

Abstract
The PTEN tumor suppressor gene is one of the most commonly mutated genes in human cancer. Because inactivation of PTEN is a somatic event, PTEN mutations represent an important genetic difference between cancer cells and normal cells and therefore a potential anticancer drug target. However, it remains a substantial challenge to identify compounds that target loss-of-function events such as mutations of tumor suppressors. In an effort to identify small molecules that preferentially kill cells with mutations of PTEN, the authors developed and implemented a high-throughput, paired cell-based screen composed of parental HCT116 cells and their PTEN gene-targeted derivatives. From 138 758 compounds tested, two hits were identified, and one, N′-[(1-benzyl-1H-indol-3-yl)methylene]benzenesulfonohydrazide (CID1340132), was further studied using a variety of cell-based models, including HCT116, MCF10A, and HEC1A cells with targeted deletion of either their PTEN or PIK3CA genes. Preferential killing of PTEN and PIK3CA mutant cells was accompanied by DNA damage, inhibition of DNA synthesis, and apoptosis. taken together, these data validate a cell-based screening approach for identifying lead compounds that target cells with specific tumor suppressor gene mutations and describe a novel compound with preferential killing activity toward PTEN and PIK3CA mutant cells.
Keywords: PTEN, PIK3CA, human somatic cell gene targeting, phenotypic screen, DNA damage, high-throughput screen, synthetic lethality
Introduction
Cancer is a Genetic Disease Caused by activating mutations in oncogenes and inactivating mutations in tumor suppressor genes. Because these mutant genes are present only in the patient's cancer cells (and not in the patient's normal tissues), they represent an appealing target for anticancer drug development. However, the majority of these cancer-causing mutations are loss-of-function events that defy standard inhibitor-based drug development strategies. As such, it is an important challenge for cancer small-molecule drug discovery to develop methods making it possible to identify compounds that selectively target cells with mutations in tumor suppressor genes.1,2
The most common strategy for meeting this challenge is to identify and inhibit a downstream gain of function. However, there are two difficulties with this approach. First, this strategy requires detailed knowledge of the signaling pathway(s) regulated by the tumor suppressor. This type of information is often not available, especially for newly discovered tumor suppressor genes. Second, because tumor suppressors often represent critical nodal points in signaling pathways, these pathways tend to diverge downstream of the tumor suppressor. One classic example is the p53 tumor suppressor, which transcriptionally regulates multiple disparate effector pathways that control various aspects of cancer biology such as cell cycle control, apoptosis, and angiogenesis. Therefore, inhibition of a single downstream effector is often insufficient to completely restrain signaling from a mutant tumor suppressor gene.
A second strategy for overcoming the “loss-of-function problem” is to perform so-called phenotypic high-throughput screening (HTS) with isogenic paired cell lines that differ only in the tumor suppressor gene of interest.3 This approach, sometimes known as screening for “synthetic lethality,” makes it possible to identify chemical compounds that are preferentially cytotoxic or cytostatic toward cells with a mutant tumor suppressor gene, without requiring any information about the gene's function. However, the lack of a discrete, purified biochemical target can complicate subsequent definition of structure–activity relationships (sar) for chemical optimization. it is worth noting, however, that Powell et al.4 and Wang et al.5 Have demonstrated that it is possible to perform effective SAR using a paired, cell-based phenotypic screen as the primary assay. because of these concerns, relatively few synthetic lethality compound screens have been performed and published (rare examples in Jennings et al.,6 Issaeva et al.,7 Dolma et al.,8 and Stockwell et al.9). Instead, most synthetic lethality screening is currently performed with shRNA or siRNA libraries to identify synthetically lethal targets, not synthetically lethal compounds. The idea is that once a target is identified, it can be purified for a standard inhibitor-based screening campaign.
In addition to concerns about the feasibility of defining SAR, it has proven challenging to develop the paired isogenic cell lines needed for tumor suppressor gene–targeted phenotypic screens. this is in large part because stable, ectopic reexpression of wild-type tumor suppressor genes in human cancer cells that lack them generally leads to cell cycle arrest or cell death—neither outcome compatible with the generation of a proliferating cell line. In an effort to overcome this problem and develop paired cell lines for phenotypic screening campaigns, we and others have developed human somatic cell gene targeting technology (reviewed in Rago et al.10 and Waldman et al.11). This technique, which is, in principal, similar to making knockout mice, makes it possible to create isogenic pairs of human cancer cells that differ only in the presence or the absence of a single wild-type tumor suppressor gene.
