

Abstract
Background
Salt stress is one of the most representative abiotic stresses that severely affect plant growth and development. MicroRNAs (miRNAs) are well known for their significant involvement in plant responses to abiotic stresses. Although miRNAs implicated in salt stress response have been widely reported in numerous plant species, their regulatory roles in the adaptive response to salt stress in radish (Raphanus sativus L.), an important root vegetable crop worldwide, remain largely unknown.
Results
Solexa sequencing of two sRNA libraries from NaCl-free (CK) and NaCl-treated (Na200) radish roots were performed for systematical identification of salt-responsive miRNAs and their expression profiling in radish. Totally, 136 known miRNAs (representing 43 miRNA families) and 68 potential novel miRNAs (belonging to 51 miRNA families) were identified. Of these miRNAs, 49 known and 22 novel miRNAs were differentially expressed under salt stress. Target prediction and annotation indicated that these miRNAs exerted a role by regulating specific stress-responsive genes, such as squamosa promoter binding-like proteins (SPLs), auxin response factors (ARFs), nuclear transcription factor Y (NF-Y) and superoxide dismutase [Cu-Zn] (CSD1). Further functional analysis suggested that these target genes were mainly implicated in signal perception and transduction, regulation of ion homeostasis, basic metabolic processes, secondary stress responses, as well as modulation of attenuated plant growth and development under salt stress. Additionally, the expression patterns of ten miRNAs and five corresponding target genes were validated by reverse-transcription quantitative PCR (RT-qPCR).
Conclusions
With the sRNA sequencing, salt-responsive miRNAs and their target genes in radish were comprehensively identified. The results provide novel insight into complex miRNA-mediated regulatory network of salt stress response in radish, and facilitate further dissection of molecular mechanism underlying plant adaptive response to salt stress in root vegetable crops.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1416-5) contains supplementary material, which is available to authorized users.
Keywords: Radish (Raphanus sativus L.), Salt stress, MicroRNA, Target gene, RT-qPCR, High-throughput sequencing
Background
Salt stress is one of the major environmental threats that negatively affect plant growth and development. Approximately 20% of agricultural land and 50% of cropland worldwide are subjected to salt exposure [1]. Irrigation water containing trace amounts of sodium chloride (NaCl) and seawater is main source of salt in soil [2]. Increasing soil salinity lowers the ability of plant to take up water, and excess ions like Na+ and Cl− absorbed by roots are harmful to growth of plant by injuring metabolic processes and decreasing photosynthetic efficiency [3]. High salinity also imposes secondary stresses like nutritional imbalance and oxidative stress that result in cell damage, yield decrease and growth inhibition. To address these challenges, genetically engineering plants to enhance salt tolerance will be a promising approach. Therefore, deciphering the physiological processes and molecular genetic mechanism related to salt-stress resistance will certainly facilitate the understanding of complex biological responses of plants against high salinity, and help in genetically engineering of stress-resistant plants.
MicroRNAs (miRNAs) are a series of endogenous small non-coding RNA molecules, which negatively regulate gene expression at transcriptional and post-transcriptional levels by modulating both mRNA degradation and translational suppression based on sequence complementarity with their target(s) [4]. In plants, a long primary transcript known as miRNA primary precursor (pri-miRNA) is transcribed from a nuclear-encoded miRNA gene. Then, a miRNA:miRNA* duplex is released from the fold-back stem loop of miRNA precursor obtained from pri-miRNA by two cuts under the guide of Dicer-like 1 (DCL1) assisted by the dsRNA binding protein HYL1 [5]. Finally, the mature miRNA is methylated by HEN1 [6], and then bound with the argonaute protein 1 (AGO1) to form a functional special structure named RNA-induced silencing complex (RISC), which targets specific mRNAs and suppresses their expression by cleavage. Aside from the roles in modulating a wide range of essential biochemical, molecular and physiological processes, many studies reported that miRNAs were involved in plant responses to a variety of abiotic stresses such as salt [7,8], drought [9,10], heat [11,12], cold [7,13] and oxidative stress [14].
Understanding the miRNA-mediated regulatory network of salt stress response will lay the foundation for unraveling the complex molecular genetic mechanism of salt-stress tolerance. A growing body of evidences suggested that miRNA-guided gene regulation could play a vital role in plant response to salt stress. Using microarray approach, several miRNAs such as miR156, miR159, miR167, miR168, miR171, miR319 and miR396 were found to be differentially expressed after salt shock in Arabidopsis [7] and Zea mays [15]. Recently, the extensive application of next generation sequencing (NGS) technology has provided unparalleled opportunities to obtain comprehensive sequencing data for the detection of salt-responsive miRNAs in various plant species. Using this approach, Dong et al. [16] identified 104 differentially expressed miRNAs in salt-stressed functional soybean nodules. Under salt stress, seven downregulated conserved miRNA families and two upregulated miRNA families were isolated in Populus tomentosa [8]. Also in Populus euphratica, 132 miRNAs showed expression alterations during salt stress [17]. In salt-stressed Caragana intermedia, the expression of seven miRNAs including cin-miR157a, cin-miR159a, cin-miR165a, cin-miR167b, cin-miR172b, cin-miR390a and cin-miR396a were induced, while cin-miR398a was repressed [18]. Additionally, many salt-responsive miRNAs were also been identified in some vegetable crops. For instance, in soybeans, 50 miRNAs were detected to be differentially expressed under salt treatment [19]. Differentially expressed miRNAs including 11 downregulated and three upregulated ones were identified after salt stress in Solanum linnaeanum using NGS technology [20]. Moreover, 11 miRNAs were identified to be differentially regulated by abiotic stresses (heat, cold, salinity and drought) in celery [21]. Totally, 42 known and 39 candidate miRNAs were differentially expressed under salt condition in broccoli [22]. Taken together, these findings implied that miRNA-mediated gene regulatory pathways could play significant roles in plant adaptive response towards salt stress.
Radish (Raphanus sativus L., 2n = 18), belonging to the Brassicaceae family, is a globally important root vegetable crop especially in Asia [23]. Using Solexa-sequencing technology, Xu et al. [24] identified 545 conserved miRNA families and 79 novel miRNAs from radish roots. More recently, some conserved and novel miRNAs associated with cadmium stress response, embryogenesis and lead stress response were also identified in radish [25-27]. Salt stress is a limiting factor for radish that adversely influences germination, fresh weight, health-promoting compounds and antioxidant activity [28]. Therefore, exploring the regulatory mechanism responsive to salt stress will be of important significance for engineering of salt-tolerant radish germplasm. However, no investigation on identification of miRNAs and their target genes responsive to salt stress in radish has been reported to date. In this study, two small RNA (sRNA) libraries from the control (NaCl-free) and salt-treated (200 mM NaCl for 48 h) radish roots were constructed and sequenced using NGS technology. The aims were to detect salt-responsive miRNAs from radish roots, explore their roles in plant response to salt stress by predicting their target transcripts, and reveal the miRNA-mediated regulatory network of salt stress response in radish. The obtained results of this study could provide valuable information for further validating the regulatory roles of salt-responsive miRNAs in radish, and facilitate dissection of molecular mechanism underlying plant adaptive response to salt stress in radish and other root vegetable crops.
Results
Overview of transcriptome and sRNA sequencing in radish
To establish an overall reference sequence database, a cDNA library constructed from radish roots was sequenced, totally 57.03 M raw reads were generated and 130,953 contigs were obtained [NCBI Sequence Read Archive (SRA) with the GenBank accession No.SRS706782]. These mRNA transcriptome sequences, together with the available GSS (Genome Survey Sequence) and EST (Expressed Sequence Tag) sequences released in NCBI database, formed the radish reference genome for identification of known and novel miRNAs in radish, as well as the prediction of miRNA corresponding target genes.
Radish seedlings under salt treatment exhibited some negatively morphological changes including chlorisis and withering of leaves, and slight inhibition of plant growth. In this study, 18.38 M and 17.49 M raw reads were generated from CK and Na200 libraries, respectively (Table 1). After filtering out adapter contamination and low-quality tags, 18.13 M (representing 3,370,688 unique sequences) and 17.24 M (representing 4,200,793 unique sequences) clean reads were acquired from CK and Na200 libraries, respectively (Tables 1 and 2). Among them, 3,947,380 (11.16%) were CK library-specific with 2,565,929 (37.92%) unique sequences, 4,563,071 (12.90%) were only derived from Na200 library with 3,396,034 (50.19%) unique sRNAs, and 26,861,034 (75.94%) were shared in both with 804,759 (11.89%) unique sequences (Table 3).
Table 1.
Summary of cleaning data from CK and Na200 sRNA libraries of radish roots
| Type | CK | Na200 | ||
|---|---|---|---|---|
| Count | Percent (%) | Count | Percent (%) | |
| Total_reads | 18,381,536 | 17,486,008 | ||
| High_quality | 18,301,525 | 100 | 17,412,468 | 100 |
| 3′adapter_null | 2,335 | 0.01 | 2,221 | 0.01 |
| Insert_null | 1,600 | 0.01 | 4,224 | 0.02 |
| 5′adapter_contaminants | 94,403 | 0.52 | 81,947 | 0.47 |
| Smaller_than_18nt | 72,800 | 0.4 | 79,371 | 0.46 |
| Poly(A) | 1,230 | 0.01 | 2,377 | 0.01 |
| clean_reads | 18,129,157 | 99.06 | 17,242,328 | 99.02 |
Table 2.
