

The nuclear receptor superfamily includes retinoid, thyroid hormone, steroid and peroxisome proliferator-activated (PPAR) receptors. Unlike plasma membrane receptors that signal through second messengers, nuclear receptors can function directly as transcription factors that control gene transcription. A historical example of nuclear receptor ligands that activate these “classical” or “genomic” pathway to inhibit pain are the steroidal anti-inflammatory drugs. More recently, two isoforms of PPAR, namely PPARα and PPARγ, have received significant interest as analgesic targets for chronic pain [4]. Administration of synthetic PPARα and PPARγ ligands reduce behavioral signs of allodynia and hyperalgesia in a number of pain models [1; 6; 13]. Similarly, antihyperalgesic effects are produced by endogenously-generated PPAR activators. Of particular importance to pain research are the fatty acid ethanolamides, palmitoylethanolamide (PEA) and oleoylethanolamide (OEA), both of which bind with high affinity to PPARα [12]. In neurons, glia, and inflammatory cells, PEA and OEA are not stored, but rather are made on demand -- endogenous levels are regulated by the relative activity of biosynthetic and degradative enzymes. Animal studies convincingly demonstrate that PEA exerts a broad spectrum pain inhibition that can be reversed with PPARα antagonists and this inhibition does not occur in deletion mutant mice lacking PPARα [6]. Fig 1A illustrates a potential mechanism through which PPARα mediates the antihyperalgesic actions of PEA.
Figure 1.
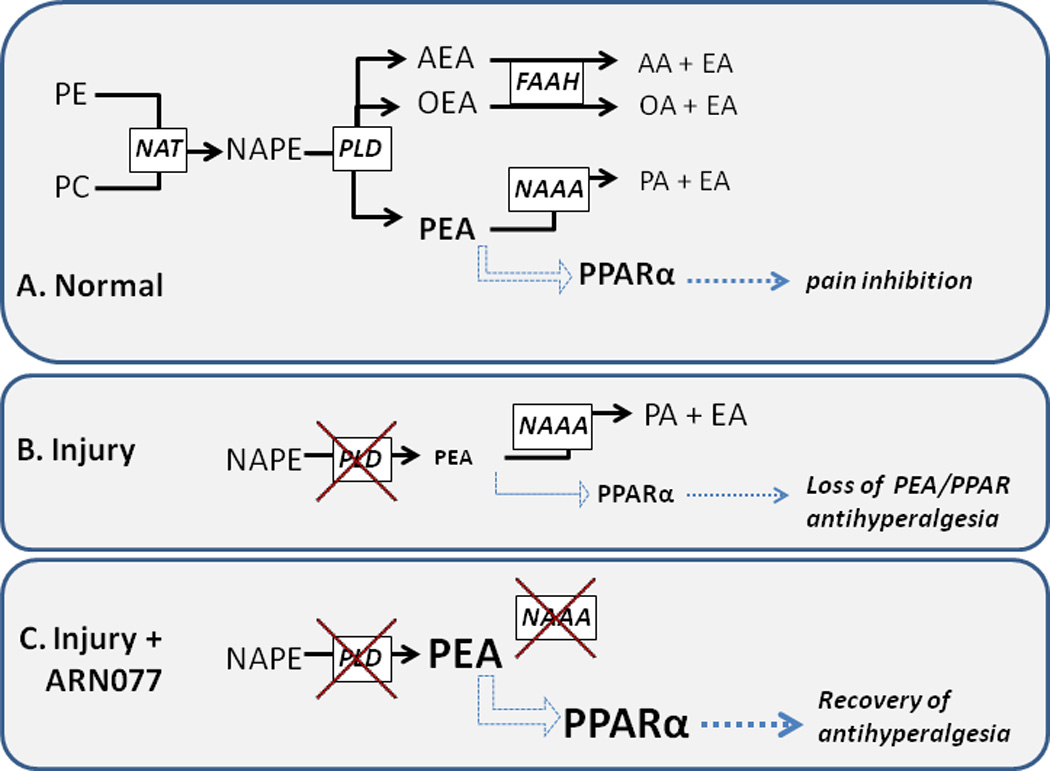
Proposed mechanism of pain inhibition by N-acylethanolamine acid amidase (NAAA). Panel A: key enzymatic pathways for fatty acid ethanolamide synthesis and degradation (solid black arrows), and a proposed mechanism of pain inhibition involving palmitoylethanolamide (PEA) activation of PPARα (blue arrows), and PPARα-mediated pain inhibition (green arrows). Panel B: Injury-induced inhibition of the enzyme that synthesizes PEA (N-acyl-phosphatidylethanolamine phospholipase D, NAPE-PLD) and ensuing loss of PEA-PPARα antihyperalgesia. Panel C: Inhibition of N-acylethanolamine acid amidase (NAAA) with the novel compound ARN077 raises PEA concentrations, thus reinstating PPARα antihyperalgesia.
