitored using FACS. Confirming the previously published data, we reported a 60% reduction in HR efficiency upon ATM inhibition (Figure 1A, second column). ATM initiates HR by stimulating extensive DSB end resection mediated by MRE11 and CtIP (1). Correspondingly, the inhibition of MRE11 nuclease activity using 25-μM Mirin resulted in a reduction in HR (Figure 1A, third column) with no effect on ATM signaling (Supplementary Figure S1B). This reduction was not due to inhibition of ATM signaling upon MRE11 inhibition. Surprisingly, a synergistic inhibitory effect was observed when both ATM and MRE11 were inhibited simultaneously (Figure 1A, fourth column). This effect was also observed in another cell line (H1299) (Supplementary Figure S1A) as well as in the previously described ATM-deficient SKX-cells (25), with an additional reduction in HR efficiency being observed when MRE11 was inhibited by Mirin (Figure 1C). Moreover, siRNA-mediated knockdown of either ATM or MRE11 (Supplementary Figure S1C) reduced HR efficiency while combined knockdown of both proteins showed a synergistic inhibitory effect on HR (Figure 1B). Consistent with this finding, we found that the simultaneous inhibition of both ATM and MRE11 resulted in a more pronounced sensitivity to MMC (Figure 1D)—which is known to reflect HR efficiency—when compared to the inhibition of either of them alone. Overall, these data show that HR is clearly more affected when ATM and MRE11 are inhibited simultaneously than when either enzyme is inhibited individually.
Figure 1.
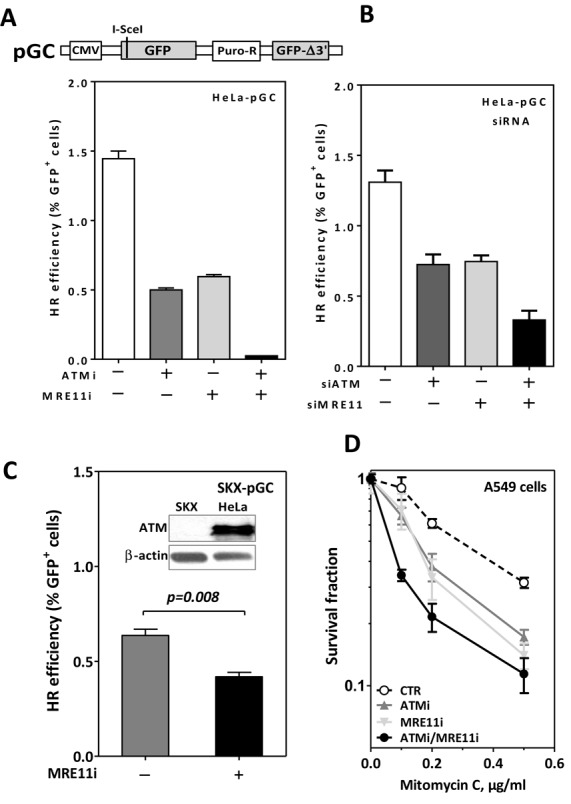
Simultaneous inactivation of ATM and MRE11 shows a synergistic inhibitory effect on HR. Schematic representation of the GFP-based HR substrate pGC ((A), upper panel). ATM and/or MRE11 were either inhibited using specific inhibitors (A) or depleted via siRNA (B) in HeLa cells harboring pGC. Cells were then transfected with I-SceI-expressing vector and the percentage of GFP+ cells (as an indication for HR efficiency) was assessed at 48-h time point post-transfection. (C) ATM-deficient SKX cells harboring pGC were treated with 25-μM Mirin and HR was measured as in (A); inset: ATM signal in SKX versus HeLa cells. (D) A549 cells were treated with the indicated concentrations of MMC after inhibition of ATM, MRE11 or both. Survival fractions were measured using the colony forming assay. Error bars represent the SEM of three independent experiments.
ATM and MRE11 work epistatically in both DSB end resection and RAD51 nucleofilament formation
Next, we sought to investigate the mechanism underlying the stronger inhibitory effect the inactivation of both ATM and MRE11 has on HR. Firstly, we tested whether this effect is related to DSB end resection, which is the initial step of HR. To this end, unsynchronized A549 cells were treated with either ATM inhibitor and/or MRE11 inhibitor for 1 h prior to irradiation with 2 Gy and the kinetics of end resection were monitored in the S/G2 cell population by enumerating RPA foci in CenpF-positive (CenpF+; Figure 2A) cells at different time points (2, 4 and 6 h). As illustrated in Figure 2B, RPA foci normally reached a peak 2 h post-IR (29.4 ± 2), then started to decline gradually before reaching a minimum of five foci at the 6-h time point. In line with its role in end resection, the inhibition of ATM led to a delay in RPA focus kinetics, with an almost four times fewer RPA foci at the 2-h time point (8 ± 1) and with a maximum of only 12 foci reached 4 h after irradiation. A similar effect on the kinetics of RPA foci was reported when MRE11 nuclease activity was inhibited by Mirin. More importantly, the simultaneous inhibition of both ATM and MRE11 showed no further change in RPA foci kinetics compared to when either enzyme was inhibited individually (Figure 2B). These data indicate that ATM and MRE11 work epistatically in DSB end resection and RPA foci formation and that the additional inhibition of HR seen after the simultaneous inhibition of these two enzymes (Figure 1) is not related to end resection.
Figure 2.