Distribution of small RNAs among different categories in radish
| Category | CK | Na200 | ||
|---|---|---|---|---|
| Unique sRNAs | Total sRNAs | Unique sRNAs | Total sRNAs | |
| Total | 3,370,688(100%) | 18,129,157(100%) | 4,200,793(100%) | 17,242,328(100%) |
| miRNA | 18,165(0.54%) | 1,619,066(8.93%) | 19,429(0.46%) | 3,221,269(18.68%) |
| rRNA | 145,346(4.31%) | 2,251,398(12.42%) | 204,170(4.86%) | 3,442,702(19.97%) |
| snRNA | 4,403(0.13%) | 15,898(0.09%) | 6,650(0.16%) | 27,950(0.16%) |
| snoRNA | 2,292(0.07%) | 4,553(0.03%) | 2,976(0.07%) | 6,963(0.04%) |
| tRNA | 10,292(0.31%) | 260,694(1.44%) | 20,295(0.48%) | 295,998(1.72%) |
| Unannotated | 3,190,190(94.65%) | 13,977,548(77.10%) | 3,947,273(93.96%) | 10,247,446(59.43%) |
Table 3.
Summary of common and specific sequences between CK and Na200 sRNA libraries
| Class | Unique sRNAs | Percentage | Total sRNAs | Percentage |
|---|---|---|---|---|
| Total_sRNAs | 6,766,722 | 100.00% | 35,371,485 | 100.00% |
| NaCl_200&CK | 804,759 | 11.89% | 26,861,034 | 75.94% |
| NaCl_200_specific | 3,396,034 | 50.19% | 4,563,071 | 12.90% |
| CK_specific | 2,565,929 | 37.92% | 3,947,380 | 11.16% |
The sRNA size distribution in both libraries was summarized in Figure 1. The most abundant sRNAs ranged from 20 to 24 nt, and the 21 nt sRNAs represented the most frequent length (41.96% in CK and 33.22% in Na200 library, respectively), which was in agreement with previous reports in other plant species including trifoliate orange [13], Populus [8,29] and grapevine [30]. Furthermore, these sRNAs were annotated into several different categories (Table 2). Of these, 18,165 (0.54%) and 19,429 (0.46%) unique sRNAs were annotated as miRNAs in CK and Na200 libraries, respectively. Additionally, a predominant proportion of unique sequences (> 90% in two libraries) were unannotated sRNAs, suggesting a broad existence of novel miRNAs in radish.
Figure 1.

Length distribution of small RNAs in CK and Na200 libraries from radish roots. Y-axis represents percentages of sRNAs identified in this study; X-axis represents the length of sRNAs. Two libraries are shown by different colors.
Identification of known miRNAs in radish
A total of 117 unique sequences belonging to 28 conserved miRNA families and 19 unique sequences representing 15 non-conserved miRNA families were identified in both libraries (Table 4 and Additional file 1). The diversity of radish miRNA families could be determined from their number of members (Figure 2). As shown, most of the conserved miRNA families had members of no less than two. Of these, the miR156/157 and miR165/166 families were the largest ones with 14 members, followed by miR167 and miR169 with nine members. However, some conserved miRNA families including miR161, miR162, miR171, miR391, miR393, miR397, miR403 and miR535, had only one member. Moreover, most of the non-conserved miRNA families contained only one member.
Table 4.
Known miRNA families and their expression abundance in CK and Na200 libraries
| Family | No. of members | miRNA reads | Total reads | Ratio (Na200/CK) | |
|---|---|---|---|---|---|
| CK | Na200 | ||||
| Conserved miRNA | |||||
| miR156/157 | 14 | 390,906 | 477,699 | 868,605 | 1.22 |
| miR158 | 3 | 699,743 | 3,592,759 | 4,292,502 | 5.13 |
| miR159 | 4 | 3,509 | 2,881 | 6,390 | 0.82 |
| miR160 | 6 | 4,119 | 3,776 | 7,895 | 0.92 |
| miR161 | 1 | 0 | 10 | 10 | _ |
| miR162 | 1 | 750 | 922 | 1,672 | 1.23 |
| miR164 | 5 | 21,869 | 70,350 | 92,219 | 3.22 |
| miR165/166 | 14 | 91,496 | 208,941 | 300,437 | 2.28 |
| miR167 | 9 | 24,223 | 19,501 | 43,724 | 0.81 |
| miR168 | 4 | 196,762 | 319,663 | 516,425 | 1.62 |
| miR169 | 9 | 10,148 | 2,552 | 12,700 | 0.25 |
| miR171 | 1 | 155 | 28 | 183 | 0.18 |
| miR172 | 6 | 1,486 | 4,762 | 6,248 | 3.20 |
| miR319 | 3 | 14,416 | 17,633 | 32,049 | 1.22 |
| miR390 | 4 | 431 | 911 | 1,342 | 2.10 |
| miR391 | 1 | 1,066 | 1,993 | 3,059 | 1.87 |
| miR393 | 1 | 15 | 13 | 28 | 0.87 |
| miR394 | 2 | 201 | 416 | 617 | 2.07 |
| miR395 | 3 | 95 | 48,134 | 48,229 | 506.67 |
| miR396 | 4 | 981 | 1,177 | 2,158 | 1.20 |
| miR397 | 1 | 15,201 | 6,493 | 21,694 | 0.43 |
| miR398 | 4 | 2,032 | 326 | 2,358 | 0.16 |
| miR399 | 5 | 249 | 150 | 399 | 0.60 |
| miR403 | 1 | 1,634 | 2,649 | 4,283 | 1.62 |
| miR408 | 4 | 269,431 | 115,871 | 385,302 | 0.43 |
| miR482 | 2 | 145 | 1,543 | 1,688 | 10.64 |
| MiR535 | 1 | 0 | 1,524 | 1,524 | _ |
| miR2111 | 4 | 341 | 122 | 463 | 0.36 |
| Non-conserved miRNA | |||||
| miR400 | 1 | 137 | 151 | 288 | 1.10 |
| miR414 | 1 | 344 | 0 | 344 | 0.00 |
| miR774 | 1 | 3,704 | 4,062 | 7,766 | 1.10 |
| miR824 | 2 | 529 | 619 | 1,148 | 1.17 |
| miR825 | 1 | 1,455 | 1,861 | 3,316 | 1.28 |
| miR827 | 1 | 275 | 230 | 505 | 0.84 |
| miR841 | 1 | 0 | 293 | 293 | _ |
| miR845 | 2 | 3,003 | 7,471 | 10,474 | 2.49 |
| miR857 | 1 | 1,413 | 819 | 2,232 | 0.58 |
| miR1511 | 2 | 10,593 | 25,438 | 36,031 | 2.40 |
| miR1520 | 1 | 17,011 | 17,004 | 34,015 | 1.00 |
| miR2118 | 1 | 27,628 | 20,177 | 47,805 | 0.73 |
| miR2615 | 1 | 7,581 | 1,074 | 8,655 | 0.14 |
| miR5298 | 2 | 21 | 39 | 60 | 1.86 |
| miR5649 | 1 | 35 | 45 | 80 | 1.29 |
Figure 2.
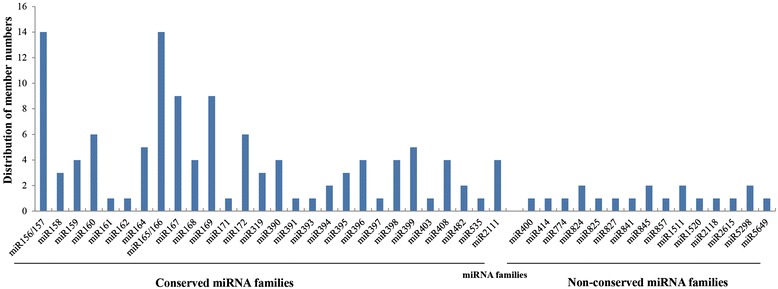
Distribution of known miRNA family size in R. sativus. Y-axis represents various known miRNA families identified in this study. X-axis represents the number of members for each miRNA family.
The number of miRNA reads in two libraries was highly variable with a ratio (Na200/CK) ranging from 0 to 506.67, and was exploited as the indicator for assessing miRNA expression level (Figure 3 and Table 4). miR158 presented the highest expression abundance with 699,743 and 3,592,759 copies in CK and Na200 libraries, respectively. Several conserved miRNA families including miR156/157, miR165/166, miR168 and miR408 also exhibited extraordinarily high abundance in both libraries, while some other miRNA families (miR164, miR167, miR169, miR319, miR395, miR397, miR845, miR1511, miR1520 and miR2118) were moderately expressed with a total reads ranging from 100,000 to 1,000,000. However, several miRNA families including miR161, miR393, miR5298 and miR5649 were detected to be expressed at an extremely low level in both libraries. Furthermore, a significant distinction in expression abundance was also observed among different members in a certain miRNA family (Additional file 1). For example, in the miR156/157 family, the read number of miR157a was 589,530, while miR156h had only 22 copies. This vast expression span among different members within a family suggested the precise expression of miRNAs under certain conditions.