PE, phosphatidylethanolamine. PC, phosphatidylcholine. NAT, N-acetyltransferase. NAPE, N-acylphosphatidylethanolamines. AEA, arachidonoylethanolamide (anandamide). OEA, oleoylethanolamide. AA, arachidonic acid. OA, oleic acid. PA, palmitic acid. EA, ethanolamine.
Palmitoylethanolamide is approved in some countries (e.g. Italy) as a dietary supplement in humans, and preliminary but intriguing clinical trials and case studies suggest that oral PEA is effective for a variety of pain syndromes [7]. Unfortunately, the analgesic potential of direct PPARα activators, synthetic or natural, has not been met. Due to the pleiotropic nature of PPAR action, currently available synthetic ligands designed to activate PPARα directly have yielded undesired off-target effects [8]. PEA is not very potent (doses close to 1 g are typically administered) and its analgesic efficacy (magnitude of pain reduction) is far from powerful, perhaps because PEA concentrations are not adequate in key target tissues. In this issue of Pain, Sasso et al. [11] provide a solution to this problem, with an approach that is designed to increase the intrinsic concentrations of PEA. Their compelling new strategy arises from a longstanding discovery that inhibition of fatty acid amine hydrolase (FAAH) increases levels of fatty acid ethanolamides (FAE), notably anandamide (Fig 1A). The anandamide, in turn, exerts an analgesic action at cannabinoids receptors. Not surprisingly, those findings led to an intensive effort towards the clinical development of FAAH inhibitors for chronic pain [2]. But in addition to FAAH, fatty acid ethanolamides can be hydrolyzed by an assortment of enzymes, notably N-acylethanolamine acid amidase (NAAA), the primary enzyme involved in the hydrolysis of PEA [15]. NAAA hydrolyzes PEA to palmitic acid and ethanolamine, with much greater efficacy and selectivity than FAAH – the latter efficiently hydrolyzes OEA in addition to anandamide (Fig 1A). However, as NAAA was only recently cloned, in 2005[14], in contrast to the many potent and selective FAAH inhibitors now available [9], NAAA inhibitors have only recently begun to emerge [3]. Sasso et al. [11] take advantage of a new, potent and selective compound, ARN077, to test the hypothesis that NAAA inhibitors can increase endogenous PEA, and thus reduce hyperalgesia.
Fatty acid ethanolamides are formed and then released from membrane glycerophospholipids through the phosphodiesterase-transacylation pathway. Fig 1A includes a simplified scheme of the most widely-accepted enzymatic pathways for FAE synthesis and degradation in neurons and immune cells. Fig 1B illustrates that inflammatory injury suppresses the enzyme that generates fatty acid ethanolamides, thus stopping the production of FAEs, including PEA [16]. As illustrated in Fig 1C, Sasso et al [11] selectively inhibits NAAA, thus reinstating PEA concentrations. The resulting increase in PEA-mediated PPARα activation then generates antihyperalgesic actions, setting the stage for the development of a new pharmacotherapeutic target for chronic pain.
In many ways, the results of Sasso et al [11] provide an instructive example of outstanding preclinical drug development, as they include: 1) measurement in skin and nerve show that the drug does what it was designed to do – namely return depleted PEA levels back to normal concentrations; 2) full time course of behavioral responses to topical ARN007, indicating a reasonably long duration of action; 3) establishment of a full dose-response relationship (1–30% topical solution) supporting a pharmacological target; 4) demonstration of antihyperalgesic effects in multiple pain models, suggesting broad-spectrum action; 5) studies in both mice and rats, which suggests generalization across species; 6) examination of multiple routes of administration, including topical administration, which suggest opportunities for clinical translation; 7) negative controls with cannabinoid receptor antagonists, which point to a PPAR rather than cannabinoid receptor mechanism of action; 8) positive controls showing that ARN007 is more efficacious than a high dose of gabapentin.
