

Abstract
Colorectal cancer (CRC) is a major cause of morbidity and mortality around the world, and approximately 5% of them develop in a context of inherited mutations leading to some form of familial colon cancer syndromes. Recognition and characterization of these patients have contributed to elucidate the genetic basis of CRC. Polyposis Syndromes may be categorized by the predominant histological structure found within the polyps. The aim of the present paper is to review the most important clinical features of the Hamartomatous Polyposis Syndromes, a rare group of genetic disorders formed by the peutz-Jeghers syndrome, juvenil polyposis syndrome and PTEN Hamartoma Tumor Syndrome (Bannayan-Riley-Ruvalacaba and Cowden Syndromes). A literature search was performed in order to retrieve the most recent and important papers (articles, reviews, clinical cases and clinical guidelines) regarding the studied subject. We searched for terms such as “hamartomatous polyposis syndromes”, “Peutz-Jeghers syndrome”, “juvenile polyposis syndrome”, “juvenile polyp”, and “PTEN hamartoma tumour syndrome” (Cowden syndrome, Bananyan-Riley-Ruvalcaba). The present article reports the wide spectrum of disease severity and extraintestinal manifestations, with a special focus on their potential to develop colorectal and other neoplasia. In the literature, the reported colorectal cancer risk for Juvenile Polyposis, Peutz-Jeghers and PTEN Hamartoma Tumor Syndromes are 39%-68%, 39%-57% and 18%, respectively. A review regarding cancer surveillance recommendations is also presented.
Keywords: Hereditary GI cancer syndromes, Peutz-Jeghers, Juvenile polyposis, Cowden syndrome, PTEN tumor
Core tip: This is a brief review about clinical presentation, diagnosis, molecular features and surveillance recommendations regarding hamartomatous polyposis syndromes: Peutz-jeghers syndrome, juvenil polyposis Syndrome and PTEN Hamartoma Tumor Syndrome (Bannayan-Riley-Ruvalacaba and Cowden Syndromes).
INTRODUCTION
Colorectal polyps may be histologically classified as neoplastic, hyperplastic, hamartomatous or inflammatory. Some of these polyps may develop sporadically or as part of a polyposis syndrome. Hereditary Polyposis Syndromes account for approximately 1% of all cases of colorectal cancer (CRC) and are associated with a broad spectrum of extra-colonic tumors. Each syndrome has its own genetic basis, polyp histology and distribution, clinical features, and malignancy risk.
Taking into account the histological nature of the polyp, the gastrointestinal syndromes may derive from adenomas (familial adenomatous polyposis, MutYH-associated polyposis), from hyperplastic polyps (serrated polyposis syndrome), from hamartomas [Peutz-Jeghers Syndrome (PJS), Juvenile Polyposis Syndrome (JPS), PTEN Hamartoma Tumor Syndrome] or from mixed polyps (Hereditary Mixed Polyposis Syndrome).
Hamartomatous polyp usually appear macroscopically as pedunculated, cherry-red lesions. They vary in size and its characteristic histological structure allows the distinction between a Peutz-Jeghers and Juvenile Polyp[1]. Peutz-Jeghers polyps (Figure 1) are tipically multilobulated with a papillary surface and branching bands of smooth muscle covered by hyperplastic glandular mucosa. A Juvenile Polyp (Figure 2) exhibits a normal epithelium with a dense stroma, an inflammatory infiltrate and a smooth surface with dilated, mucus-filled cystic glands in the lamina propria. For this reason, it might be difficult to distinguish it from an inflammatory polyp.
Figure 1.
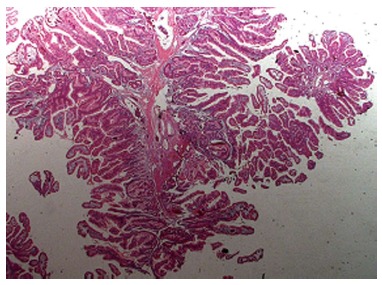
Histological features of a Peutz-Jeghers polyp. Note that they are typically multilobulated with a papillary surface and branching bands of smooth muscle covered by hyperplastic glandular mucosa.
Figure 2.
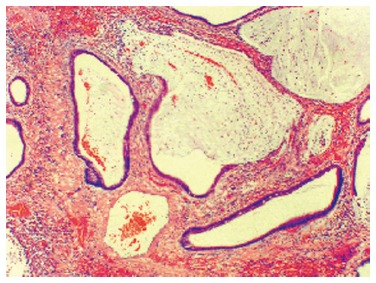
A Juvenile Polyp exhibiting a normal epithelium with a dense stroma, an inflammatory infiltrate and a smooth surface with dilated, mucus-filled cystic glands in the lamina propria.