Figure 3.

Abundance of each known miRNA family in R. sativus. Y-axis represents various known miRNA families and X-axis represents the abundance of each miRNA family in this study.
Identification of novel miRNA candidates
In the present study, based on the recent annotated criteria of novel miRNAs [31], 68 sRNA unique sequences belonging to 51 miRNA families were identified as putative novel miRNAs. Of these, seven novel miRNAs were detected to have miRNA*s (complementary miRNA sequences), and many miRNA*s were sequenced only once (Table 5 and Additional file 2), which might be partially attributed to that most of the miRNA*s had been degraded in miRNA pathway [13]. Among these novel miRNAs, most of them were CK-specific or Na200-specific, which accounted for 31.65% and 35.44%, respectively. In addition, the novel mature miRNAs displayed a main length distribution ranging from 21 nt to 24 nt, and 21 nt miRNAs accounted for the highest proportion (65.43%). Regarding their expression abundance, only 20 novel miRNAs (29.41%) had been sequenced more than 100 copies in each sRNA library. Moreover, the precursors of 68 potential novel miRNAs were also predicted, with minimum free energy (MFE) values ranging from −98.7 to −18.0 kcal/mol and an average length of 148 nt (Additional file 3).
Table 5.
Novel miRNAs with their complementary miRNA*s under salt stress in radish
| miRNA | Sequence (5′–3′) | Length (nt) | Precursor length (nt) | Count | MFE (kcal/mol) | Arm | miRNA location | |
|---|---|---|---|---|---|---|---|---|
| CK | Na200 | |||||||
| rsa-mir2-5p | AAAUCAUACUUUCAUUGAUA | 20 | 185 | 277 | 0 | −71.3 | 5′ | CL10794.Contig2 |
| rsa-mir2-3p | UCAAUGAAAGGUAUGAUUCCC | 21 | 185 | 277 | 1,228 | −71.3 | 3′ | CL10794.Contig2 |
| rsa-mir4-5p | ACGUUUCUCGAACUCAAGACC | 21 | 107 | 0 | 3 | −64.5 | 5′ | FY453420 |
| rsa-mir4-3p | UCUUGAGUUCGAGGGACGCCA | 21 | 107 | 115 | 193 | −64.5 | 3′ | FY453420 |
| rsa-mir11-5p | AGGCGAUGAUGGAUACCGAGAA | 22 | 91 | 310 | 0 | −31.8 | 5′ | CL2420.Contig13 |
| rsa-mir11-3p | CUCGGUAGCGAUGGUUCAAUCUCG | 24 | 91 | 1 | 0 | −31.8 | 3′ | CL2420.Contig13 |
| rsa-mir13-5p | AUAUACUGAAGUUUAUACUCU | 21 | 208 | 33 | 57 | −33.5 | 5′ | EY928450 |
| rsa-mir13-3p | AUCAUAAAAUCUUCAUUAUCUAG | 23 | 208 | 1 | 1 | −33.5 | 3′ | EY928450 |
| rsa-mir22-5p | UGGUGCAGGUCGGGAACUGAU | 21 | 110 | 13 | 0 | −57.7 | 5′ | EY910368 |
| rsa-mir22-3p | CGGAUCCCGCCUUGUAUCAAG | 21 | 110 | 1 | 0 | −57.7 | 3′ | EY910368 |
| rsa-mir24-5p | CGGUUAGCUUGGAAGCCAAAA | 21 | 178 | 0 | 1 | −41.2 | 5′ | CL10961.Contig1 |
| rsa-mir24-3p | UUGUUUUCUGAGAAAAUGGGC | 21 | 178 | 0 | 10 | −41.2 | 3′ | CL10961.Contig1 |
| rsa-mir35b-5p | UCGACGGGAAGGGGCUUUCUCU | 22 | 72 | 0 | 1 | −20 | 5′ | CL7248.Contig2 |
| rsa-mir35b-3p | GGAAUGUUGUUUGGCUCGAAG | 21 | 72 | 94 | 64 | −20 | 3′ | CL7248.Contig2 |
MFE (kcal/mol), minimal folding free energy.
Identification of salt-responsive miRNAs in radish
To identify the differentially expressed miRNAs under salt stress, the analysis of differential expression patterns was performed by statistical comparison between two libraries. In total, 49 known and 22 novel miRNAs were identified to be differentially regulated under salt stress (Additional file 4), and their expression alterations were showed in Figure 4. Among them, 41 miRNAs (31 known and 10 novel ones) were upregulated, and 30 miRNAs (18 known and 12 novel ones) were repressed by salt stress. Of these, 23 miRNAs including 16 known and seven novel miRNAs were markedly differentially expressed with an absolute value of log2 ratio (Na200/CK) > 10. In addition, many of these salt-responsive miRNAs, such as miR160b/d-3p, miR166g/h-3p, miR535b and rsa-mir3, were confined to be expressed in CK or Na200 library, suggesting that these miRNAs might be thoroughly induced or repressed by salt stress. Further analysis also indicated that different members in a certain miRNA family might have similar or disparate expression patterns after salt exposure. For example, miR172a and miR172e-3p were significantly upregulated, whereas miR172c was downregulated, providing further evidence on the complexity of miRNA regulatory roles.
Figure 4.
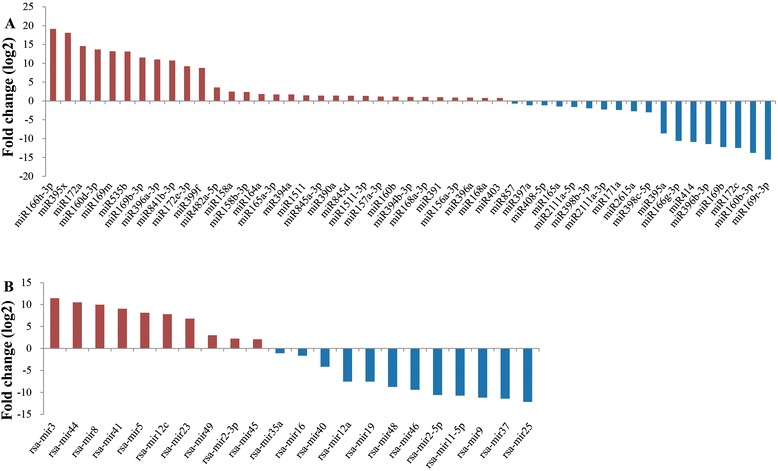
Comparative relative expression of differentially expressed miRNAs. Y-axis represents various differentially expressed miRNAs under salt stress in radish; X-axis represents the fold change value of each miRNA between Na200 and CK libraries. A represents differentially expressed known miRNAs and B represents differentially expressed novel miRNAs. The miRNAs with positive fold change value are upregulated, and the ones with negative fold change value are downregulated.
Prediction and annotation of target genes for salt-responsive miRNAs
Predicting the target genes of miRNAs would be essential for better understanding of the biological functions for these salt-responsive miRNAs. As a result, 581 genes were predicted to be targets for 65 salt-responsive miRNAs (Table 6 and Additional file 5). All target sequences were successfully classified into three GO (Gene Ontology) ontologies using blast2go program, including cellular components, molecular functions and biological processes (Figure 5). As shown, the main terms were “cell” (GO: 0005623), “cell part” (GO: 0044464) and “organelle” (GO: 0043226) in the cellular component category. For their molecular functions, the “binding” (GO: 0005488) and “catalytic activity” (GO: 0003824) were the most abundant subcategories. The predominant terms implicated in biological processes were “biological regulation” (GO: 0065007), “cellular progress” (GO: 0009987), “developmental progress” (GO: 0032502), “metabolic progress” (GO: 0008152) and “response to stimulus” (GO: 0050896).
Table 6.