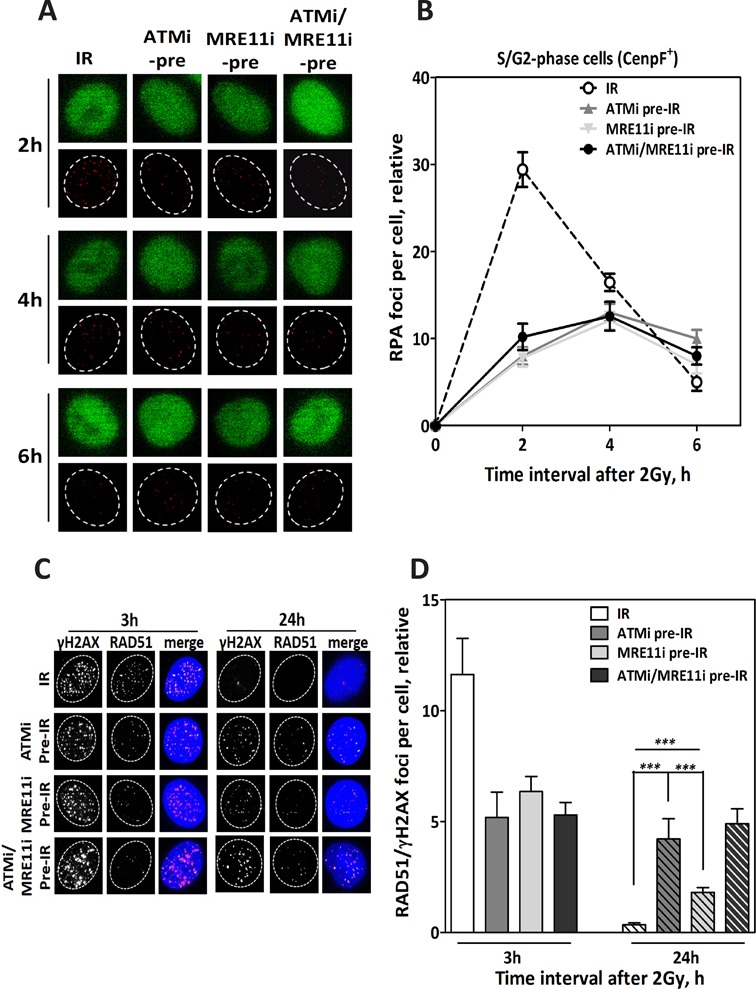
ATM and MRE11 work epistatically in the DSB end resection and RAD51 nucleofilament formation steps of HR. (A) Representative IF photos for RPA foci in CenpF+ cells (S/G2) after inhibition of ATM (ATMi pre-IR), MRE11 (MRE11i pre-IR) or both (ATMi/MRE11i pre-IR) prior to irradiation with 2 Gy. (B) Quantification of RPA foci CenpF+ cells (n = 100) at the indicated time points after 2 Gy. Inhibition of ATM or MRE11 led to a reduction in the number of RPA foci at the indicated time points compared to cells irradiated alone (IR). (C) Representative IF photos for RAD51 foci in A549 cells after inhibition of ATM (ATMi pre-IR), MRE11 (MRE11 pre-IR) or both (ATMi/MRE11i pre-IR) prior to irradiation with 2 Gy. (D) Quantification of RAD51 foci co-localized with γH2AX foci in 100 cells. Inhibition of ATM, MRE11 or both led to a reduced number of RAD51 foci compared to cells irradiated alone (IR) at the 3-h time point. At the 24-h time point, the number of residual RAD51 foci was significantly higher upon ATM inhibition than after MRE11 inhibition. Simultaneous inhibition of both enzymes led to a slight increase in RAD51 compared to ATM inhibition alone. In all cases, the number of foci measured in non-irradiated cells was subtracted from that observed in irradiated cells. Error bars represent the SEM of three independent experiments. P value less than 0.001 was designated with three (***) asterisks.
After DSB end resection and RPA loading, RAD51 is recruited to the resected ends to replace RPA and allowing for the formation of RAD51 nucleofilament, which then mediates homology search and strand invasion (12). Therefore, the additional reduction of HR upon the simultaneous inhibition of ATM and MRE11 might be related to RAD51 nucleofilament formation. In order to test this possibility, unsynchronized A549 cells were treated with the two inhibitors (Ku55933 and Mirin) 1 h prior to 2 Gy either alone or simultaneously. RAD51 foci that co-localized with γH2AX (Figure 2C) were then counted 3 h after IR. Upon inhibition of either ATM or MRE11, the number of RAD51 foci was found to decrease 2-fold at this time point. Importantly, no further reduction in RAD51 foci formation was reported when both enzymes were inhibited simultaneously, indicating that ATM and MRE11 work epistatically in RAD51 nucleofilament formation step. Essentially similar results were obtained after transient knockdown of ATM and MRE11 (Supplementary Figure S2). Altogether, these data demonstrate that the pronounced inhibitory effect observed on HR following the simultaneous inhibition of ATM and MRE11 is not related to RAD51 nucleofilament formation.
It is worth noting that although RAD51 nucleofilament formation was deficient upon MRE11 inhibition, most of the loaded RAD51 foci (measured at 3-h time point) disappeared 24 h after IR (Figure 2D). In contrast, the inhibition of ATM delayed the decline of RAD51 foci until this time point, which indicates an involvement of ATM, but not MRE11, in HR after RAD51 nucleofilament formation.
ATM but not MRE11 is involved in HR after RAD51 nucleofilament formation
In order to verify the involvement of ATM in HR after RAD51 nucleofilament formation, ATM and MRE11 were inhibited either individually or simultaneously 2 h after IR, a time point when end resection has initiated (19). We subsequently monitored RAD51 foci co-localized with γH2AX at 3 and 24 h after IR. As anticipated, the kinetics of RPA foci measured in S/G2 cells (identified as CenpF+) up to 6 h after irradiation with 2 Gy were not affected when ATM and MRE11 were inhibited either individually or simultaneously (Figure 3A and B). Moreover, the inhibition of either ATM or MRE11 2 h post-IR did not affect the formation of RAD51 foci measured 3 h after IR (Figure 3C and D). However, a significantly higher number of residual RAD51/γH2AX foci were observed 24 h after IR following inhibition of ATM, but not of MRE11 (P < 0.001 and P = 0.56, respectively; Figure 3C and D). Combining both inhibitors had the same effect of ATMi alone.
Figure 3.