The clinical significance of the Hamartomatous Polyposis Syndromes lies on their association with colorectal and other extracolonic malignancies (gastrointestinal, urogenital, breast and thyreoid)[2]. Thus, knowledge of their genetic basis and clinical expressions help establish diferential diagnosis and allow the construction of screening, surveillance and treatment recomendations, that should differ from the general population.
Genetic data and prevalence of PJS, JPS and PTEN Hamartoma Tumor Syndrome (Bannayan-Riley-Ruvalacaba and Cowden Syndromes) are presented in Table 1.
Table 1.
Genetic features and prevalence of pure Hamartomatous Polyposis Syndromes
| Syndrome | Mode of inheritance | Gene | Incidence |
| Juvenile Polyposis | AD | SMAD4/DPC4 BMPR1A | 1:100 to 1:160 thousand |
| Peutz-Jeghers | AD | STK11/LKB1 | 1:60 mil a 1:300 thousand |
| BRRS | AD | PTEN | Rare |
| Cowden | AD | PTEN, SDH and KLLN epimutations | 1:200 thousand |
BRRS: Bannayan-Riley-Ruvalacaba syndrome; AD: Autosomal dominant; SDH: Succinate dehydrogenase (B and C subunits); KLLN: p53 target gene.
The aim of the present paper was to review the most important clinical features of the Hamartomatous Polyposis Syndromes, focusing on their potential to develop neoplasia, especially colorectal. This review was based on a literature search in order to retrieve the most recent and important papers (articles, reviews, clinical cases and clinical guidelines) regarding the subject. We searched for terms such as “hamartomatous polyposis syndromes”, “PJS”, “JPS”, “juvenile polyp”, and “PTEN hamartoma tumour syndrome” (Cowden syndrome, Bananyan-Riley-Ruvalcaba).
Table 2 presents the main clinical features and the reported malignancies described in association with these syndromes, revealing how heterogeneous this group is regarding polyp distribution and neoplasia risks.
Table 2.
Clinical features and colon cancer risk in Hamartomatous Polyposis Syndromes, according to literature series
| Syndrome | Main clinical features polyp distribution | Increased risk of other tumors | Colon cancer risk |
| Juvenile Polyposis | Juvenile polyps Distribution: large bowel (mainly), small bowel, stomach | Gastric and colorectal | 39%-68% |
| Peutz-Jeghers | Peutz-Jeghers polyps Typical melanotic oral and dermic pigmentations Distribution: small bowel, large bowel, stomach | Gastric, small bowel, pancreas, colorectal, ovary, uterus, breasts, sex cords | 39%-57% |
| PTEN | Mucocutaneous tumors (multiple trichilemmomas) Distribution: Small bowel, large bowel, stomach | Breast, thyroid, retina and uterus cancer | 18% |
The Hereditary Mixed Polyposis Syndrome is not discussed here cause this entity encompass polyps with distinct histologies (adenomas, serrated, hyperplastic, juvenile, mixed juvenile-adenomatous or hyperplastic adenomatous)[3]. In the same context, other syndromes where hamartomatous polyps are present (Multiple endocrine neoplasia type 2B, Gorlin, Neurofibromatosis type 1, Birt-Hogg-Dubbé and Cronkhite-Canada) have not either been included in this revision.
PJS
History and genetics
The association of mucosal pigmentation and gas trointestinal polyposis was first described by the English Sir Jonathan Huchinson in 1896. Although this condition has received many denominations throughout time, it was only after the work of the Dutch Peutz[4] (1886-1957) in 1921 and the American Jeghers[5] (1944), who firmed the disease features, that this association was nominated PJS.
Gastrointestinal polyps from PJS present distinct features from those found in other Hamartomatous Syndromes, such as the presence of a muscular component infiltrating the conective tissue in a pattern of ramification. Although a good pathologist should be able suggest the diagnosis based on histology, the establishment of a hamartomatous polyposis syndrome should be based on molecular features, as clinical manifestations may differ slightly.
PJS is inherited by an autosomal-dominant gene that is responsible either by the polyposis and the pigmentation. Nevertheless, some isolated cases have been reported. The genetic mutation occurs in a supressor gene that codifies the serina/threonina kinase (LKB1 ou STK11), located in chromossome 19p13.3[6]. Germline mutations of this gene lead to hamartoma formation, and other somatic mutations may transform hamartomas into adenomas and subsequently carcinomas[7]. The multiple mutations identified in gene LKB1 are responsible by the phenotypic variability of PJS, including the development of aggressive cases and other that never developed cancer.
Clinical features
PJS is characterized by the triad mucocutaneous melanic pigmentation, intestinal polyposis and familial history. Diagnostic criteria of PJS include two or more hamartomatous polyps in the gastrointestinal tract or one confirmed Peutz-Jeghers polyp with a family history of PJS or typical perioral pigmentation[8].