The target genes for some salt-responsive known miRNAs
| miRNA | Target gene No. | Description | Gene name |
|---|---|---|---|
| miR156/157 | CL5609.Contig1 | ethylene-responsive transcription factor ERF113 | Rap2.6 L |
| CL967.Contig2 | glutamine synthetase | GS2 | |
| EX767226 | squamosa promoter-binding-like protein 13 | SPL13 | |
| FD556392 | squamosa promoter-binding-like protein 2 | SPL2 | |
| FD946000, FD946993, FD946993, FD560558, FD988312, EY898725, EX756914, EV551892, EV528056, Rsa#S43017568 | squamosa promoter-binding-like protein 3 | SPL3 | |
| FD557561, EX762240 | squamosa promoter-binding-like protein 5 | SPL5 | |
| EY930450 | squamosa promoter-binding-like protein 6 | SPL6 | |
| EX886942, EW715846,EX771535, EV548910 | squamosa promoter-binding-like protein 9 | SPL9 | |
| miR159 | EY949798, EY938664, Rsa#S42037487 | myb domain protein 101 | MYB101 |
| EY896930 | myb domain protein 65 | MYB65 | |
| FD584389, EY935636, FD977876, Rsa#S42034459, Rsa#S42591074 | putative ubiquitin-conjugating enzyme E2 17 | UBC17 | |
| miR160 | FD576484, FD550653, EV524607, Rsa#S42581764 | auxin response factor 16 | ARF16 |
| EX896877 | auxin response factor 17 | ARF17 | |
| miR164 | EX773809 | NAC domain containing protein 80 | NAC80 |
| EW715661, EV566600 | NAC domain containing protein 100 | NAC100 | |
| miR165/166 | Unigene8382 | defensin-like protein 2 | LCR69 |
| Unigene16151 | ATP synthase subunit G protein | ||
| miR169 | EV547500, EV543544 | glutamate decarboxylase 5 | GAD5 |
| Unigene27845 | histone acetyltransferase HAC1 | HAC1 | |
| EX750227 | nuclear transcription factor Y subunit A-2 | NF-YA2 | |
| FD989248, EV526819 | nuclear transcription factor Y subunit A-3 | NF-YA3 | |
| miR172 | EX761783 | AP2-like ethylene-responsive transcription factor SNZ | SNZ |
| EW732550 | AP2-like ethylene-responsive transcription factor TOE2 | TOE2 | |
| EY906836, FD572123 | Floral homeotic protein APETALA 2 | AP2 | |
| EY910663 | sulfate transporter 1.3 | SULTR1;3 | |
| miR395 | EX903518, FY437914, FY449298, FY448599, FY444096, FY449933, FY444103, EV539245 | ATP sulfurylase 1 | APS1 |
| FY443904, FY441630, FY445587, FY444933,FY443821, FY442807, FY442799, EY933376, EY916371 | sulfate adenylyltransferase | APS4 | |
| miR396 | EW733962, EY907316 | transcription factor bHLH74 | |
| EW733484, FY442946, FD941766, FD563793, EW713404, EW713403, FD971887, FD555982, FD551358, FD541643, EY908741, EX771641, EV550709, EV536225, EV535300, EV528362, EV524610 | atypical CYS HIS rich thioredoxin 5 | ACHT5 | |
| EY930318 | L-ascorbate peroxidase 1 | APX1 | |
| miR397 | FY438692 | laccase 2 | LAC2 |
| FD987950, FY452752 | laccase 11 | LAC11 | |
| FD989705, EV528485 | laccase 17 | LAC17 | |
| Unigene4183 | Lectin-domain containing receptor kinase A4.3 | LECRKA4.3 | |
| miR398 | FD972015, FD549426, FD544311, EX773977, EX757683, EX749645, EV525782 | superoxide dismutase [Cu-Zn] | CSD1 |
| miR399 | FD560927, EX890146 | putative ubiquitin-conjugating enzyme E2 24 | PHO2/UBC24 |
| miR403 | EW726356, EX749374, Rsa#S41987411 | Argonaute family protein | AGO2 |
| miR414 | Unigene3598 | ABC transporter G family member 10 | |
| FD967208 | AP2-like ethylene-responsive transcription factor SNZ | SNZ | |
| CL5793.Contig1 | CBL-interacting serine/threonine-protein kinase 21 | CIPK21 | |
| Unigene10439 | pentatricopeptide repeat-containing protein | ||
| EV527283, EY913653 | potassium transporter | KUP3p | |
| EX886639 | scarecrow-like protein 13 | SCL13 | |
| miR482 | FD580320 | MATE efflux family protein | FRD3 |
| miR841 | Unigene15520 | bZIP transcription factor family protein |
Detailed information of target genes for all salt-responsive miRNAs was listed in Additional file 5.
Figure 5.
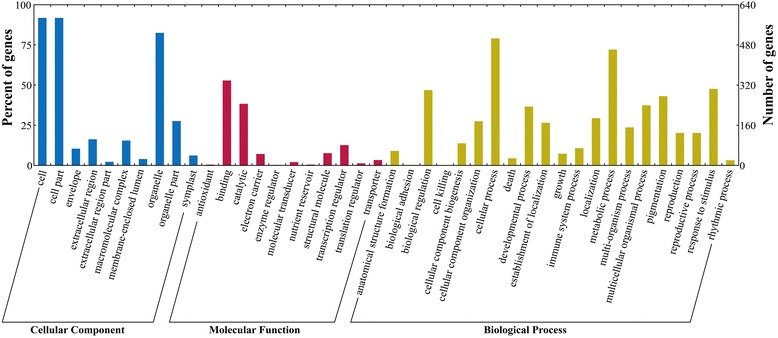
Gene ontology classification of potential target genes for differentially expressed miRNAs. Blue, red and yellow represent three GO ontologies: cellular component, molecular function and biological progress, respectively.
Many of the predicted target genes were homologous to those encoding some stress-related transcription factors (TFs), including SPB-like proteins (SPLs), myb domain proteins (MYBs), auxin response factor (ARF) family, APETALA2 (AP2), NAC domain-containing proteins (NACs), nuclear transcription factor Y (NF-YA and NF-YB) and bZIP (Table 6 and Additional file 5). Moreover, several target genes encoding important enzymes or functional proteins playing roles in diverse metabolic pathways, such as argonaute (AGO2), glutamine synthetase (GS2), glutamate decarboxylase 5 (GAD5), argininosuccinate synthase, S-adenosylmethionine (SAM)-dependent methyltransferase, pentatricopeptide repeats (PPRs) and histone acetyltransferase (HAC1), were also identified. By annotation of targets, a few transcripts were found likely to participate in plant abiotic stress responses. For instance, a miR414 target was CBL-interacting serine/threonine-protein kinase 21 (CIPK21), which was related to stress signal perception and transduction [32]; miR397a targeted laccases (LACs), which were involved in lignification and thickening of the cell wall [33]. In general, these results implied that these putative target genes might be implicated in diverse biological processes under salt stress in radish.
RT-qPCR validation
To verify the results of deep sequencing and detect the dynamic expression profiles of salt-responsive miRNAs at different stages of salt treatment (0, 3, 6, 12, 24, 48 and 96 h), the expression of ten salt-responsive miRNAs were analyzed with RT-qPCR (Figure 6). As expected, the obtained data suggested that all examined miRNAs shared a coincidental expression change between sRNA sequencing and RT-qPCR. For known miRNAs, miR166g-3p had a downregulated expression pattern, except that it slightly increased at 1 h of salt treatment. The expression of downregulated miR169b fell at first then abruptly increased at 12 h, and again gradually decreased to a low level. In contrast to miR169b, miR841b-3p showed an opposite expression trend with increasing treatment time. miR172c was downregulated and maintained at a quite low level at all treatment stages. With the increase of treatment time, the expression of miR397a was initially restricted, but then gradually restored to the initial level. For the novel miRNAs, rsa-mir3 expression increased until 24 h of salt treatment. rsa-mir9 and rsa-mir48 shared a similar expression pattern, but they reached their maxima at 3 h and 6 h, respectively. rsa-mir12a was downregulated at nearly all treatment stages, except that it abruptly elevated at 24 h. The level of rsa-mir23 gradually increased to its maximum at 12 h, and then dramatically restored to the initial level.
Figure 6.
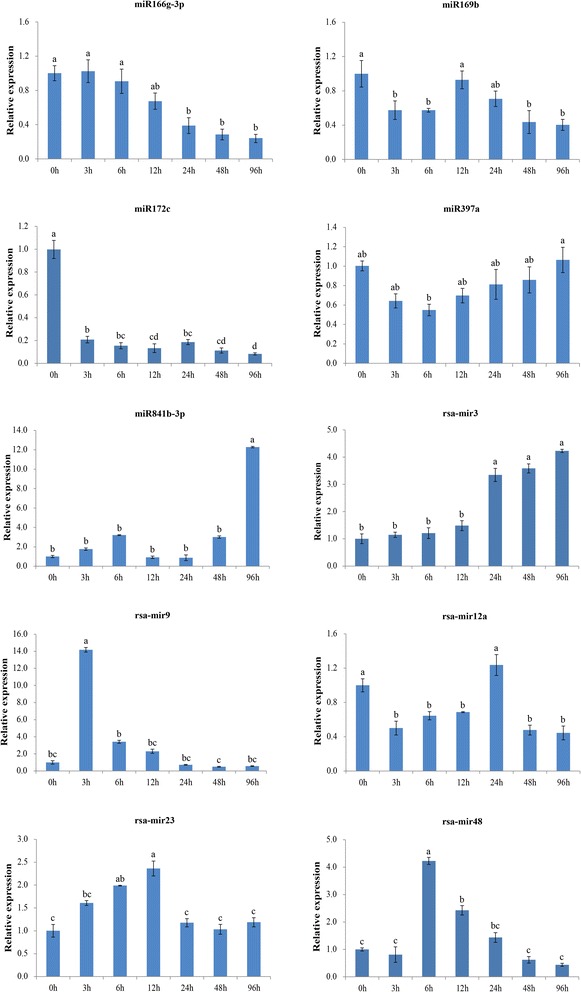
RT-qPCR analysis of several salt stress-responsive miRNAs. The expression level in the untreated sample (0 h) was set to a value of 1. Each bar shows the mean ± SE of triplicate assays.