PTEN is one such tumor suppressor gene for which the availability of targeted therapeutics would be extremely valuable. PTEN, located on chromosome 10q, is one of the most commonly deleted and mutated genes in human cancer.12,13 The PTEN gene encodes a PIP3 lipid phosphatase known to regulate a variety of downstream pathways, including checkpoint control, cell growth, sensitivity to apoptosis, and others. We and others have previously reported the use of human somatic cell gene targeting to create isogenic sets of human cancer cells that differ only in the presence or absence of their endogenous wild-type PTEN genes.14–16
To identify compounds that are selectively cytotoxic or cytostatic toward cells with mutational inactivation of the PTEN tumor suppressor gene, we performed a high-throughput screen of 138 758 compounds in HCT116 PTEN+/+ and PTEN−/− cells. N′-[(1-benzyl-1H-indol-3-yl)methylene]benzenesulfonylhydrazide (CID1340132) was identified as a compound that preferentially inhibited the growth of cells with PTEN deletion, as well as in cells expressing mutant PIK3CA. Furthermore, CID1340132 induced DNA damage, inhibited cellular proliferation, and induced apoptosis. As such, in this study, we provide validation for the general approach of using gene-targeted human cell lines for the identification of tumor suppressor–specific compounds, and we further characterize one potentially promising PTEN-specific lead compound discovered in this screening campaign.
Materials and Methods
Cell lines
HCT116 PTEN−/− cells, HEC1a PTEN−/− cells, MCF10A PTEN−/− cells, and HCT116 PIK3CA knockout (KO) cells were created by human somatic gene targeting and have been previously described.14–17 All parental cells were obtained from the American Type Culture Collection (Manassas, VA), which authenticates all human cell lines by DNA fingerprinting, and independent evidence of authenticity is also provided by cytogenetic and immunophenotypic tests. All experiments were performed on cell lines that had been passaged <6 months after receipt. HCT116 and Hec1a cells were maintained in McCoy's 5A medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. MCF10A PTEN+/+ and PTEN−/− cells were maintained in Dulbecco's modified Eagle's medium (DMEM)/F-12 medium (Invitrogen) supplemented with 5% horse serum, 20 ng/mL epidermal growth factor (EGF; sigma, St. Louis, MO), 10 μg/ mL insulin (Invitrogen), 0.5 μg/mL hydrocortisone (Sigma), and 1% penicillin/streptomycin. All cells were cultured at 37°c in 5% CO2.
High-throughput drug screening
HCT116 PTEN+/+ and PTEN−/− cells were seeded at a density of 1250 cells per well in 384-well, black, clear-bottom tissue culture plates, in 20 μL McCoy's 5A medium with 10% FBS. After 48 h, 5 μL of complete medium containing compounds from the national institutes of Health (NIH) Roadmap compound library at 5× the final dose was added to the plates, resulting in a final culture volume of 25 μL. The final concentration of each compound was 10 μM in 0.1% DMSO. in addition, 0.1% DMSO was used as a negative control, and 20 μM Hyamine 1662 (Sigma) in 0.1% DMSO was used as a positive control. After 60 h, 25 μL of CellTiter-Glo reagent (Promega, Madison, WI) was added to each well, and plates were incubated at room temperature for 10 min in the dark, followed by analysis with a PerkinElmer (Waltham, MA) EnVision luminescence microplate reader. Data were validated by Z′ factor analysis, which was calculated using the following equation: Z′ factor = 1 – [(3σs + 3σb)/(μs – μb)], where σ is the SD of signal (σs) or background (σb), and μ is the mean.
Confirmatory growth inhibition assays
Cells were seeded in 96-well plates at a density of 3000 cells per well in 80 μL medium. One day after plating, various concentrations of CID1340132 in 20 μL medium were added to the plates. Cells treated with medium containing 0.1% DMSO were used as a negative control. After incubation for 72 h, 100 μL of CellTiter-Glo reagent (Promega) was added to each well for 10 min, after which luminescence was measured using the Victor2 1420 Multilabel Counter (Wallac; PerkinElmer). Percentage growth inhibition was calculated as [100 – (measurement of treatment – background)/(measurement of control – background) × 100]. IC50 values were determined after fitting growth inhibition curves to dose–response curves using GraphPad Prism software (GraphPad Software, La Jolla, CA).