The pigmentation is manifested by dark black or blue spots around the lips, eyes and extremities (hands and feet), but are also found in the neck, thorax and perineum. They are formed by smooth melanin deposits in a round or oval shape, rarely confluent, with a 1 cm maximal diameter (Figure 3). They may appear since the neonatal period or even after the begining of the gastrointestinal symptoms[9].
Figure 3.
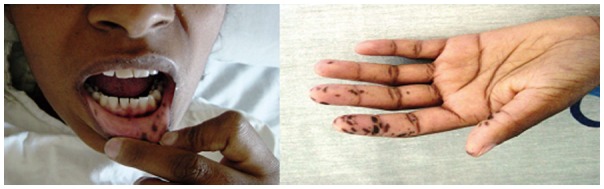
Mucocutaneous pigmentation in Peutz-Jeghers Syndrome.
The most important clinical manifestations are secondary to the polyps, that may affect the small bowel (70%-95%), colon (27%), stomach (25%) and colorectum (24%-50%); the jejunum is more commonly involved than duodenum and ileum[10]. Gastrointestinal symptoms usually develop during the second and third decades, with abdominal pain resulting from hiperperistalism or polyp invagination. PJS polyps may may also cause obstruction, prolapse through the rectum, bleeding and anemia. Isolated polyps may rarely develop in the absence of other clinical features and are not associated with gastrointestinal cancer risk[11].
Risk of malignancy
Since its classical description in 1944[5], numerous cases of PJS associated with gastrointestinal (duodenum, jejunum, pancreas, stomach and colon) or extra-intestinal carcinomas (breast, ovary, cervix, thiroid, lung, pancreas and testicles) have been reported[2]. The supposed carcinogenesis is based on the controversial idea that the hamartomas may develop carcinomas as adenomatous and malignant alteration have been described in hamartomas[12,13].
It’s been estimated that lifetime risk of any gastrointestinal cancer approaches 70% (mainly colorectal at 39% and pancreatic at 36%). Additional tumors (breast, sex chord in females, adenoma malignum of the cervix, Sertoli cell tumors of the tests, etc.) increase patient’s lifetime risk to near 90%[14,15].
In a Dutch group of 133 PJS from 54 families, Van Lier et al[16] found 37% cancers, and CRC was the most common malignancy (14%). Compared to the general population, this report confirms a 9 fold increased cancer risk, a higher risk among women (20 fold) compared to men (5 fold), a 3.5 fold increased mortality rate and that gastrointestinal cancers develop at young age. In a recent paper, Beggs et al[17] reported a high rate of extracolonic tumors such as gastric (29%), small bowel (13%), pancreatic (36%), breast (54%), ovarian (21%), lung (15%), cervical (10%) and uterine/testicular (9% each).
In another paper[18], CRC turned to be the most common luminal gastrointestinal cancer (17/40) among 419 patients with 297 documented mutations, with a cumulative risk of 3%, 5%, 15% and 39% at ages 40, 50, 60 and 70 years, respectively (Table 3). The risk of developing cancer at any site was four fold that observed in the general population. In females with PJS, the risk of breast cancer was also increased six fold over the population and is comparable to the BRCA mutations.
Table 3.
Cumulative cancer risk by site and age in Peutz-Jeghers Syndrome (Hearle et al[18])
| Cancer/Age | 20 yr | 30 yr | 40 yr | 50 yr | 60 yr | 70 yr |
| All cancers | 2 | 5 | 17 | 31 | 60 | 85 |
| Gastrointestinal | - | 1 | 9 | 15 | 33 | 57 |
| Breast | - | - | 8 | 13 | 31 | 45 |
| Gynecological | - | 1 | 3 | 8 | 18 | 18 |
| Pancreas | - | - | 3 | 5 | 7 | 11 |
| Lung | - | - | 2 | 4 | 13 | 17 |
Similarly, in a metanalysis to evaluate the risk of many tumors, Giardiello et al[19] grouped 107 men and 106 women from 79 families, and reported estimated cumulative cancer risks of 54% for breast, 39% for colorectal, 36% for pancreas, 29% for stomach and 21% for ovarian cancer by 64 years of age.
Management of PJS is based on the treatment of symptomatic benign conditions, large polyps and surveillance of malignant tumors. For this reason, endoscopic resection of polyps larger than 1.5 cm is advisible, even in asymptomatic patients. Patients scheduled to a conservative follow-up must undergo periodic examination after 30 years of age, with bienal evaluation of superior and inferior digestive tract, anual pelvic, testicular and abdominal ultrasound (mainly for pancreas) and anual mammography after 25 years Family member should be equally examinated[20].