Furthermore, the expression patterns of five corresponding target genes, namely SAM-dependent methyltransferase gene (unigene23846 targeted by miR166g-3p), NF-YA3 (FD989248 targeted by miR169b), SNZ (EX761783 targeted by miR172c), RHF2A (FY444261 targeted by rsa-mir23) and thioesterase gene (EX908068 targeted by rsa-mir48), were also examined to confirm the dynamic correlation between the miRNAs and their target genes under salt stress. The results revealed an approximately negative correlation between the expression of miRNAs and their corresponding targets (Figure 7). For instance, the salt-stressed downregulation of miR166g-3p and miR172c led to upregulation of SAM-dependent methyltransferase gene and SNZ, respectively. However, the expression of RHF2A was found to be restrained by upregulated rsa-mir23 in the early stage of salt exposure.
Figure 7.

RT-qPCR analysis of several miRNA-targets. The expression level in the untreated sample (0 h) was set to a value of 1. Each bar shows the mean ± SE of triplicate assays. Blue and red represent miRNA(s) and their corresponding target gene(s), respectively.
Discussions
As one of the most serious abiotic stresses worldwide, salt stress poses an increasingly severe threat to plant growth and development. In plants, to cope with salt stress, the modulation of numerous stress-responsive genes at transcriptional and posttranscriptional levels is activated. MicroRNAs are well known as ubiquitous regulators of gene expression and play vital roles in plant responses to abiotic and biotic stresses [34]. In recent years, increasing reports have demonstrated that miRNA-guided gene regulation plays a significant role in salt stress response in various plant species, such as Arabidopsis [7], P. tomentosa [8], soybean [19] and S. linnaeanum [20]. Radish is an important root vegetable crop worldwide. Although several studies of miRNAs associated with some important horticulture traits have been conducted in radish [25-27], no study on identification of salt-responsive miRNAs and their target genes has been reported to date.
sRNA sequencing and identification of known miRNAs in radish
In the present study, a total of 3.37 M and 4.20 M unique sRNA sequences were obtained from CK and Na200 libraries, respectively, suggesting adequate sequencing depth for further analysis. 21 nt sRNAs might play more prevailing roles in salt stress response due to their most abundant expression in both libraries (Figure 1), which was consistent with previous studies in several tree and vine species including Populus [8,29], trifoliate orange [13], and grapevine [30]. In addition, a striking divergence also existed in expression patterns among diverse types of sRNAs, which showed that the levels of 21 nt and 22 nt sRNAs were markedly decreased under salt stress, whereas those of 20 nt, 23 nt and 24 nt sRNAs were significantly increased (Figure 1). Due to the identified sRNAs with different sizes represented diverse functions in regulating gene expression, a more extensive modulation of gene expression by sRNAs might exist during response to salt stress.
The diversity of radish miRNA families could be determined by the abundance and number of members. In this study, conserved miRNA families had relatively higher expression and number of family members when compared with non-conserved ones (Figures 2 and 3), which was in agreement with previous findings in radish [25,26]. Moreover, it was also inferred that the read number of miRNAs varied from one to millions of copies reflected their diverse expression levels in radish. For example, miR156/157, miR158, miR166, miR168 and miR408 had extraordinarily high number of reads, suggesting these miRNAs might be expressed at a higher level, whereas miR161 and miR393 showed low abundance with less than 100 reads, therefore, possibly were expressed at a lower level.
Characterization of salt-responsive miRNAs in radish
A number of miRNAs were regulated by salt stress in diverse plant species, such as Arabidopsis [7], Z. mays [15] and S. linnaeanum [20]. In Arabidopsis, several upregulated miRNAs (miR156, miR158, miR159, miR165, miR168, miR169, miR171, miR319, miR393, miR394, miR396 and miR397) and downregulated miR398 were detected under salt stress using the miRNA-microarray technology [7]. By the same method, Ding et al. [15] also reported that miR156, miR164, miR167 and miR396 were downregulated, while miR162, miR168, miR395 and miR474 were upregulated in salt-stressed Z. mays. Moreover, by NGS technology, 11 downregulated miRNAs (miR156b/c, miR162b, miR167a/b, miR171b/e, miR172a, miR319a, miR399b and miR5300) and three upregulated miRNAs (miR164c, miR166d and miR397a) were identified in S. linnaeanum under NaCl exposure [20]. Many miRNAs were evolutionarily conserved across different plant species, which indicated that the conserved regulation of miRNAs played a vital role in plant response to salt stress. In the present study, 49 known miRNAs belonging to 28 miRNA families were differentially expressed and considered as salt-responsive miRNAs (Figure 4a and Additional file 4). However, several salt-responsive miRNA families including miR156, miR168, miR319, miR391, miR403 and miR857, did not show significant alterations of expression in the presence of salt stress in radish, although some of them were detected to be significantly altered by salt stress in other plant species [7,15]. This discrepancy suggested that these miRNAs potentially expressed in a species-specific manner under salt stress.
Additionally, it was worth noting that some salt-responsive miRNAs identified in this study might be fine-tuned across distinct biotic and abiotic stresses. For instance, miR169 and miR319 were widely reported to play important roles in ABA, drought and salt stress responses [4,35,36], and miR398 was considered as a bridge linking plant responses to oxidative stress and other stresses such as water deficit, salt stress, ABA stress, UV stress, nutrient deficiency and bacterial infection [35,37]. This observation was likely to be attributed to the shared regulatory genes modulated by these stress-related miRNAs across diverse stress responses, indicating that the inferred cross-regulation of miRNAs might link plant responses to various stresses [4]. However, further efforts are still needed to precisely confirm the roles of these salt-responsive miRNAs and explore the regulatory mechanism underlying these functions in plant adaptive response to salt stress.
miRNA-mediated regulatory network of salt stress response in radish
microRNAs function in gene modulation by regulating specific mRNA transcripts for degradation in plants. Recently, the NGS technology coupled with bioinformatics analysis have been widely applied to identify miRNAs and their corresponding target genes responsive to salt stress in plants [8,16,18,20]. In this study, a number of miRNAs were identified to be salt-responsive in radish, and many of them might play crucial roles in regulatory network of salt stress response by regulating specific stress-related genes. On basis of these results, a putative model of miRNA-target interactions involved in plant response to salt stress was put forward here, which presented the proposed regulation cascades after salt exposure in radish root cells (Figure 8).
Figure 8.
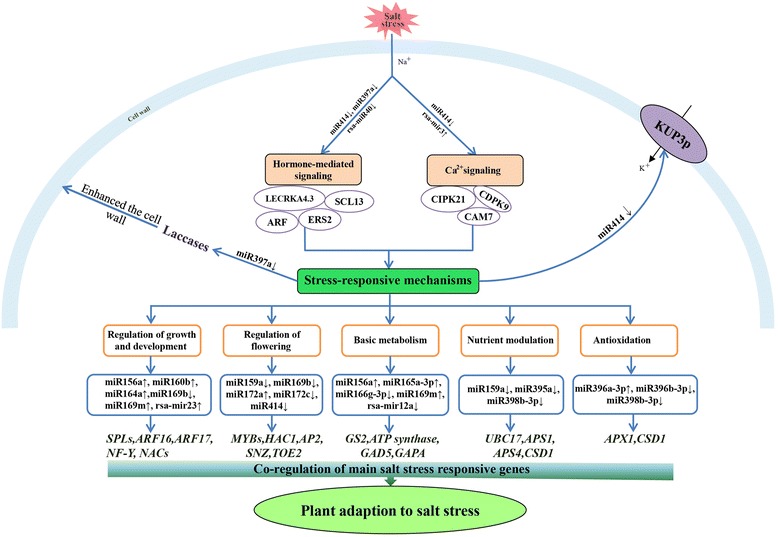
The proposed model of miRNA-mediated regulatory network associated with salt stress response in radish. The potential interactions between salt-responsive miRNAs and their corresponding target genes were shown.
In the present study, target prediction for 65 salt-responsive miRNAs revealed that many targets were TF genes including SPLs, MYBs, ARFs, AP2, NACs and NF-Y (Table 6 and Figure 8), which were reported to function in activating stress-responsive genes [38]. Many target genes might play important roles in plant responses to abiotic stresses. For example, miR169-targeted nuclear factor Y subunit A (NF-YA), which conditioned whole plant growth through modifying carbohydrate metabolism and cell elongation, was widely regulated under drought and salt stress [36,39]. Moreover, a set of evidences also supported the involvement of miR164 in stress responses, regulating the miRNA-mediated cleavage of NAC. NACs were widely modulated by various abiotic stresses like salinity, cold, ABA or drought [40-42], and also integrated responses to environmental stimuli into regulation of plant development processes [43-45].