Western blot analysis
Cells were lysed in lysis buffer (Cell Signaling Technologies, Danvers, MA) containing 20 mM Tris-Hcl (pH 7.5), 150 mM nacl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin, and proteinase inhibitor (Roche, Basel, Switzerland) and phosphatase inhibitor cocktails (sigma). Cell lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes. After probing with primary antibodies, the membranes were incubated with horseradish peroxidase–conjugated secondary antibody and visualized by ECL (Pierce, Rockford, IL). Antibodies specific for total Akt, P-Akt-Ser473, caspase-3, Cyclin D1, p27KIP1, and PARP were obtained from Cell Signaling Technologies. Antibodies specific for p21WAF1/CIP1 were obtained from Zymed (Carlsbad, CA), p53 (clone DO-1) from calbiochem (San Diego, CA), and α-tubulin from NeoMarkers (Fremont, CA). All primary antibodies were used at 1:1000 dilution.
Sub-G1 fraction analysis
Sub-G1 fractions were determined by flow cytometric analysis of nuclear DNA content following staining with pro-pidium iodide. Briefly, cells were treated with CID1340132, harvested and fixed in 70% ethanol, and stained with phosphate-buffered saline (PBS) containing 0.1% Triton X-100, 50 μg/mL RNase, and 50 μg/mL propidium iodide for 60 min at room temperature. Fluorescence was measured on a FACSort flow cytometer (Becton Dickinson, Franklin Lakes, NJ), and data were analyzed using ModFit software (Verity Software House, Topsham, ME).
BrdU incorporation assay
Cells were pulsed with 10 μmol/L bromodeoxyuridine (BrdU) for 1 h, then trypsinized, fixed, and stained using the BrdU Flow Kit (PharMingen, San Diego, CA) following the manufacturer's instructions. Cells were counterstained with propidium iodide, and fluorescence was measured on a FACsort flow cytometer (Becton Dickinson). Data were analyzed using FCs express v.3 software (DeNovo Software, Los Angeles, CA). In total, 20 000 events were collected, and doublet/triplet discrimination was performed by area width analysis.
γ-H2AX staining
HCT116 cells and gene targeted derivatives were grown on 0.01% poly-L-Ornithine (Sigma)–coated cover slips and treated with 20 μM CID1340132 or 0.1% DMSO. After 24 h, cells were fixed with cold methanol for 20 min on ice and then blocked with 10% normal goat serum for 1 h at room temperature. Cells were then immunostained with antibody against γ-H2AX (clone JBW301) from Upstate Biotechnology (Lake PlaCID, NY; 1:500), and stained cells were detected with FITC-conjugated secondary antibody. Hoechst 33342 staining was used to detect nuclei. γ-H2AX foci were visualized under an Olympus Laser confocal microscope (Olympus, Center Valley, Pa). Nuclei with more than five foci were counted as γ-H2aX foci-positive cells, and a minimum of 100 nuclei were analyzed to estimate γ-H2AX foci formation frequency.
Comet assay
HCT116 cells and gene-targeted derivatives were treated with various concentrations of CID1340132 or vehicle alone for 30 h. Wells were washed with PBS, and attached cells were harvested in PBS containing 20 mM edta by scraping. cells were washed once with cold PBS containing 20 mM EDTA and were suspended at 1 × 105 cells/mL in cold PBS. Comet assays were performed using the OxiSelect Comet Assay Kit (cell Biolabs, San Diego, CA) following the manufacturer's instructions. Briefly, 10 μL of cell suspension was mixed with 100 μL Comet Agarose, and 75 μL of the mixture was added to OxiSelect Comet Slides. Slides were placed at 4°c for 15 min to solidify the agarose and immersed in lysis buffer provided in the kit for 1 h at 4°c, followed by electrophoresis at 14 V, 300 ma in alkaline electrophoresis buffer for 30 min. After electrophoresis, slides were rinsed three times with water, fixed with 70% ethanol, and air-dried. Slides were then stained with Vista Green DNA Dye and observed under a Nikon E600 Epifluorescence microscope (Nikon, Tokyo, Japan). Pictures of nuclei were recorded, and tail moments of at least 100 random nuclei were evaluated using the Comet Assay IV software (Perceptive Instruments, Suffolk, UK).