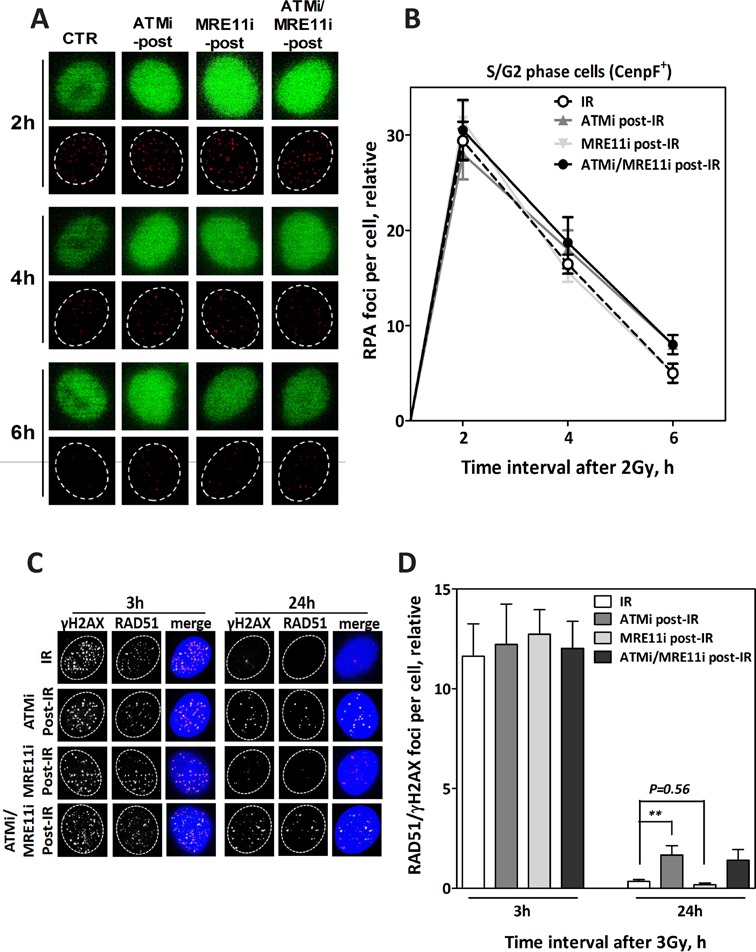
ATM but not MRE11 functions in HR after end resection and RAD51 nucleofilament formation. (A) Representative IF photos for RPA foci in CenpF+ cells (S/G2) after inhibition of ATM (ATMi post-IR), MRE11 (MRE11i post-IR) or both (ATMi/MRE11i post-IR) 2 h after irradiation with 2 Gy. (B) Quantification of RPA foci in CenpF+ cells (n = 100) at the indicated time points after 2 Gy. Inhibition of ATM or MRE11 post-IR did not affect the number of RPA foci at the indicated time points compared to the control cells (CTR). (C) Representative IF photos for RAD51 foci in A549 cells after inhibition of ATM (ATMi post-IR), MRE11 (MRE11i post-IR) or both (ATMi/MRE11i post-IR) 2 h after irradiation with 2 Gy. (D) Quantification of RAD51 foci co-localized with γH2AX foci in 100 cells. Inhibition of ATM, MRE11 or both post-IR had no effect on the number of RAD51 foci compared at 3-h time point to solely irradiated cells (IR). At the 24-h time point post-IR, a significantly higher number of residual RAD51 foci remained upon inhibition of ATM but not MRE11 compared to the control. In all cases, the number of foci measured in non-irradiated cells was subtracted from that observed in irradiated cells. Error bars represent the SEM of three independent experiments. P value less than 0.01 was designated with two (**) asterisks.
Similar effects of ATM and MRE11 inhibition were also found with respect to cellular radiosensitivity (Supplementary Figure S3). No change in cellular radiosensitivity was shown when MRE11 was inhibited either 2 or 4 h after IR, while the inhibition of ATM applied at the same time intervals after IR clearly enhanced cellular radiosensitivity.
The greater number of residual RAD51/γH2AX foci reported after ATM inhibition post-IR might alternatively have resulted from secondary DSBs arising from perturbed replication forks (26). To exclude this possibility, the polymerase inhibitor aphidicolin (APH) was added to inhibit replication and hence the formation of secondary DSBs (27). RAD51 foci were then counted at different time points after exposure to 2 Gy in S/G2 cells (CenpF+ cells; Supplementary Figure S4A). After irradiation alone, there was a steep increase in RAD51 foci, with a peak being reached at the 4-h time point (22.1 ± 1.2 foci per cell), followed by a continuous decline down to seven foci per cell at the 8-h time point after IR (Supplementary Figure S4B). Adding ATMi prior to IR clearly affected these kinetics, with the foci showing only a moderate increase at the 2-h time point (5.6 ± 0.45) without decline up to 8 h (5.5 ± 0.57). Importantly, and in agreement with the above data (Figure 3D), the inhibition of ATM 2 h post-IR did not alter the induction of RAD51 foci at the 4-h time point, but did clearly affect its decline, as shown by the significantly larger number of RAD51 foci remaining at 8 h (11.78 ± 0.69 compared to 7 ± 0.56; P < 0.001). Since replication was blocked by APH, this higher number of RAD51 foci cannot be attributed to stalled replication forks and secondary DSBs, but rather indicates a compromised HR.
ATM is involved in HR after end resection and RAD51 nucleofilament formation in both S and G2 phases
Next, we examined the effects of ATM inhibition on HR—before and after IR specifically in S and G2 phase. Unsynchronized A549 cells were pulse-labeled with EdU before being irradiated with 2 Gy. RAD51 foci kinetics were subsequently monitored in either S-phase (EdU+/CenpF+) or G2-phase cells (EdU−/CenpF+) as previously described (6,23) (Supplementary Figure S5A and B). In G2-phase cells, the kinetics of RAD51 foci measured after 2 Gy (Figure 4A) were similar to those shown above (Supplementary Figure S4B), with a maximum being reached at 4 h and continuous decline thereafter. Adding ATMi pre-IR strongly affected these kinetics, with only a small increase in RAD51 foci followed by a plateau up to 8 h after IR (Figure 4A). In contrast, when ATMi was applied 2 h post-IR, no change was observed in the induction of RAD51 foci, with a maximum level identical to that of control cells (i.e. at 4-h time point). Importantly, however, the decline in RAD51 was clearly impaired, with a significantly larger number of RAD51 foci remaining at 6-, 8- and 10-h time points (P = 0.003,P = 0.002 andP = 0.002, respectively) after IR. This increase in the number of remaining RAD51 foci was associated with a substantial increase in the number of residual γH2AX foci at the 10-h time point (P = 0.003) (Figure 4B).