Additionally, different members of auxin response factors (ARF) family (ARF16 and ARF17), which were reported to participate in auxin signaling pathways and be negative regulators of growth and development [4,46], were also identified for salt-responsive miR160b in radish. Similar findings of miR160-mediated ARF regulation were also reported in salt-stressed P. tomentosa [8], drought-stressed P. persica [10] and cold-stressed trifoliate orange [13]. These observations implied miR160-regulated ARFs might play an important role in plant responses to various abiotic stresses by fine-tuning plant growth and development under stress conditions. Other genes including miR156/157-targeted SPLs (SPL2, SPL3, SPL5, SPL6, SPL9 and SPL13) and miR172-targeted AP2, which were involved in regulating plant growth and development, were also identified in this study. It was reported that miR156-regulated SPLs and miR172-targeted AP2 conjointly conditioned the transitions among different developmental stages including embryogenesis, vegetative and reproductive growth, and participated in determining floral organ identity [47,48]. Similar to SPLs and AP2, miR159-regulated MYBs (MYB65 and MYB101) were also considered to modulate plant growth and development especially flowering under salt stress. It was reported that overexpressed miR159 resulted in a delayed flowering state concomitant with a repression of its target gene, GAMYB in gloxinia [17]. Given that plant growth and development including bolting and flowering were usually adversely-influenced under stress conditions, it might indicate the involvement of these miRNA-target transcripts in the network of genes regulated by salt-responsive miRNAs in radish (Figure 8).
Apart from key TFs, a number of genes which encode important enzymes or functional proteins, such as APX1, CSD1, APSs (APS1 and APS4), LACs (LAC2, LAC11 and LAC17), UBCs (UBC17 and UBC24), Ca2+-mediated signal-related proteins (CAM7, CIPK21 and CDPK9), were also considered to play important roles in salt stress response. Among them, CIPK21, CAM7 and CDPK9 (targeted by miR414, rsa-mir3 and rsa-mir5, respectively) were reported to cooperatively perform their functions to initiate the stimulus-specific downstream signal transduction [32]. miR397-targeted LACs encoding laccases were related to lignification and thickening of the cell wall in secondary cell growth [33], and accordingly enhanced the ability to alleviate stress damage. Furthermore, several target genes were found to be cross-regulated by miRNAs in response to salt stress and other abiotic stresses including oxidative stress and nutrient stress. As represented in this study, miR398b-3p targeted CSD1 encoding superoxide dismutase [Cu-Zn], which was reported to participate in scavenging excess reactive oxygen species (ROS) in plants exposed to salt stress [49]. Additionally, L-ascorbate peroxidase encoded by miR396-regulated APX1 was also a significant ROS scavenging enzyme, which was involved in regulation of intracellular ROS expression levels [50]. Several similar miRNA-targets had also been identified in plants exposed to nutrient deficiency. For instance, miR395 was widely reported to participate in catalyzing inorganic sulfate assimilation by suppressing the expression of ATP sulfurylases (APSs) [51], and miR398-mediated CSD1 regulation was responsible for maintaining copper concentration in plant cells [52]. Moreover, miR399 was also found to regulate phosphorus homeostasis by modulating the expression of ubiquitin-conjugating E2 enzyme (UBC) [53,54]. On account of the fact that prolonged salt stress could usually lead to some secondary stresses such as oxidative stress and nutrition disorder [55], the results indicated that these miRNA-regulated target genes might play significant roles in plant adaptive response to salt stress, as an indispensable part of regulatory network responsive to salt stress in radish (Figure 8).
Conclusions
The application of sRNA sequencing technology combined with bioinformatics analysis provides an unprecedented opportunity to obtain comprehensive understanding of novel and salt-responsive miRNAs in radish. A total of 49 known and 22 potential novel miRNAs were differentially expressed under salt stress. Prediction and analysis of target genes for these salt-responsive miRNAs demonstrated that numerous transcription and regulatory factors (enzymes) primarily functioned in a variety of biological pathways under stress conditions, including stress-related signal sensing and transduction, regulation of ion homeostasis, basic metabolic processes, secondary stress responses, as well as modulation of attenuated plant growth and development. Given that plant responses to abiotic stresses consist of many complex biochemical processes in which different components operate together, this investigation could advance our insights into the miRNA-mediated regulatory network of salt stress response, and the results will facilitate further dissection of molecular mechanism underlying plant response to salt stress in radish and other root vegetable crops.
Methods
Plant materials and salt stress treatment
The seeds of advanced inbred line “NAU-YH” were surface-sterilized with 1% sodium hypochlorite, and then germinated at 25°C for 2 d in the dark. Germinated seeds were cultured in individual pots containing loam soil and grown in a growth chamber with a 16-h light at 25°C and 8-h dark at 18°C cycle. Three-week-old seedlings with thickened flesh roots were transferred into modified half-strength hoagland nutrient solution as previously described [56]. For the salt-treated group, the radish seedlings were treated with 200 mM NaCl for 3 h, 6 h, 12 h, 24 h, 48 h and 96 h, respectively. Seedlings cultured under NaCl-free solution were used as control. Fresh roots at different salt-treated time points were immediately collected and stored at −80°C for further use.
Small RNA (sRNA) library construction and sequencing
Total RNA was extracted from the control (NaCl-free, CK) and salt-stressed (200 mM NaCl for 48 h, Na200) radish roots with TRIzol reagent (Invitrogen, USA) following the manufacturer’s instructions. Two sRNA libraries were constructed according to previously reported procedures [25,57]. Briefly, sRNA fractions of 18–30 nt isolated and purified by 15% denaturing polyacrylamide gel electrophoresis were ligated with specialized adaptors to the 5’ and 3’ ends (Illumina) using T4 RNA ligase. They were then reverse transcribed to cDNA using SuperScript II Reverse Transcriptase (Invitrogen), followed by PCR amplification. The final PCR products were purified and subjected to deep sequencing using Solexa sequencer (Illumina) HiSeq2000 at the Beijing Genomics Institute (BGI), Shenzhen, China.
Identification of known and novel miRNAs
After removing undesirable raw reads including low-quality reads, adapter reads, contaminants and reads either shorter than 15 nt or longer than 30 nt, the remaining unique sequences were mapped to the radish reference genome which consisted of radish GSS, EST and transcriptome sequences, to analyze the expression and distribution of sRNAs on genome using SOAP2 program [25,26]. Perfectly matched sequences were retained for following analysis. By querying against the NCBI Genbank (http://www.ncbi.nlm.nih.gov/genbank/) and Rfam (10.1) (http://www.sanger.ac.uk/resources/databases/rfam.html) databases, the sRNA sequences matching rRNA, tRNA, snRNA, snoRNA as well as sequences containing poly (A) tails were excluded. The remaining unique sequences were aligned against miRBase 20.0 (http://www.mirbase.org/index.shtml) to identify radish known miRNAs. The matched sequences with no more than two mismatches were considered as known miRNAs. Then, unannotated unique sequences were mapped to the radish reference sequences to uncover novel miRNAs from radish, according to previous criteria [31], using MIREAP software (http://sourceforge.net/projects/mireap/). The sRNA secondary structures were also formed by Mfold (http://mfold.rna.albany.edu/?q=DINAMelt/Quickfold).
Differential expression analysis of miRNAs responsive to salt stress
The counts of identified miRNAs in two libraries were normalized as transcripts per million (TPM) according to the formula: Normalised expression = actual miRNA count/total count of clean reads × 1,000,000. The normalized values of miRNAs with abundance of zero were modified to 0.01 for further analysis. The remaining normalized reads were used to calculate the p-value and the change in expression abundance. The differential expression of miRNAs between two libraries was calculated as: Fold-change = log2 (Na200/CK). The p-value was obtained according to previously reported methods [19]. The miRNAs with values of log2 ratio (Na200/CK) > 0.5 and < −0.5, along with the p-value of < 0.05, were considered as upregulated and downregulated during salt stress, respectively.
Prediction of putative miRNA targets for salt-responsive miRNAs
The putative target genes were predicted by the plant small RNA target analysis server as described by Barvkar et al. [58] and Zhai et al. [26]. The criteria for target prediction were based on previous studies following the alignment between each miRNA and their targets [59,60], including: (1) no more than four mismatches; (2) no more than two adjacent mismatches in miRNA/target duplex; (3) no more than one mismatch at positions 1–9; (4) no mismatches at positions 10–11, and (5) no more than 2.5 mismatches in positions 1–12 of the miRNA/target duplex (5’ end of the miRNA). To understand the biological functions of the target genes, the Blast2GO program was applied to obtain the GO annotations on the basis of the BLAST searches against the available Nr database in NCBI [24,26,61].
Validation of miRNAs and their potential target genes by RT-qPCR
Small RNAs were extracted from samples exposed to 200 mM NaCl at different time points (0, 3, 6, 12, 24, 48 and 96 h) using a small RNA isolation kit (TaKaRa, Dalian, China) according to the manufacturer’s instructions. Then, the small RNAs were reverse-transcribed to cDNAs using the One Step PrimeScript® miRNA cDNA Synthesis Kit following the manufacturer’s protocols. For RT-qPCR validation of target genes, total RNAs were isolated as described above and reverse-transcribed into first-strand cDNA using the Superscript III First-Strand Synthesis System (Invitrogen, USA). The primer sequences of miRNAs and their target genes were shown in Additional file 6. The RT-qPCR analysis was carried out on an iCycler iQ real-time PCR detection system (BIO-RAD) using SYBR® Premix Ex Taq™ II (TaKaRa). Each PCR reaction was carried out in a total volume of 20 μl containing 2 μl of cDNA, 10 μl of 2 × SYBR Green PCR Master Mix, and 0.2 μM primer pairs. The PCR amplified condition was performed following the previous reports [25,26]. The 5.8S rRNA was used as the internal reference gene for normalization. All reactions were run in triplicate and the data were statistically analyzed by SAS Version 9.0 software (SAS Institute, NC, USA) using Duncan’s multiple range test at the P < 0.05 level of significance.