Statistical analysis
All quantitative data obtained after the high-throughput screen were analyzed by two-tailed unpaired student t test or one-way analysis of variance (ANOVA) using GraphPad Prism software. Errors and error bars represent SEM.
Results
Validation of a phenotypic cell-based screen for PTEN-targeted compounds
We have previously described the creation of an isogenic set of HCT116 colon cancer cells with targeted deletion of both wild-type alleles of PTEN. Deletion of PTEN from HCT116 cells led to a substantial increase in phosphorylated Akt, an inability to restrain cell size during cell cycle arrest, and activation of p53.14,17
Initial optimization and validation studies were performed to convert this cell system into a high-throughput, cell-based screen. First, we determined the optimal number of cells to plate in a 384-well plate so the cells would continue proliferating for 5 days after plating. The timeframe was designed to make it possible to allow the cells to adhere for 48 h after plating, add compounds, and incubate for an additional 60 h. The objective was to plate few enough cells so they are dividing throughout a 60-h assay (making it possible to detect compounds with cytostatic activity) but a large enough number of cells to produce a favorable signal-to-noise ratio.
To determine an optimal number of cells for the screen, various numbers of HCT116 PTEN+/+ and HCT116 PTEN−/− cells were plated in 384-well plates and relative cell number assessed with the CellTiter-Glo assay after 5 days of incubation. Based on these data, 1250 HCT116 PTEN+/+ and 1500 HCT116 PTEN−/−cells were plated per well for subsequent experiments.
Next, we tested the screen against a small number of well-characterized compounds: wortmannin, hygromycin, LY294002, and geneticin (Fig. 1A). We expected that wortmannin, hygromycin, and LY294002 were likely to demonstrate roughly similar cytotoxicity against both cell lines. In contrast, we expected geneticin to have enhanced cytotoxicity against HCT116 PTEN+/+ parental cells because the targeting vector used to create the PTEN−/− derivatives conferred resistance to geneticin. These compounds were tested at a 4 log range of concentrations in triplicate in a 60-h assay to demonstrate that the assay was robust and yielded reproducible responses for the compounds tested (Fig. 1A). Using the data from these experiments, Z factors were calculated.18 For HCT116 PTEN+/+ cells, Z′ = 0.660; for HCT116 PTEN−/− cells, Z′ = 0.787, which are indicative of a highly robust assay suitable for HTS.
Fig. 1.

Validation and implementation of PTEN-targeted cell-based screen. (A) Four test compounds were used to test the robustness of the screen and determine Z′. Geneticin was used as a positive control because the HCT116 PTEN+/+ cells are G418S, whereas the PTEN−/−cells are G418R. (B) Cytotoxicity profiles of 138758 compounds against PTEN+/+ and PTEN−/− isogenic cells. Compounds were tested at a single concentration of 10 μM in a 60-h assay. (C) Chemical structure of CID1340132.
Screen implementation
The full screen was then implemented at the southern research Molecular Libraries Screening Center in Birmingham, Alabama (Table 1, Table 2, and Fig. 1B). A total of 138 758 compounds derived from the NIH small-molecule repository were tested at a final concentration of 10 μM, as well as cell viability and number measured 60 h later using the CellTiter-Glo assay (Promega). A scatter graph of growth inhibition in PTEN+/+ versus PTEN−/− cells from the single-dose screen is depicted in Figure 1B. In total, 2052 compounds were identified with cytotoxic or cytostatic activity toward HCT116 PTEN+/+ cells (defined as IC50 <10 μM), and 1661 compounds were identified with cytotoxic or cytostatic activity toward HCT116 PTEN−/− cells (Table 1). Of these putatively active compounds, 1658 were retested in a 10-point dose response (30–0.06 μM), and 565 and 559 were confirmed to have IC50 values <10 μM in the HCT116 PTEN+/+ and PTEN−/− cells, respectively. Twelve compounds were selected for follow-up based on a PTEN+/+ ic50/PTEN−/− IC50 value >5 (Table 2). Fresh powders were purchased for these compounds and manually retested to eliminate false positives, leaving two hits for further study: CID1340132 and CID6143250.