Figure 4.
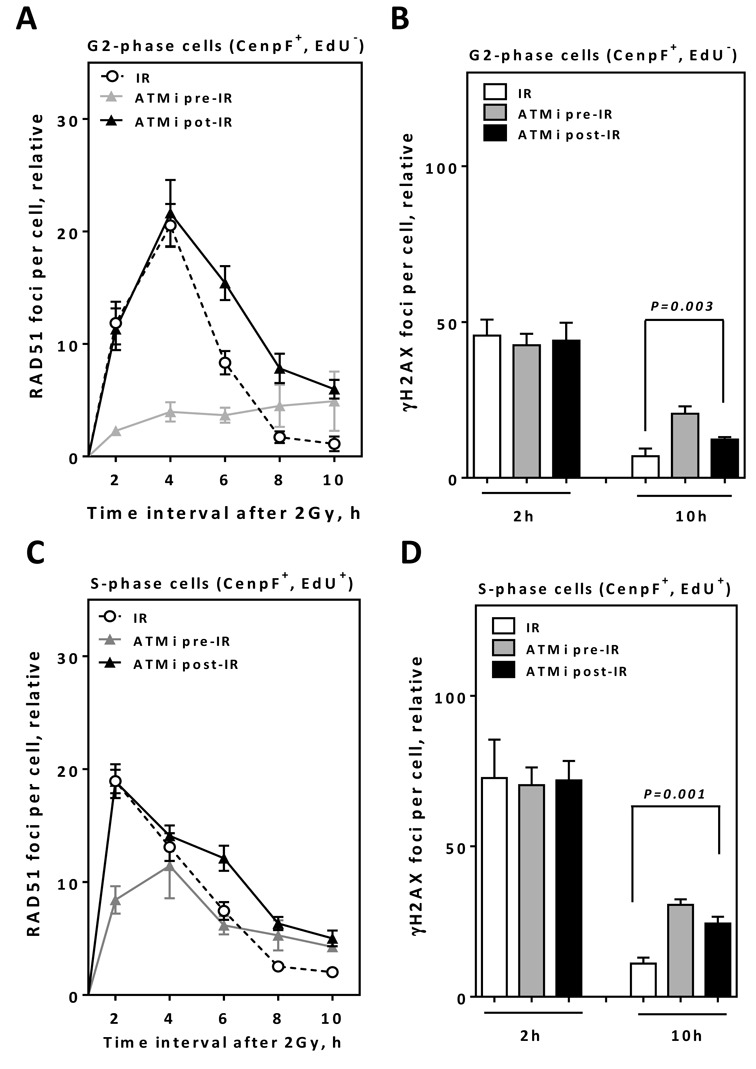
ATM inhibition after end resection results in higher numbers of residual RAD51/γH2AX foci in both S and G2 cells. (A) A549 cells were EdU pulse labeled, irradiated with 2 Gy and the kinetics of RAD51 foci numbers were monitored in CenpF+/EdU− G2-cells at the indicated time points. (B) γH2AX foci were evaluated at the indicated time points in CenpF+/EdU− G2-cells. (C) The kinetics of RAD51 foci numbers were monitored in CenpF+/EdU+ S-cells at the indicated time points. (D) γH2AX foci were evaluated in CenpF+/EdU+ S-cells at the indicated time points. In all cases, the number of foci measured in non-irradiated cells was subtracted from that observed in irradiated cells. Error bars represent the SEM of three independent experiments.
In S-phase cells, RAD51 foci increased rapidly in number, with a peak at 2 h after IR (18.9 ± 1.5). The foci then start to decline in number and reaching a minimum at the 10-h time point (2.0 ± 0.13). The fact that the maximum number of RAD51 foci was reached 2 h earlier than in G2-phase cells (Figure 4A) suggests that either end resection is faster in S-phase or that a sub-fraction of DSBs in S-phase might not require end resection before RAD51 loading. Notably, the effect of ATM inhibition on RAD51 foci kinetics in S-phase prior to IR was less pronounced than that observed in the G2 phase (Figure 4A and C), probably because through initiation of HR by ATR in S-phase (7). However, the number of RAD51 foci remaining 10 h after IR was higher than that observed for control cells (P = 0.001). Importantly, the inhibition of ATM 2 h post-IR resulted in a comparable number of RAD51 foci at 2-h time point compared to uninhibited cells (P = 0.699), but evoked significantly higher number of foci remaining at the 6-, 8- and 10-h time points (P = 0.002, P < 0.0001 and P = 0.0001, respectively) (Figure 4C). The increased number of residual RAD51 foci was associated with a substantial increase in residual γH2AX foci at the 10-h time point (P = 0.001) (Figure 4D). Altogether, these data suggest not only that the residual RAD51 foci observed in Supplementary Figure S4 are not an indirect effect of cell synchronization (i.e. by APH), but more importantly suggest the requirement of ATM for the later steps in HR in both S and G2 phases.
Role of ATM in HC DSB repair is functions independently of its role on HR after RAD51 nucleofilament formation
Previously, it was demonstrated that heterochromatic DSBs are generally repaired more slowly than euchromatic DSBs and that ATM is specifically required for DSB repair within HC through the phosphorylation and removing of KAP1 from the HC (28). Thus, we tested whether the impairment in HR reported upon ATM inactivation after end resection is related to ATM's role in the repair of HC-DSBs. To address this point, KAP1 was depleted in A549 cells using siRNA (Supplementary Figure S6A) as previously described (19) and RAD51 foci were monitored 4 and 8 h after IR and upon inhibition of ATM either—before or after IR. The siRNA-mediated KAP1 knockdown did not alter the number of RAD51 foci at either the 4- or 8-h time point after IR (Supplementary Figure S6B). In KAP1-depleted cells, the inhibition of ATM pre-IR led to a decrease in the number of RAD51 foci at the 4-h time point compared to uninhibited cells (Supplementary Figure S6B). Interestingly, however, this number was higher when compared to ATM inhibition alone (12.7 ± 0.88 versus 5.6 ± 0.44, respectively). This finding confirms previous data showing that the negative effect of ATM inhibition on HR can be partly rescued when KAP1 is depleted (19). No decline in the number of RAD51 foci up to 8-h time point was observed when ATM was inhibited prior to exposure to IR in KAP1-depleted cells. Upon inhibition of ATM post-end resection, there was no difference between KAP1 normal and depleted cells. In both cases, the loading of RAD51 foci (measured at 4 h) was not affected, but importantly, a greater number of residual RAD51 foci remained at the 8-h time point (P = 0.01). Altogether, these data reveal that the role of ATM in HR after the presynapsis stage is not related to its function on HC via KAP1.