JPS
Genetics and history
JPS is a rare genetic disease that exhibits incomplete penetrance and heterogeneity, with positive familiar history appearing in only 20% to 50% of patients. There were described germinative mutations in the SMAD4 (MADH4) (chromosome 18q21.1) and in the BMPR1A (chromosome 10q 21-22) genes[21,22]. The genetic mutations have not been identified in all cases of JPS. SMAD4 mutations are more common and predispose to polyposis in the upper digestive tract[23]. BMPR1A mutations are found in 40%-100% of families without SMAD4 mutation.
Pathological features of polyps in children were described many years ago, at the same time when the term juvenile polyp was coined by Horrilleno et al[24] in 1957. But it was Morson in 1962 who established those polyps as hamartomas[25], and McColl et al[26] in 1964 defined the JPS as a distinct entity.
Clinical features
When discovered as isolated sigmoid or rectal lesions during infancy, Juvenile polyps may cause bleeding, hematochezia, intussusception, or even self-amputation (Figure 4). In this cases, the risk of malignization is very low. Once recognized, they should undergo endoscopic resection.
Figure 4.
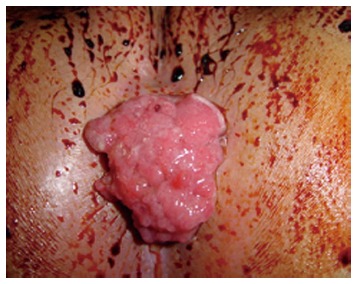
Prolapsed polyp through the anus in a patient with Juvenile Polyposis.
On the other hand, development of JPS is much more less frequent, being characterized by numerous hamartomatous polyps in the intestine and other parts of the gastrointestinal tract. Diagnostic criteria include: (1) more than 5 juvenile polyps in the colorectum; and (2) multiple juvenile polyps throughout the gastrointestinal tract or one or more polyp and a positive family history of juvenile polyposis[27-29].
During infancy, the polyposis may affect all the digestive tract, and the prognosis is dependent on this involvement (referred as JP of infants). These cases are not associated with familiar history[28]. Within the other forms of the disease, the polyposis may appear during the second or third decades, more rarely (15%) in adults. Within the gastrointestinal tract, the most affected sites are the colorectum (98%), stomach (14%), jejunum/ileum (7%) and duodenum (2%)[29]. Similarly, in 262 patients with PJS, Höfting et al[30] reported colorectal, gastric and intestinal lesions in 98%, 13.6% and 8.8% of them, respectively.
Some patients may refer familar history suggesting an autosomal dominant pattern of inheritance[31]. Some congenital abnormalities have been described in 15%-20% (midgut malrotation, cardiac anomalies, cleft palate, supranumerary teeth, macrocephaly, hydrocephalus, polydactyly, mesenteric lymphangioma, etc.), mainly in patients not referring familiar history. SMAD4 mutations are associated with JPS and hereditary hemorrhagic telangiectasia, and some carriers may present symptoms from both conditions. Conective tissue disorders have been documented in approximately one-fifth of these patients, such as enlarged aortic root, aortic and mitral insufficiency, aortic dissection and others[32].
Risk of malignancy
Carcinomas from many locations have been reported within a wide variation of lifetime cumulative cancer risk[33,34]. The estimated lifetime risk of gastrointestinal cancer in JPS family members varies from 9% to 50%[22]. Although most of these tumors consist of colon cancer, tumors arising in the stomach, upper gastrointestinal tract and pancreas have also been reported. The estimated risk for CRC is 17%-22% by age 35[35] and a lifetime risk of gastric and duodenal cancer of 10%-21%[15,36].
Specialized centres have reported adenomatous features or adenomas associated with juvenile polyps in 2 a 15% of the patients, suggesting a possible histogenical mechanism to carcinogenesis[33,37,38]. Otherwise, it is not known if those adenomas are formed through a total conversion of a juvenile polyp or if they represent “de novo” lesions.
Isolated juvenile polyps should be endoscopically or surgically excised, depending on location. In PJS patients, regular endoscopic examinations is considered a more conservative approach after 15 years of age. There is a tendency to manage the patient according with symptoms severity and polyp features (number, accelerated growing and displasia). In the case of few polyps, polypectomy is indicated. A prophylactic colectomy (Ileal-rectal anastomosis or pouch surgery has been advocated by others, especially in patients with adenomatous features, displasia and a strong history of CRC[39,40].
Some studies showed that up to half of patients required a completion proctectomy after initial total colectomy. Annual endoscopic surveillance of the rectum and ileal mucosa is advisable after surgery in order to detect recurrent polyps. First-degree relatives must be screened by colonoscopy from the second decade of life up to the age of 70[15,22,31].