The finding that palmitoylethanolamide produces its antihyperalgesic effects through a PPARα mechanism was initially quite puzzling. The rapid actions in vivo described previously and also in the Sasso et al [11] report do not fit with the time required for gene transcription following nuclear receptor activation. Responses mediated by the genomic pathways require time for protein synthesis, and therefore typically occur in hours to weeks, not minutes. On the other hand recent measurements of potassium chloride induced depolarization-induced Ca transients in cultured dorsal root ganglion neurons indicate that PEA and ARN077 can directly inhibit activation of sensory neurons [5]. The PPARα antagonist GW6471 blocked these actions. This finding suggests a ligand-dependent, transcription-independent receptor mechanism. Still, some important unresolved questions remain. These questions relate to the exact identity, signaling mechanisms and cellular location of the non-genomic receptors. Do they couple to G-proteins in the plasma membrane, as is the case for the non-genomic estrogen receptor, G protein-coupled estrogen receptor 1 (GPER-1, formerly GRP30) [10]. These questions await study; the development of new reagents, such as ARN077, will be critical towards the determination of specific molecular mechanisms by which PEA rapidly inhibits pain.
Footnotes
The author has no conflicts of interest
REFERENCES
- 1.Churi SB, Abdel-Aleem OS, Tumber KK, Scuderi-Porter H, Taylor BK. Intrathecal rosiglitazone acts at peroxisome proliferator-activated receptor-gamma to rapidly inhibit neuropathic pain in rats. J Pain. 2008;9(7):639–649. doi: 10.1016/j.jpain.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Marzo V. Inhibitors of endocannabinoid breakdown for pain: not so FA(AH)cile, after all. Pain. 2012;153(9):1785–1786. doi: 10.1016/j.pain.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 3.Duranti A, Tontini A, Antonietti F, Vacondio F, Fioni A, Silva C, Lodola A, Rivara S, Solorzano C, Piomelli D, Tarzia G, Mor M. N-(2-oxo-3-oxetanyl)carbamic acid esters as N-acylethanolamine acid amidase inhibitors: synthesis and structure-activity and structure-property relationships. Journal of medicinal chemistry. 2012;55(10):4824–4836. doi: 10.1021/jm300349j. [DOI] [PubMed] [Google Scholar]
- 4.Fehrenbacher JC, Loverme J, Clarke W, Hargreaves KM, Piomelli D, Taylor BK. Rapid pain modulation with nuclear receptor ligands. Brain Res Rev. 2009;60(1):114–124. doi: 10.1016/j.brainresrev.2008.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khasabova IA, Xiong Y, Coicou LG, Piomelli D, Seybold V. Peroxisome proliferator-activated receptor alpha mediates acute effects of palmitoylethanolamide on sensory neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32(37):12735–12743. doi: 10.1523/JNEUROSCI.0130-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319(3):1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- 7.Marini I, Bartolucci ML, Bortolotti F, Gatto MR, Bonetti GA. Palmitoylethanolamide versus a nonsteroidal anti-inflammatory drug in the treatment of temporomandibular joint inflammatory pain. Journal of orofacial pain. 2012;26(2):99–104. [PubMed] [Google Scholar]
- 8.Menendez-Gutierrez MP, Roszer T, Ricote M. Biology and therapeutic applications of peroxisome proliferator- activated receptors. Current topics in medicinal chemistry. 2012;12(6):548–584. doi: 10.2174/156802612799436669. [DOI] [PubMed] [Google Scholar]
- 9.Piscitelli F, Di Marzo V. "Redundancy" of endocannabinoid inactivation: new challenges and opportunities for pain control. ACS chemical neuroscience. 2012;3(5):356–363. doi: 10.1021/cn300015x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nature reviews Endocrinology. 2011;7(12):715–726. doi: 10.1038/nrendo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasso Antinociceptive effects of the N-Acetylethanolamine acid amidase inhibitor ARN077 in rodent pain models. Pain. doi: 10.1016/j.pain.2012.10.018. THE ARTICLE OF THIS COMMENTARY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, Mor M, Russo R, Maccarrone M, Antonietti F, Duranti A, Tontini A, Cuzzocrea S, Tarzia G, Piomelli D. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci U S A. 2009;106(49):20966–20971. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor BK, Dadia N, Yang CB, Krishnan S, Badr M. Peroxisome proliferator-activated receptor agonists inhibit inflammatory edema and hyperalgesia. Inflammation. 2002;26(3):121–127. doi: 10.1023/a:1015500531113. [DOI] [PubMed] [Google Scholar]
- 14.Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. The Journal of biological chemistry. 2005;280(12):11082–11092. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- 15.Ueda N, Tsuboi K, Uyama T. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA) Progress in lipid research. 2010;49(4):299–315. doi: 10.1016/j.plipres.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Zhu C, Solorzano C, Sahar S, Realini N, Fung E, Sassone-Corsi P, Piomelli D. Proinflammatory stimuli control N-acylphosphatidylethanolamine-specific phospholipase D expression in macrophages. Molecular pharmacology. 2011;79(4):786–792. doi: 10.1124/mol.110.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]