Inhibition of ATM following end resection leads to HR deficiency
To further confirm that the inhibition of ATM following end resection results in HR deficiency, we made use of the pDSRed-I-SceI-GR plasmid (29) (Figure 5A, upper panel), which expresses an I-SceI endonuclease fused to a DSRed domain. I-SceI is also fused to a glucocorticoid receptor (GR) ligand-binding domain, which allows for the nuclear localization of the plasmid upon addition of triamcinolone acetonide (TA). Having established that MRE11 is involved exclusively in the end resection step, we first sought to determine the exact length of time that MRE11 is actually needed for HR, a time point which indicates the completion of end resection. To this end, HeLa cells carrying pGC were transfected with pDSRed-ISceI-GR and 24 h later, MRE11 nuclease activity was inhibited either 1 h prior to or 2, 6 and 12 h after TA treatment. As shown in Figure 5A, HR was significantly suppressed upon inhibition of MRE11 prior to TA treatment (0.05 ± 0.009 compared to 0.218 ± 0.01; P = 0.001). The inhibitory effect on HR was partly relieved when MRE11 was inhibited 2 and 6 h post-TA treatment, and was completely abrogated when MRE11 was inhibited 12 h after exposure to TA. These data indicate that DSB end resection by MRE11 is fully completed 12 h after the addition of TA. Importantly, when ATM was inhibited at the 12-h time point following treatment with TA, HR was still found to be reduced by more than 50% (Figure 5A, black columns). In line with the data in Figure 1, ATM inhibition prior to DSB induction strongly reduced HR. Overall, these data clearly confirm that in contrast to MRE11, ATM continues to be involved in HR after DSB end resection.
Figure 5.
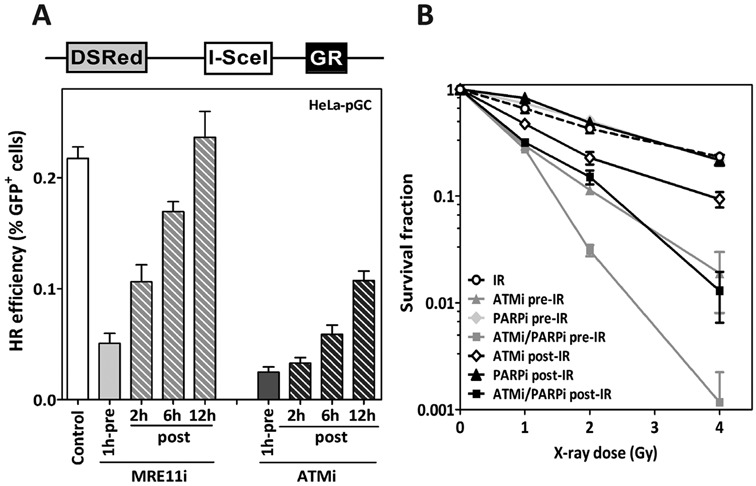
ATM inhibition after end resection leads to HR deficiency and increases radiosensitization after PARP inhibition. (A) Upper panel: Schematic representation of the inducible I-SceI-expressing vector pDSRed-I-SceI-GR. Addition of triamcinolone (TA) leads to the nuclear localization of the I-SceI enzyme. Lower panel: HeLa cells harboring the pGC reporter were transfected with pDSRed-I-SceI-GR. Twenty-four hours later, MRE11 or ATM was inhibited either 2 h prior to TA treatment (pre) or 2 , 6 or 12 h after TA (post). GFP+ cells were evaluated 48 h after I-SceI-transfection as an indication of HR efficiency. (B) Radiosensitivity of exponentially growing A549 cells was measured by colony forming assay after inhibition of ATM (10-μM KU55933) and/or PARP (1-μM Olaparib) either prior to (-pre) or after (-post) the indicated X-ray doses. Error bars represent the SEM of three independent experiments.
HR in G2 leads to SCEs (22). Indeed, SCE is significantly reduced upon inhibition of ATM at 2-h time point after 2 Gy (Supplementary Figure S7A and B). HR-deficient cells can be selectively radiosensitized by inhibition of PARP (30). To further validate the HR deficiency mediated by ATM inhibition post-DSB induction, ATM and PARP were inhibited either individually or simultaneously 1 h prior to or 2 h after exposure to IR, with the effects on cell survival subsequently being monitored by colony assay. As illustrated in Figure 5B, the inhibition of ATM pre-IR enhanced the radiosensitivity of A549 cells. The inhibition of PARP alone by 1-μM Olaparib did not affect the radiosensitivity of A649 cells, as previously described (27), but did clearly enhance cellular radiosensitivity when combined with ATM inhibitor prior to IR. A similar but less pronounced effect on cellular radiosensitivity was found when both ATM and PARP were inhibited (Figure 5B) post-IR. The Olaparib-mediated radiosensitization effect was not associated by any increase in apoptosis level (Supplementary Figure S8). This observation confirms that the inhibition of ATM after exposure to IR leads to HR deficiency and, as a consequence, cells can be radiosensitized by PARPi.