PTEN HAMARTOMATOUS TUMOR SYNDROME
Genetics and clinical features
PTEN Hamartomatous Tumor Syndrome (PHTS) groups patients diagnosed with either Cowden (CS) or Bannayan-Riley-Ruvalcaba syndromes (BRRS). Both are inherited in an autosomal dominant pattern and develop due to mutations of the PTEN gene (phosphatase and tensin homolog), a tumor suppressor gene located on 10q23.3. PTEN mutations have been recently found in only 25% of CS patients. Other patients were described as having SDH gene mutations (succinate dehydrogenase B and C) or KLLN epimutations in 10% and 30% of the cases, respectively[41].
While BRRS is usually diagnosed during infancy, CS prevails in adults. Mucocutaneous features allow early recognition of CS, manifesting before the neoplastic changes. They appear in 80% of the patients and are represented by multiple facial triquilemomas, oral mucosa papilomatosis and hand queratosis (Figure 5). Colorectal polyps are small, sessile and asymptomatic, being found in 35%-65% of patients[42].
Figure 5.
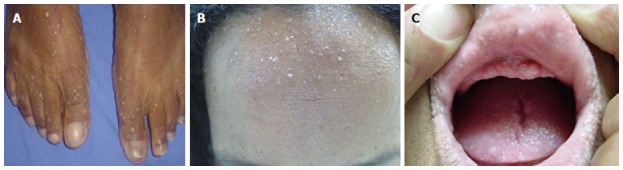
Feet queratosis (A), multiple facial triquilemomas (B) and oral mucosa papilomatosis (C) in a patients with Cowden’s Syndrome.
Cowden’s syndrome should be screened for the development of various cancers, such as thyroid (10%), breasts (30%-50%), endometrium and colorectal. Less than 10% of patients develop Central Nervous System tumors[43].
BRRS is characterized by intestinal polyposis (45% of patients) associated with dermatological lesions (pigmented macules of the glans penis)[44]. Extraintestinal manifestations have been described such as macrocephaly, subcutaneous lipomas, vascular malformations, high birth weight and central nervous system anomalies[45].
Cancer risks in PHTS
CRC risk in PHTS has been evaluated in the past few years. In a study of 127 patients with PTEN mutations (62 colonoscopies), Heald et al[46] found a wide spectrum of polyps and 13% CRC diagnosed in patients under 50 years of age. In a multi-national cohort of 3399 patients with CS (368 with PTEN mutations), Tan et al[47] reported a significantly increased incidence of CRC (10 fold), breast (20 fold), thyroid (50 fold), endometrium (40 fold), kidney (30 fold) and melanoma (8 fold).
In a group of 156 patients from 101 families with PTEN mutations, Nieuwenhuis et al[48] reported a cumulative risk of 70% for benign gastrointestinal polyps and 18% for CRC at age 60, respectively. This three to four-fold increase in CRC risk led the authors to recommend colonoscopy after 40 years of age.
Recommendations for screening and surveillance
Besides rare, recognition and screening of any Hamartomatous Polyposis Syndromes is a great deal for the patient, as these disorders may manifest important complications due to polyp bleeding or intestinal obstruction. Family members at risk should be fully evaluated after the second decade of life even if they are asymptomatic.
Once diagnosis is established, upper and lower endoscopic investigation (as well as radiological images) should be performed every 2 to 5 years[42,46]. Moreover, especial attention should be driven to extraintestinal malignancies at risk such as breasts, thyroid, uterus and others[47].
Gastrointestinal surveillance aims to reduce the polyp burden, its complications and cancer development. Furthermore, polyp management may reduce surgical intervention and prevent resection or emergency surgery, as demonstrated for PJS[49]. As the chance of malignant degeneration of colonic polyps has also been recognized in all hamartomatous polyposis syndromes, screening colonoscopy should be advised for all patients. Current recommendations for screening and surveillance according to recent publications[17,40,48,50,51] are resumed in Table 4.
Table 4.
| Syndrome | Screening | Work-up | Interval |
| Peutz-Jeghers | 18-25 yr | Endoscopy (upper/lower) | 2-3 yr |
| 25 yr | MRI and mammography | Annual | |
| 10 yr | Testicular examination | Annual | |
| 30 yr | MRI or CT (pancreas) | 1-2 yr | |
| Juvenile Polyposis | 15-18 yr | Upper endoscopy | 1-3 yr |
| Colonoscopy | 1-3 yr | ||
| Upper endoscopy and video capsule endoscopy for HHT | 3 yr | ||
| PTEN | After 25 yr | Colonoscopy Mamography/thyroid US | 3-5 yr Annual |
US: Ultrasound; CT: Computorized tomography; MRI: Magnetic resonance imaging; HHT: Hereditary hemorrhagic telangiectasia.