Acknowledgements
This work was in part supported by grants from the NSFC (31171956, 31372064), Key Technology R & D Program of Jiangsu Province (BE2013429), JASTIF [CX (12) 2006, CX (13) 2007], JKL-HCGI(2014017) and the PAPD.
Abbreviations
- miRNA
MicroRNA
- SPL
Squamosa promoter binding-like protein
- ARF
Auxin response factor
- NF-Y
Nuclear transcription factor Y
- RT-qPCR
Reverse-transcription quantitative PCR
- RISC
RNA-induced silencing complex
- NGS
Next generation sequencing
- EST
Expressed Sequence Tag
- GSS
Genome survey sequence
- MFE
Minimum free energy
- GO
Gene Ontology
- MYB
Myb domain protein
- AP2
APETALA2
- LAC
Laccase
- ROS
Reactive oxygen species
- snRNA
Small nuclear RNA
- snoRNA
Small nucleolar RNA
- TPM
Transcripts per million
Additional files
The detailed information of known miRNAs identified in radish.
The detailed information of novel miRNA candidates identified in radish.
The secondary structures of identified potential novel miRNAs in radish.
Summary of differentially expressed miRNAs under salt stress in radish.
Targets and functional annotations for the salt-responsive miRNAs in radish.
The miRNAs, targets and their primer sequences for RT-qPCR validation.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XS, LX and LL conceived and designed the research. XS, RY and XL conducted experiments. YW and YG contributed powerful analytical tools. XS and KZ analyzed data, and XS and LX wrote the manuscript. YW, XZ, RW and CL helped with the revise of manuscript. All authors read and approved the final manuscript.
Contributor Information
Xiaochuan Sun, Email: 2013204023@njau.edu.cn.
Liang Xu, Email: nauxuliang@njau.edu.cn.
Yan Wang, Email: wangyan8217@126.com.
Rugang Yu, Email: yurugang@126.com.
Xianwen Zhu, Email: xianwen.zhu@ndsu.edu.
Xiaobo Luo, Email: 2013104069@njau.edu.cn.
Yiqin Gong, Email: jwcgyq@njau.edu.cn.
Ronghua Wang, Email: 2012104070@njau.edu.cn.
Cecilia Limera, Email: cnlimera@gmail.com.
Keyun Zhang, Email: keyunzhang@njau.edu.cn.
Liwang Liu, Email: nauliulw@njau.edu.cn.
References
- 1.Munns R, Tester M. Mechanisms of salinity tolerance. Annu Rev Plant Biol. 2008;59:651–81. doi: 10.1146/annurev.arplant.59.032607.092911. [DOI] [PubMed] [Google Scholar]
- 2.Tester M, Davenport R. Na+ tolerance and Na+ transport in higher plants. Ann Bot. 2003;91(5):503–27. doi: 10.1093/aob/mcg058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mäser P, Eckelman B, Vaidyanathan R, Horie T, Fairbairn DJ, Kubo M, et al. Altered shoot/root Na+ distribution and bifurcating salt sensitivity in Arabidopsis by genetic disruption of the Na+ transporter AtHKT1. FEBS Lett. 2002;531(2):157–61. doi: 10.1016/S0014-5793(02)03488-9. [DOI] [PubMed] [Google Scholar]
- 4.Sunkar R, Li YF, Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012;17(4):196–203. doi: 10.1016/j.tplants.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Kurihara Y, Takashi Y, Watanabe Y. The interaction between DCL1 and HYL1 is important for efficient and precise processing of pri-miRNA in plant microRNA biogenesis. RNA. 2006;12(2):206–12. doi: 10.1261/rna.2146906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, et al. Methylation as a crucial step in plant microRNA biogenesis. Science. 2005;307(5711):932–5. doi: 10.1126/science.1107130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu HH, Tian X, Li YJ, Wu CA, Zheng CC. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA. 2008;14(5):836–43. doi: 10.1261/rna.895308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren Y, Chen L, Zhang Y, Kang X, Zhang Z, Wang Y. Identification and characterization of salt-responsive microRNAs in Populus tomentosa by high-throughput sequencing. Biochimie. 2013;95(4):743–50. doi: 10.1016/j.biochi.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 9.Li B, Qin Y, Duan H, Yin W, Xia X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J Exp Bot. 2011;62(11):3765–79. doi: 10.1093/jxb/err051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eldem V, Akçay UÇ, Ozhuner E, Bakır Y, Uranbey S, Unver T. Genome-wide identification of miRNAs responsive to drought in peach (Prunus persica) by high-throughput deep sequencing. PLoS One. 2012;7(12):e50298. doi: 10.1371/journal.pone.0050298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu X, Wang H, Lu Y, de Ruiter M, Cariaso M, Prins M, et al. Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J Exp Bot. 2011;63(2):1025–38. doi: 10.1093/jxb/err337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Ren Y, Zhang Y, Xu J, Sun F, Zhang Z, et al. Genome-wide identification and expression analysis of heat-responsive and novel microRNAs in Populus tomentosa. Gene. 2012;504(2):160–5. doi: 10.1016/j.gene.2012.05.034. [DOI] [PubMed] [Google Scholar]
- 13.Zhang XN, Li X, Liu JH. Identification of conserved and novel cold-Responsive microRNAs in trifoliate orange (Poncirus trifoliata (L.) Raf.) using high-throughput sequencing. Plant Mol Biol Rep. 2014;32(2):328–41. doi: 10.1007/s11105-013-0649-1. [DOI] [Google Scholar]
- 14.Sunkar R, Kapoor A, Zhu JK. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell. 2006;18(8):2051–65. doi: 10.1105/tpc.106.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y. Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot. 2009;103(1):29–38. doi: 10.1093/aob/mcn205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong Z, Shi L, Wang Y, Chen L, Cai Z, Wang Y, et al. Identification and dynamic regulation of microRNAs involved in salt stress responses in functional soybean nodules by high-throughput sequencing. Int J Mol Sci. 2013;14(2):2717–38. doi: 10.3390/ijms14022717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Bian H, Song D, Ma S, Han N, Wang J, et al. Flowering time control in ornamental gloxinia (Sinningia speciosa) by manipulation of miR159 expression. Ann Bot. 2013;111(5):791–9. doi: 10.1093/aob/mct034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu J, Li W, Yang W, Qi L, Han S. Identification of microRNAs in Caragana intermedia by high-throughput sequencing and expression analysis of 12 microRNAs and their targets under salt stress. Plant Cell Rep. 2013;32(9):1339–49. doi: 10.1007/s00299-013-1446-x. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Dong Y, Yin H, Wang N, Yang J, Liu X, et al. Characterization of the stress associated microRNAs in Glycine max by deep sequencing. BMC Plant Biol. 2011;11(1):170. doi: 10.1186/1471-2229-11-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhuang Y, Zhou XH, Liu J. Conserved miRNAs and their response to salt stress in wild Eggplant Solanum linnaeanum roots. Int J Mol Sci. 2014;15(1):839–49. doi: 10.3390/ijms15010839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang Q, Wang F, Li MY, Tan HW, Ma J, Xiong AS. High-throughput analysis of small RNAs and characterization of novel microRNAs affected by abiotic stress in a local celery cultivar. Sci Hortic. 2014;169:36–43. doi: 10.1016/j.scienta.2014.02.007. [DOI] [Google Scholar]
- 22.Tian Y, Tian Y, Luo X, Zhou T, Huang Z, Liu Y, et al. Identification and characterization of microRNAs related to salt stress in broccoli, using high-throughput sequencing and bioinformatics analysis. BMC Plant Biol. 2014;14(1):226. doi: 10.1186/s12870-014-0226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, He Q. China radish. Beijing: Scientific and Technical Documents Publishing House; 2005. [Google Scholar]
- 24.Xu L, Wang Y, Xu Y, Wang L, Zhai L, Zhu X, et al. Identification and characterization of novel and conserved microRNAs in radish (Raphanus sativus L.) using high-throughput sequencing. Plant Sci. 2013;201:108–14. doi: 10.1016/j.plantsci.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Xu L, Wang Y, Zhai L, Xu Y, Wang L, Zhu X, et al. Genome-wide identification and characterization of cadmium-responsive microRNAs and their target genes in radish (Raphanus sativus L.) roots. J Exp Bot. 2013;64:4271–87. doi: 10.1093/jxb/ert240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhai L, Xu L, Wang Y, Huang D, Yu R, Limera C, et al. Genome-wide identification of embryogenesis-associated microRNAs in radish (Raphanus sativus L.) by high-throughput sequencing. Plant Mol Biol Rep. 2014;32:900–15. doi: 10.1007/s11105-014-0700-x. [DOI] [Google Scholar]
- 27.Wang Y, Liu W, Shen H, Zhu X, Zhai L, Xu L, et al. Identification of radish (Raphanus sativus L.) miRNAs and their target genes to explore miRNA-mediated regulatory networks in lead (Pb) stress responses by high-throughput sequencing and degradome analysis. Plant Mol Biol Rep. 2014. doi:10.1007/s11105-014-0752-y.