ATR can partly compensate for the role of ATM in HR prior to, but not after, RAD51 nucleofilament formation
Both ATM and ATR are members of PI3K kinase family and can function redundantly in the DDR after DSB induction by IR (7). Although a strong reduction in HR efficiency has been reported upon ATM inhibition prior to the induction of DSBs, cells are still able to perform HR to some extent (Figure 1). Therefore, we asked whether ATR can even partly alleviate the deficiency in HR resulting from ATM inhibition. In line with our previous data (6), ATR inactivation by a specific inhibitor (0.02-μM VE-821) led to an inhibition of HR in HeLa-pGC cells, though to a lesser extent than after ATM inactivation (0.81 ± 0.09 and 0.29 ± 0.006, respectively). The combined inactivation of both kinases showed a more pronounced suppressive effect on HR (0.07 ± 0.02), indicating that both ATM and ATR can partly compensate for the other's absence, with the absence of both resulting in a more severe inhibitory effect on HR (Figure 6A).
Figure 6.
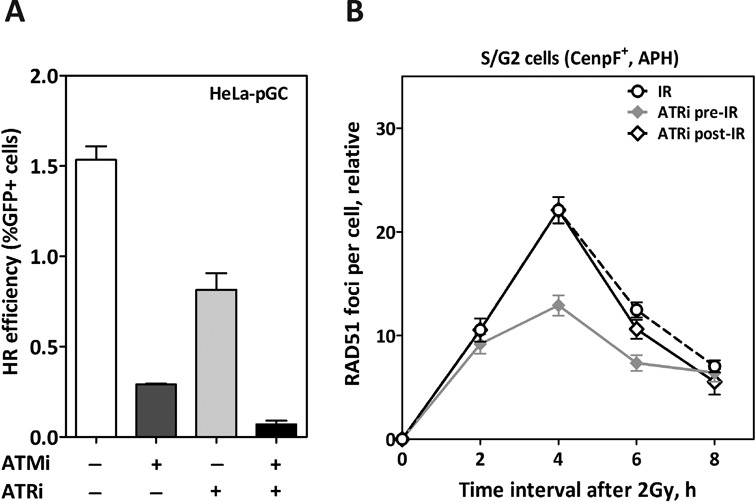
ATR can partially compensate for ATM before but not after RAD51 nucleofilament formation. (A) HR efficiency was measured as in Figure 1A in HeLa cells after the inhibition of ATM (10-μM KU55933), ATR (0.02-μM VE-821) or both. (B) The kinetics of RAD51 foci numbers were monitored in CenpF+ cells after inhibition of ATR and 1 h prior to (ATRi pre-IR) or 4 h after (ATRi post-IR) irradiation. Five-micromolar aphidicolin (APH) was added to block the transition of S-phase cells into G2. In all cases, the number of foci measured in non-irradiated cells was subtracted from that observed in irradiated samples. Error bars represent the SEM of three independent experiments.
Next, we asked whether ATR compensates for the absence of ATM in the early or later steps of HR. To this end, RAD51 foci kinetics were measured in the S/G2 phase upon ATR inhibition either before or after the irradiation of A549 cells with 2 Gy. ATR inhibition 2 h prior to IR did not alter the number of RAD51 foci seen during the first 2 h after IR, but clearly led to a reduction in the number of RAD51 foci at 4-h time point (2-fold). RAD51 foci then declined to reach similar numbers of uninhibited cells at the 8-h time point (P = 0.56). This indicates that ATR is required for the early steps of HR, i.e. for RAD51 nucleofilament formation. In order to investigate whether ATR is also involved in later steps of HR after RAD51 nucleofilament formation, ATR inhibitor (ATRi) was added 4 h post-IR, a time point at which the number of RAD51 foci normally reaches its peak, indicating that RAD51 nucleofilament formation is almost complete. Our data revealed no change in the declining number of RAD51 foci at 6 and 8 h upon ATR inhibition post-IR as compared to uninhibited cells (P = 0.11 and P = 0.09, respectively), indicating that ATR is not required for later steps of HR. Altogether, these data demonstrate that ATR may partially compensate for the absence of ATM in early but not in later steps of HR.
DISCUSSION
The current work reports for the first time that ATM is involved in HR not only in presynapsis through the stimulation of DSB end resection, but importantly also in the later steps of HR after end resection and RAD51 nucleofilament formation (Figure 7).
Figure 7.
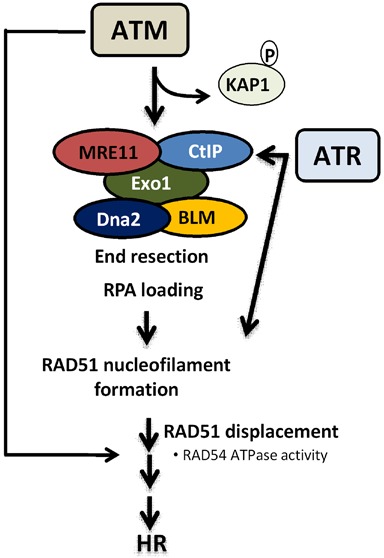
Proposed model for the role of ATM in HR. ATM is indeed required for the presynapsis stage of HR: (i) to phosphorylate and remove KAP1, facilitating the recruitment of HR proteins; (ii) to regulate the end resection step by stimulating the nucleolytic activity of MRE11/CtIP to generate 3′-ssDNA overhangs and facilitate RAD51 nucleofilament formation; (iii) in addition, we describe here for the first time that ATM is also required for HR after RAD51 nucleofilament formation. In the absence of ATM, ATR can only participate in the early steps of HR, possibly through the stimulation of end resection and RAD51 nucleofilament formation.
ATM is critical for the HR pathway during the presynapsis stage to (i) prepare the chromatin structure by opening the HC area, thus facilitating the recruitment of HR proteins, and (ii) to directly regulate the end resection step through the stimulation of enzymatic activity and recruitment of nucleases such as MRE11 and CtIP to generate 3′-ssDNA overhangs and hence facilitate RAD51 nucleofilament formation (14,18,31). We report here that ATM is additionally involved in the later steps of HR after end resection and RAD51 loading (i.e. after presynapsis). This is evidenced by the findings that RAD51 loading is not affected upon inhibition of ATM after end resection has been initiated. Instead, cells exhibited (i) a greater number of residual RAD51 foci co-localized with γH2AX foci after 2 Gy (Figure 3D), (ii) a clear impairment in RAD51 displacement from the DSB sites in both the S and G2 phases (Supplementary Figure S2 and Figure 4), (iii) a substantial reduction in HR efficiency as measured by the specific HR repair pGC substrate (Figure 5B), (iv) a reduced SCE (Supplementary Figure S7) and (v) a radiosensitization mediated by PARP inhibition (Figure 5B).