Surveillance of the breast, colon and rectum and the small intestines should be established for PJS patients[51]. After comparing surveillance programs already published, Beggs et al[17] proposed to postpone the gastrointestinal screening till the late teens, with repeated exams each three years till 50 years of age (and each 1-2 years thereafter). Colonoscopy should be performed every 2-5 years from 25 years of age.
Recommendations regarding JPS families include colonoscopy every 1-2 years starting at 15-18 years and upper endoscopy with a 1-2 year interval from 25 years of age[22,52]. The group from the St. Mark’s Hospital[53] showed that colonic polyps predominated in the right colon and that carpeting disease represents a special concern. They recommend upper and lower gastrointestinal endoscopy every 1-3 years starting at 12 years. Moreover, they advise annul full blood count and cardiovascular examination and screening for HHT (hereditary-hemorrhagic telangiectasia) symptoms (mainly A-V malformations) in SMAD4 mutation carriers.
Finally, PTEN-mutations carriers are suggested to perform dermatological examination, neurological, psychological testing, and thyroid ultrasound from the late teens. After 30 years, women should undergo annual mammogram, endometrial examination and transvaginal ultrasound[47]. Biannual colonoscopy is advised after 40 years of age[48].
Footnotes
Conflict-of-interest: All authors declare no conflicts of interest (including commercial, personal, political, intellectual, or religious interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 24, 2014
First decision: December 17, 2014
Article in press: February 9, 2015
P- Reviewer: Barreto S, Lu F, Riss S S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Shussman N, Wexner SD. Colorectal polyps and polyposis syndromes. Gastroenterol Rep (Oxf) 2014;2:1–15. doi: 10.1093/gastro/got041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marsh D, Zori R. Genetic insights into familial cancers-- update and recent discoveries. Cancer Lett. 2002;181:125–164. doi: 10.1016/s0304-3835(02)00023-x. [DOI] [PubMed] [Google Scholar]
- 3.Stojcev Z, Borun P, Hermann J, Krokowicz P, Cichy W, Kubaszewski L, Banasiewicz T, Plawski A. Hamartomatous polyposis syndromes. Hered Cancer Clin Pract. 2013;11:4. doi: 10.1186/1897-4287-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peutz JLA. Very remarkable case of familial polyposis of mucous membrane of intestinal tract and nasopharynx accompanied by peculiar pigmentations of skin and mucous membrane. Ned Maandschr Gneesk. 1921;10:134–146. [Google Scholar]
- 5.Jeghers H. Pigmentation of the skin. N Engl J Med. 1944;231:181–184. [Google Scholar]
- 6.Hemminki A, Tomlinson I, Markie D, Järvinen H, Sistonen P, Björkqvist AM, Knuutila S, Salovaara R, Bodmer W, Shibata D, et al. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat Genet. 1997;15:87–90. doi: 10.1038/ng0197-87. [DOI] [PubMed] [Google Scholar]
- 7.Miyaki M, Iijima T, Hosono K, Ishii R, Yasuno M, Mori T, Toi M, Hishima T, Shitara N, Tamura K, et al. Somatic mutations of LKB1 and beta-catenin genes in gastrointestinal polyps from patients with Peutz-Jeghers syndrome. Cancer Res. 2000;60:6311–6313. [PubMed] [Google Scholar]
- 8.Tomlinson IP, Houlston RS. Peutz-Jeghers syndrome. J Med Genet. 1997;34:1007–1011. doi: 10.1136/jmg.34.12.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habr-Gama A, Gama-Rodrigues JJ, Warde P. Síndrome de Peutz-Jeghers. Apresentação de dois casos. Arq Gastroent S Paulo. 1975;12:53–62. [Google Scholar]
- 10.Corredor J, Wambach J, Barnard J. Gastrointestinal polyps in children: advances in molecular genetics, diagnosis, and management. J Pediatr. 2001;138:621–628. doi: 10.1067/mpd.2001.113619. [DOI] [PubMed] [Google Scholar]
- 11.Oncel M, Remzi FH, Church JM, Goldblum JR, Zutshi M, Fazio VW. Course and follow-up of solitary Peutz-Jeghers polyps: a case series. Int J Colorectal Dis. 2003;18:33–35. doi: 10.1007/s00384-002-0411-x. [DOI] [PubMed] [Google Scholar]
- 12.Campos FG, Borba M, Habr-Gama A, Pinotti HW. Síndrome de Peutz-Jegher e Carcinoma Colônico. Relato de Um Caso e Revisão Bibliográfica. Rev Bras Coloproct. 1993;12:91–96. [Google Scholar]