- 28.Yuan G, Wang X, Guo R, Wang Q. Effect of salt stress on phenolic compounds, glucosinolates, myrosinase and antioxidant activity in radish sprouts. Food Chem. 2010;121(4):1014–9. doi: 10.1016/j.foodchem.2010.01.040. [DOI] [Google Scholar]
- 29.Barakat A, Wall PK, DiLoreto S, Carlson JE. Conservation and divergence of microRNAs in Populus. BMC Genomics. 2007;8(1):481. doi: 10.1186/1471-2164-8-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, et al. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010;62(6):960–76. doi: 10.1111/j.0960-7412.2010.04208.x. [DOI] [PubMed] [Google Scholar]
- 31.Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, et al. Criteria for annotation of plant MicroRNAs. Plant Cell. 2008;20(12):3186–90. doi: 10.1105/tpc.108.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastava A, Rai A, Patade V, Suprasanna P. Calcium signaling and its significance in alleviating salt stress in plants. In: Ahmad P, Azooz MM, Prasad MNV, editors. Salt stress in plants: signalling, omics and adaptations. New York: Springer; 2013. pp. 197–218. [Google Scholar]
- 33.Lu S, Li Q, Wei H, Chang MJ, Tunlaya AS, Kim H, et al. Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc Natl Acad Sci U S A. 2013;110(26):10848–53. doi: 10.1073/pnas.1308936110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kruszka K, Pieczynski M, Windels D, Bielewicz D, Jarmolowski A, Szweykowska KZ, et al. Role of microRNAs and other sRNAs of plants in their changing environments. J Plant Physiol. 2012;169(16):1664–72. doi: 10.1016/j.jplph.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Contreras CC, Palomar M, Arteaga VM, Reyes JL, Covarrubias AA. Non-coding RNAs in the plant response to abiotic stress. Planta. 2012;236(4):943–58. doi: 10.1007/s00425-012-1693-z. [DOI] [PubMed] [Google Scholar]
- 36.Zhao B, Ge L, Liang R, Li W, Ruan K, Lin H, et al. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol Biol. 2009;10(1):29. doi: 10.1186/1471-2199-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu C, Ding Y, Liu H. MiR398 and plant stress responses. Physiol Plant. 2011;143(1):1–9. doi: 10.1111/j.1399-3054.2011.01477.x. [DOI] [PubMed] [Google Scholar]
- 38.Woodrow P, Pontecorvo G, Ciarmiello LF, Annunziata MG, Fuggi A, Carillo P. Transcription factors and genes in abiotic stress. In: Venkateswarlu B, Shanker AK, Shanker C, Maheswari M, editors. Crop stress and its management: Perspectives and strategies. New York: Springer; 2012. pp. 317–57. [Google Scholar]
- 39.Stephenson TJ, McIntyre CL, Collet C, Xue GP. Genome-wide identification and expression analysis of the NF-Y family of transcription factors in Triticum aestivum. Plant Mol Biol. 2007;65(1–2):77–92. doi: 10.1007/s11103-007-9200-9. [DOI] [PubMed] [Google Scholar]
- 40.Fujita M, Fujita Y, Maruyama K, Seki M, Hiratsu K, Ohme TM, et al. A dehydration-induced NAC protein, RD26, is involved in a novel ABA-dependent stress-signaling pathway. Plant J. 2004;39(6):863–76. doi: 10.1111/j.1365-313X.2004.02171.x. [DOI] [PubMed] [Google Scholar]
- 41.Fang Y, You J, Xie K, Xie W, Xiong L. Systematic sequence analysis and identification of tissue-specific or stress-responsive genes of NAC transcription factor family in rice. Mol Genet Genomics. 2008;280(6):547–63. doi: 10.1007/s00438-008-0386-6. [DOI] [PubMed] [Google Scholar]
- 42.Puranik S, Sahu PP, Srivastava PS, Prasad M. NAC proteins: regulation and role in stress tolerance. Trends Plant Sci. 2012;17(6):369–81. doi: 10.1016/j.tplants.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Hao YJ, Wei W, Song QX, Chen HW, Zhang YQ, Wang F, et al. Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J. 2011;68(2):302–13. doi: 10.1111/j.1365-313X.2011.04687.x. [DOI] [PubMed] [Google Scholar]
- 44.Kim SG, Kim SY, Park CM. A membrane-associated NAC transcription factor regulates salt-responsive flowering via FLOWERING LOCUS T in Arabidopsis. Planta. 2007;226(3):647–54. doi: 10.1007/s00425-007-0513-3. [DOI] [PubMed] [Google Scholar]
- 45.Yang SD, Seo PJ, Yoon HK, Park CM. The Arabidopsis NAC transcription factor VNI2 integrates abscisic acid signals into leaf senescence via the COR/RD genes. Plant Cell. 2011;23(6):2155–68. doi: 10.1105/tpc.111.084913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rubio SI, Cuperus JT, Weigel D, Carrington JC. Regulation and functional specialization of small RNA-target nodes during plant development. Curr Opin Plant Biol. 2009;12(5):622–7. doi: 10.1016/j.pbi.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Zhu QH, Helliwell CA. Regulation of flowering time and floral patterning by miR172. J Exp Bot. 2011;62(2):487–95. doi: 10.1093/jxb/erq295. [DOI] [PubMed] [Google Scholar]
- 48.Schwab R. Roles of miR156 and miR172 in reproductive development. In: Sunkar R, editor. MicroRNAs in plant development and stress responses. Springer: New York; 2012. pp. 69–81. [Google Scholar]
- 49.Jagadeeswaran G, Saini A, Sunkar R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta. 2009;229(4):1009–14. doi: 10.1007/s00425-009-0889-3. [DOI] [PubMed] [Google Scholar]
- 50.Maruta T, Inoue T, Noshi M, Tamoi M, Yabuta Y, Yoshimura K, et al. Cytosolic ascorbate peroxidase 1 protects organelles against oxidative stress by wounding- and jasmonate-induced H2O2 in Arabidopsis plants. Biochim Biophys Acta-Gen Subj. 2012;1820(12):1901–7. doi: 10.1016/j.bbagen.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Matsui A, Nguyen AH, Nakaminami K, Seki M. Arabidopsis non-coding RNA regulation in abiotic stress responses. Int J Mol Sci. 2013;14(11):22642–54. doi: 10.3390/ijms141122642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamasaki H, Abdel-Ghany SE, Cohu CM, Kobayashi Y, Shikanai T, Pilon M. Regulation of copper homeostasis by micro-RNA in Arabidopsis. J Biol Chem. 2007;282(22):16369–78. doi: 10.1074/jbc.M700138200. [DOI] [PubMed] [Google Scholar]
- 53.Fujii H, Chiou TJ, Lin SI, Aung K, Zhu JK. A miRNA involved in phosphate-starvation response in Arabidopsis. Curr Biol. 2005;15(22):2038–43. doi: 10.1016/j.cub.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 54.Hackenberg M, Shi BJ, Gustafson P, Langridge P. Characterization of phosphorus-regulated miR399 and miR827 and their isomirs in barley under phosphorus-sufficient and phosphorus-deficient conditions. BMC Plant Biol. 2013;13(1):214. doi: 10.1186/1471-2229-13-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chinnusamy V, Zhu J, Zhu JK. Salt stress signaling and mechanisms of plant salt tolerance. In: Setlow JK, editor. Genetic engineering: Principles and Methods. New York: Springer; 2006. pp. 141–77. [DOI] [PubMed] [Google Scholar]
- 56.Xu L, Wang L, Gong Y, Dai W, Wang Y, Zhu X, et al. Genetic linkage map construction and QTL mapping of cadmium accumulation in radish (Raphanus sativus L.) Theor Appl Genet. 2012;125(4):659–70. doi: 10.1007/s00122-012-1858-y. [DOI] [PubMed] [Google Scholar]
- 57.Hafner M, Landgraf P, Ludwig J, Rice A, Ojo T, Lin C, et al. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods. 2008;44(1):3–12. doi: 10.1016/j.ymeth.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barvkar VT, Pardeshi VC, Kale SM, Qiu S, Rollins M, Datla R, et al. Genome-wide identification and characterization of microRNA genes and their targets in flax (Linum usitatissimum) Planta. 2013;237(4):1149–61. doi: 10.1007/s00425-012-1833-5. [DOI] [PubMed] [Google Scholar]
- 59.Allen E, Xie Z, Gustafson AM, Carrington JC. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121(2):207–21. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 60.Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D. Specific effects of microRNAs on the plant transcriptome. Dev Cell. 2005;8(4):517–27. doi: 10.1016/j.devcel.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 61.Galla G, Barcaccia G, Ramina A, Collani S, Alagna F, Baldoni L, et al. Computational annotation of genes differentially expressed along olive fruit development. BMC Plant Biol. 2009;9(1):128. doi: 10.1186/1471-2229-9-128. [DOI] [PMC free article] [PubMed] [Google Scholar]