It has been previously reported that ATM is specifically required for HR taking place within HC via the phosphorylation and hence transient removal of KAP1 from the HC, thus facilitating the recruitment of CtIP/MRE11 and in turn initiating HR (28). In line with this, knockdown of KAP1 partially rescued the deficiency in RAD51 loading in ATM-deficient cells ((28) and Supplementary Figure S6). Here we show that ATM's role in HC-associated HR is dispensable for the later function of ATM in HR, as the siRNA-mediated depletion of KAP-1 did not alleviate the need for ATM in later steps of HR after RAD51 nucleofilament formation (Supplementary Figure S6).
There are at least two possibilities to explain the function of ATM in the later steps of HR. ATM might be involved in the phosphorylation of RAD51 via c-Abl, which has been shown to be associated with and activated by ATM (32–35). So far, the biological effects of this phosphorylation are not yet clear, as c-Abl-mediated phosphorylation negatively affected RAD51′s activity in one set of experiments reported (35), but enhanced its association with RAD52 (a putative modulator of RAD51) in another (34). Alternatively, ATM might directly or indirectly regulate RAD51 displacement from break ends to allow for further steps in HR (i.e. postsynapsis). One promising candidate for this function is RAD54, which is known to stabilize the RAD51 nucleofilament in an early step of HR (36) and later to remove RAD51 from DNA ends (37,38). The later function was described to specifically depend on its ATPase activity (39), which is also required for its own displacement from the ends (40). Interestingly, it has been demonstrated that RAD54 accumulates significantly in ATM-deficient cells (39), indicating a role for ATM in regulating the ATPase activity of RAD54. Consequently, upon inactivation of ATM, RAD51 cannot be efficiently displaced from the break ends due to impaired RAD54 function. In line with this assumption, Kirshner et al. (10) reported that RAD54 knockout can rescue the survival of ATM-deficient cells after IR.
In addition, we demonstrate here that after ATM inhibition, ATR can partly alleviate the deficiency in end resection and RAD51 loading, supporting a previously described model in which ATM and ATR collaborate together to maintain the activity of CtIP for efficient end resection during HR (41). Indeed, ATR was shown to promote a robust DSB end resection through the phosphorylation of CtIP at T818, even in ATM-deficient cells (41). Consistent with this, the simultaneous down-regulation of ATM and ATR completely abolished HR efficiency as measured by a plasmid assay and RAD51 foci after IR (Figure 6 and (6)). In contrast, we found that ATR was not involved in the later steps of HR, as evidenced by normal clearance of RAD51 after ATR was inhibited following completion of RAD51 nucleofilament formation (Figure 6B).
In summary, these data place ATM in two stages in HR. In addition to its known function in the initiation of HR through the activation of the end resection step, ATM also plays a role in HR in the later steps (i.e. after end resection and RAD51 nucleofilament formation), probably during RAD51 removal.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank Jennifer Volquardsen and Alexandra Zielinski for their valuable technical support.
FUNDING
Funding for open access charge: UKE-Hamburg and BMBF(# 02NUK035B).
Conflict of interest statement. None declared.
REFERENCES
- 1.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 2.Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 3.Khanna K.K., Lavin M.F., Jackson S.P., Mulhern T.D. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ. 2001;8:1052–1065. doi: 10.1038/sj.cdd.4400874. [DOI] [PubMed] [Google Scholar]
- 4.Foray N., Priestley A., Alsbeih G., Badie C., Capulas E.P., Arlett C.F., Malaise E.P. Hypersensitivity of ataxia telangiectasia fibroblasts to ionizing radiation is associated with a repair deficiency of DNA double-strand breaks. Int. J. Radiat. Biol. 1997;72:271–283. doi: 10.1080/095530097143266. [DOI] [PubMed] [Google Scholar]
- 5.Helleday T., Lo J., van Gent D.C., Engelward B.P. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007;6:923–935. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Kocher S., Rieckmann T., Rohaly G., Mansour W.Y., Dikomey E., Dornreiter I., Dahm-Daphi J. Radiation-induced double-strand breaks require ATM but not Artemis for homologous recombination during S-phase. Nucleic Acids Res. 2012;40:8336–8347. doi: 10.1093/nar/gks604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serrano M.A., Li Z., Dangeti M., Musich P.R., Patrick S., Roginskaya M., Cartwright B., Zou Y. DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair. Oncogene. 2013;32:2452–2462. doi: 10.1038/onc.2012.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huber A., Bai P., de Murcia J.M., de Murcia G. PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair. 2004;3:1103–1108. doi: 10.1016/j.dnarep.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Menisser-de Murcia J., Mark M., Wendling O., Wynshaw-Boris A., de Murcia G. Early embryonic lethality in PARP-1 Atm double-mutant mice suggests a functional synergy in cell proliferation during development. Mol. Cell. Biol. 2001;21:1828–1832. doi: 10.1128/MCB.21.5.1828-1832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirshner M., Rathavs M., Nizan A., Essers J., Kanaar R., Shiloh Y., Barzilai A. Analysis of the relationships between ATM and the Rad54 paralogs involved in homologous recombination repair. DNA Repair. 2009;8:253–261. doi: 10.1016/j.dnarep.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Kocher S., Spies-Naumann A., Kriegs M., Dahm-Daphi J., Dornreiter I. ATM is required for the repair of Topotecan-induced replication-associated double-strand breaks. Radiother. Oncol. 2013;108:409–414. doi: 10.1016/j.radonc.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 12.Mazon G., Mimitou E.P., Symington L.S. SnapShot: homologous recombination in DNA double-strand break repair. Cell. 2010;142:646.e1. doi: 10.1016/j.cell.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeda S., Nakamura K., Taniguchi Y., Paull T.T. Ctp1/CtIP and the MRN complex collaborate in the initial steps of homologous recombination. Mol. Cell. 2007;28:351–352. doi: 10.1016/j.molcel.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Huertas P., Jackson S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265:1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- 17.Paques F., Haber J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Bosch M., Lohman P.H., Pastink A. DNA double-strand break repair by homologous recombination. Biol. Chem. 2002;383:873–892. doi: 10.1515/BC.2002.095. [DOI] [PubMed] [Google Scholar]