- 13.Golberg JE, Rafferty JF. Other polyposis syndromes. Clin Colon Rectal Surg. 2002;15:113–119. [Google Scholar]
- 14.McGrath DR, Spigelman AD. Preventive measures in Peutz-Jeghers syndrome. Fam Cancer. 2001;1:121–125. doi: 10.1023/a:1013896813918. [DOI] [PubMed] [Google Scholar]
- 15.Dunlop MG. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polypolis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut. 2002;51 Suppl 5:V21–V27. doi: 10.1136/gut.51.suppl_5.v21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Lier MG, Westerman AM, Wagner A, Looman CW, Wilson JH, de Rooij FW, Lemmens VE, Kuipers EJ, Mathus-Vliegen EM, van Leerdam ME. High cancer risk and increased mortality in patients with Peutz-Jeghers syndrome. Gut. 2011;60:141–147. doi: 10.1136/gut.2010.223750. [DOI] [PubMed] [Google Scholar]
- 17.Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, Bertario L, Blanco I, Bülow S, Burn J, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975–986. doi: 10.1136/gut.2009.198499. [DOI] [PubMed] [Google Scholar]
- 18.Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott RJ, Lim W, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12:3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 19.Giardiello FM, Welsh SB, Hamilton SR. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987;316:1511–1514. doi: 10.1056/NEJM198706113162404. [DOI] [PubMed] [Google Scholar]
- 20.Wirtzfeld DA, Petrelli NJ, Rodriguez-Bigas MA. Hamartomatous polyposis syndromes: molecular genetics, neoplastic risk, and surveillance recommendations. Ann Surg Oncol. 2001;8:319–327. doi: 10.1007/s10434-001-0319-7. [DOI] [PubMed] [Google Scholar]
- 21.Eng C, Ji H. Molecular classification of the inherited hamartoma polyposis syndromes: clearing the muddied waters. Am J Hum Genet. 1998;62:1020–1022. doi: 10.1086/301847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998;5:751–756. doi: 10.1007/BF02303487. [DOI] [PubMed] [Google Scholar]
- 23.Friedl W, Uhlhaas S, Schulmann K, Stolte M, Loff S, Back W, Mangold E, Stern M, Knaebel HP, Sutter C, et al. Juvenile polyposis: massive gastric polyposis is more common in MADH4 mutation carriers than in BMPR1A mutation carriers. Hum Genet. 2002;111:108–111. doi: 10.1007/s00439-002-0748-9. [DOI] [PubMed] [Google Scholar]
- 24.Horrilleno EG, Eckert C, Ackerman LV. Polyps of the rectum and colon in children. Cancer. 1957;10:1210–1220. doi: 10.1002/1097-0142(195711/12)10:6<1210::aid-cncr2820100619>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 25.Morson BC. Some pecularities in the histology of intestinal polyps. Dis Colon Rectum. 1962;5:337–344. [Google Scholar]
- 26.Mccoll I, Busxey HJ, Veale AM, Morson BC. Juvenile polyposis coli. Proc R Soc Med. 1964;57:896–897. [PubMed] [Google Scholar]
- 27.Gardner EJ. A genetic and clinical study of intestinal polyposis: a predisposing factor for carcinoma of the colon and rectum. Am J Genet. 1951;3:167–176. [PMC free article] [PubMed] [Google Scholar]
- 28.Sachatello CR, Hahn IS, Carrington CB. Juvenile gastrointestinal polyposis in a female infant: report of a case and review of the literature of a recently recognized syndrome. Surgery. 1974;75:107–114. [PubMed] [Google Scholar]
- 29.Chow E, Macrae F. A review of juvenile polyposis syndrome. J Gastroenterol Hepatol. 2005;20:1634–1640. doi: 10.1111/j.1440-1746.2005.03865.x. [DOI] [PubMed] [Google Scholar]
- 30.Höfting I, Pott G, Stolte M. The syndrome of juvenile polyposis. Leber Magen Darm. 1993;23:107–108, 111-112. [PubMed] [Google Scholar]
- 31.Desai DC, Neale KF, Talbot IC, Hodgson SV, Phillips RK. Juvenile polyposis. Br J Surg. 1995;82:14–17. doi: 10.1002/bjs.1800820106. [DOI] [PubMed] [Google Scholar]
- 32.Wain KE, Ellingson MS, McDonald J, Gammon A, Roberts M, Pichurin P, Winship I, Riegert-Johnson DL, Weitzel JN, Lindor NM. Appreciating the broad clinical features of SMAD4 mutation carriers: a multicenter chart review. Genet Med. 2014;16:588–593. doi: 10.1038/gim.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jass JR, Williams CB, Bussey HJ, Morson BC. Juvenile polyposis--a precancerous condition. Histopathology. 1988;13:619–630. doi: 10.1111/j.1365-2559.1988.tb02093.x. [DOI] [PubMed] [Google Scholar]