- 19.Geuting V., Reul C., Lobrich M. ATM release at resected double-strand breaks provides heterochromatin reconstitution to facilitate homologous recombination. PLoS Genet. 2013;9:e1003667. doi: 10.1371/journal.pgen.1003667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shibata A., Moiani D., Arvai A.S., Perry J., Harding S.M., Genois M.M., Maity R., van Rossum-Fikkert S., Kertokalio A., Romoli F., et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasten-Pisula U., Menegakis A., Brammer I., Borgmann K., Mansour W.Y., Degenhardt S., Krause M., Schreiber A., Dahm-Daphi J., Petersen C., et al. The extreme radiosensitivity of the squamous cell carcinoma SKX is due to a defect in double-strand break repair. Radiother. Oncol. 2009;90:257–264. doi: 10.1016/j.radonc.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Conrad S., Kunzel J., Lobrich M. Sister chromatid exchanges occur in G2-irradiated cells. Cell Cycle. 2011;10:222–228. doi: 10.4161/cc.10.2.14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beucher A., Birraux J., Tchouandong L., Barton O., Shibata A., Conrad S., Goodarzi A.A., Krempler A., Jeggo P.A., Lobrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mansour W.Y., Schumacher S., Rosskopf R., Rhein T., Schmidt-Petersen F., Gatzemeier F., Haag F., Borgmann K., Willers H., Dahm-Daphi J. Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Acids Res. 2008;36:4088–4098. doi: 10.1093/nar/gkn347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mansour W.Y., Bogdanova N.V., Kasten-Pisula U., Rieckmann T., Kocher S., Borgmann K., Baumann M., Krause M., Petersen C., Hu H., et al. Aberrant overexpression of miR-421 downregulates ATM and leads to a pronounced DSB repair defect and clinical hypersensitivity in SKX squamous cell carcinoma. Radiother. Oncol. 2013;106:147–154. doi: 10.1016/j.radonc.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Wyman C., Kanaar R. DNA double-strand break repair: all's well that ends well. Annu. Rev. Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 27.Kotter A., Cornils K., Borgmann K., Dahm-Daphi J., Petersen C., Dikomey E., Mansour W.Y. Inhibition of PARP1-dependent end-joining contributes to Olaparib-mediated radiosensitization in tumor cells. Mol. Oncol. 2014;8:1616–1625. doi: 10.1016/j.molonc.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodarzi A.A., Noon A.T., Deckbar D., Ziv Y., Shiloh Y., Lobrich M., Jeggo P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 29.Rieckmann T., Kriegs M., Nitsch L., Hoffer K., Rohaly G., Kocher S., Petersen C., Dikomey E., Dornreiter I., Dahm-Daphi J. p53 modulates homologous recombination at I-SceI-induced double-strand breaks through cell-cycle regulation. Oncogene. 2013;32:968–975. doi: 10.1038/onc.2012.123. [DOI] [PubMed] [Google Scholar]
- 30.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 2011;5:387–393. doi: 10.1016/j.molonc.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain S., Sugawara N., Lydeard J., Vaze M., Tanguy Le Gac N., Haber J.E. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev. 2009;23:291–303. doi: 10.1101/gad.1751209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shafman T., Khanna K.K., Kedar P., Spring K., Kozlov S., Yen T., Hobson K., Gatei M., Zhang N., Watters D., et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387:520–523. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- 33.Baskaran R., Wood L.D., Whitaker L.L., Canman C.E., Morgan S.E., Xu Y., Barlow C., Baltimore D., Wynshaw-Boris A., Kastan M.B., et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387:516–519. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- 34.Chen G., Yuan S.S., Liu W., Xu Y., Trujillo K., Song B., Cong F., Goff S.P., Wu Y., Arlinghaus R., et al. Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J. Biol. Chem. 1999;274:12748–12752. doi: 10.1074/jbc.274.18.12748. [DOI] [PubMed] [Google Scholar]
- 35.Yuan Z.M., Huang Y., Ishiko T., Nakada S., Utsugisawa T., Kharbanda S., Wang R., Sung P., Shinohara A., Weichselbaum R., et al. Regulation of Rad51 function by c-Abl in response to DNA damage. J. Biol. Chem. 1998;273:3799–3802. doi: 10.1074/jbc.273.7.3799. [DOI] [PubMed] [Google Scholar]
- 36.Mazin A.V., Alexeev A.A., Kowalczykowski S.C. A novel function of Rad54 protein. Stabilization of the Rad51 nucleoprotein filament. J. Biol. Chem. 2003;278:14029–14036. doi: 10.1074/jbc.M212779200. [DOI] [PubMed] [Google Scholar]
- 37.Solinger J.A., Kiianitsa K., Heyer W.D. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol. Cell. 2002;10:1175–1188. doi: 10.1016/s1097-2765(02)00743-8. [DOI] [PubMed] [Google Scholar]
- 38.Kiianitsa K., Solinger J.A., Heyer W.D. Terminal association of Rad54 protein with the Rad51-dsDNA filament. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9767–9772. doi: 10.1073/pnas.0604240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morrison C., Sonoda E., Takao N., Shinohara A., Yamamoto K., Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 2000;19:463–471. doi: 10.1093/emboj/19.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agarwal S., van Cappellen W.A., Guenole A., Eppink B., Linsen S.E., Meijering E., Houtsmuller A., Kanaar R., Essers J. ATP-dependent and independent functions of Rad54 in genome maintenance. J. Cell Biol. 2011;192:735–750. doi: 10.1083/jcb.201011025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peterson S.E., Li Y., Wu-Baer F., Chait B.T., Baer R., Yan H., Gottesman M.E., Gautier J. Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol. Cell. 2013;49:657–667. doi: 10.1016/j.molcel.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.