- 34.Brosens LA, Langeveld D, van Hattem WA, Giardiello FM, Offerhaus GJ. Juvenile polyposis syndrome. World J Gastroenterol. 2011;17:4839–4844. doi: 10.3748/wjg.v17.i44.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005;100:476–490. doi: 10.1111/j.1572-0241.2005.40237.x. [DOI] [PubMed] [Google Scholar]
- 36.Järvinen H, Franssila KO. Familial juvenile polyposis coli; increased risk of colorectal cancer. Gut. 1984;25:792–800. doi: 10.1136/gut.25.7.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bussey HJR. Familial polyposis coli: family studies, histopathology, diferencial diagnosis and results of treatment. Baltimore, Maryland: Johns Hopkins University Press; 1975. [Google Scholar]
- 38.Agnifili A, Verzaro R, Gola P, Marino M, Mancini E, Carducci G, Ibi I, Valenti M. Juvenile polyposis: case report and assessment of the neoplastic risk in 271 patients reported in the literature. Dig Surg. 1999;16:161–166. doi: 10.1159/000018711. [DOI] [PubMed] [Google Scholar]
- 39.Järvinen HJ. Genetic testing for polyposis: practical and ethical aspects. Gut. 2003;52 Suppl 2:ii19–ii22. doi: 10.1136/gut.52.suppl_2.ii19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cichy W, Klincewicz B, Plawski A. Juvenile polyposis syndrome. Arch Med Sci. 2014;10:570–577. doi: 10.5114/aoms.2014.43750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J Clin Endocrinol Metab. 2011;96:E2063–E2071. doi: 10.1210/jc.2011-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Omundsen M, Lam FF. The other colonic polyposis syndromes. ANZ J Surg. 2012;82:675–681. doi: 10.1111/j.1445-2197.2012.06140.x. [DOI] [PubMed] [Google Scholar]
- 43.Fistarol SK, Anliker MD, Itin PH. Cowden disease or multiple hamartoma syndrome--cutaneous clue to internal malignancy. Eur J Dermatol. 2002;12:411–421. [PubMed] [Google Scholar]
- 44.Cohen MM. Bannayan-Riley-Ruvalcaba syndrome: renaming three formerly recognized syndromes as one etiologic entity. Am J Med Genet. 1990;35:291–292. doi: 10.1002/ajmg.1320350231. [DOI] [PubMed] [Google Scholar]
- 45.Hanssen AM, Fryns JP. Cowden syndrome. J Med Genet. 1995;32:117–119. doi: 10.1136/jmg.32.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heald B, Mester J, Rybicki L, Orloff MS, Burke CA, Eng C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology. 2010;139:1927–1933. doi: 10.1053/j.gastro.2010.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400–407. doi: 10.1158/1078-0432.CCR-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nieuwenhuis MH, Kets CM, Murphy-Ryan M, Colas C, Möller P, Hes FJ, Hodgson SV, Olderode-Berends MJ, Aretz S, Heinimann K, et al. Is colorectal surveillance indicated in patients with PTEN mutations? Colorectal Dis. 2012;14:e562–e566. doi: 10.1111/j.1463-1318.2012.03121.x. [DOI] [PubMed] [Google Scholar]
- 49.Latchford AR, Neale K, Phillips RK, Clark SK. Peutz-Jeghers syndrome: intriguing suggestion of gastrointestinal cancer prevention from surveillance. Dis Colon Rectum. 2011;54:1547–1551. doi: 10.1097/DCR.0b013e318233a11f. [DOI] [PubMed] [Google Scholar]
- 50.Canzonieri C, Centenara L, Ornati F, Pagella F, Matti E, Alvisi C, Danesino C, Perego M, Olivieri C. Endoscopic evaluation of gastrointestinal tract in patients with hereditary hemorrhagic telangiectasia and correlation with their genotypes. Genet Med. 2014;16:3–10. doi: 10.1038/gim.2013.62. [DOI] [PubMed] [Google Scholar]
- 51.Jelsig AM, Qvist N, Brusgaard K, Nielsen CB, Hansen TP, Ousager LB. Hamartomatous polyposis syndromes: a review. Orphanet J Rare Dis. 2014;9:101. doi: 10.1186/1750-1172-9-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cairns SR, Scholefield JH, Steele RJ, Dunlop MG, Thomas HJ, Evans GD, Eaden JA, Rutter MD, Atkin WP, Saunders BP, et al. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002) Gut. 2010;59:666–689. doi: 10.1136/gut.2009.179804. [DOI] [PubMed] [Google Scholar]
- 53.Latchford AR, Neale K, Phillips RK, Clark SK. Juvenile polyposis syndrome: a study of genotype, phenotype, and long-term outcome. Dis Colon Rectum. 2012;55:1038–1043. doi: 10.1097/DCR.0b013e31826278b3. [DOI] [PubMed] [Google Scholar]