Table 1. Initial Cytotoxicity Screen.
| HCT116PTEN+/+ | HCT116PTEN−/− | |||||
|---|---|---|---|---|---|---|
| Compounds Tested | IC50 <10 μM | % | Compounds Tested | IC50 <10 μM | % | |
| Single-dose screen | 138 758 | 2052 | 1.5 | 138 758 | 1661 | 1.2 |
| Ten-point dose screen | 1658 | 565 | 34.1 | 1658 | 559 | 33.7 |
Table 2. Subsequent Differential Cytotoxicity Screen.
| Compounds Tested | ΔIC50(PTEN+/+/PTEN−/−)>5 | % | |
|---|---|---|---|
| Ten-point dose screen | 1658 | 12 | 0.7 |
| Manual test | 12 | 2 | 16.7 |
Validation of CID1340132 as a novel compound that selectively inhibits the growth of HCT116 PTEN−/− cells
We next performed dose–response evaluations of these two compounds to confirm the results obtained from the primary screen and to demonstrate that the differential growth inhibitory effect was not a clone-specific artifact specific to a single clone of gene-targeted cells. As such, these experiments were performed using multiple independently derived clones of HCT116 PTEN−/− and PTEN+/+ cells.
Of the two hits, CID1340132 (N′-[(1-benzyl-1H-indol-3-yl) methylene] benzenesulfonylhydrazide) was most robust and selective in preferentially inhibiting the growth of HCT116 PTEN−/− cells as compared with PTEN+/+ cells. As such, we focused our subsequent efforts on characterizing this compound. The chemical structure of CID1340132 is shown in Figure 1C, and growth inhibition curves for HCT116 PTEN+/+ and PTEN−/− cells are depicted in Figure 2A.
Fig. 2.

CID1340132 selectively inhibits cell viability in PTEN−/− cells. Effect of CID1340132 on cell viability of (A) HCT116, (B) Hec1a, and (C) MCF10A PTEN+/+ and PTEN−/−. (D) Effect of CID1340132 on cell viability of HCT116 cells with targeted deletion of either wild-type or oncogenic PIK3CA.
To determine if the preferential growth inhibitory activity of CID1340132 toward HCT116 PTEN−/− cells was generalizable to model systems derived from other tumor types, the compound was tested against several additional isogenic sets of PTEN gene-targeted human cancer cells. The cell lines included Hec1a PTEN+/+ and PTEN−/− cells (derived from endometrial cancer) and McF10a PTEN+/+ and PTEN−/− cells (derived from normal breast epithelium). In each case, the PTEN−/− cells were more sensitive to CID1340132 than their respective parental PTEN+/+ cells (Fig. 2B,C). Relative PTEN+/+ ic50/PTEN−/− IC50 for Hec1a and McF10a cells was 2.1 (25.2 ± 1.5 μM vs. 14.2 ± 1.5 μM) and 8.0 (49.5 ± 6.3 μM vs. 5.5 ± 2.3 μM), respectively.
CID1340132 is also selective toward cells expressing mutant PIK3CA
Next, we determined if CID1340132 was preferentially growth inhibitory toward cancer cells with activation of the PI3K signaling pathway via a different mechanism. Other than inactivation of PTEN, the most common mechanism for direct activation of PI3K signaling in human cancer cells is oncogenic activation of the PIK3CA oncogene.19,20 HCT116 cells harbor an endogenous activating allele of PIK3CA, and we and others have previously described the creation of HCT116 cell derivatives in which either the oncogenic or the wild-type allele of PIK3CA had been deleted via human somatic cell gene target-ing.17,21 Therefore, CID1340132 was tested against this isogenic set of PIK3CA gene-targeted HCT116 cells. We found that cells harboring mutant PIK3CA were substantially more sensitive to CID1340132 (IC50 3.9 ± 0.4 μM) than their isogenic counterparts harboring only wild-type PIK3CA (IC50 14.9 ± 0.4 μM; Fig. 2D).
CID1340132 induces apoptosis of PTEN and PIK3CA mutant cells
To identify the molecular mechanism(s) explaining the selectivity of CID1340132 toward PTEN and PIK3CA mutant cells, we tested whether CID1340132 induces apoptosis. Isogenic cells expressing either endogenous wild-type or endogenous mutant PIK3CA were treated with 0 to 20 μM CID1340132 for 48 h. Cellular apoptosis, as indicated by the presence of sub-G1 cells, was determined by flow cytometry. As shown in Figure 3A, CID1340132 induced the accumulation of sub-G1 cells in a dose-dependent manner in mutant PIK3CA cells but not in wild-type PIK3CA cells. Similar results were obtained using the isogenic set of Hec1a PTEN+/+ and PTEN−/− cells (Fig. 3B).
Fig. 3.
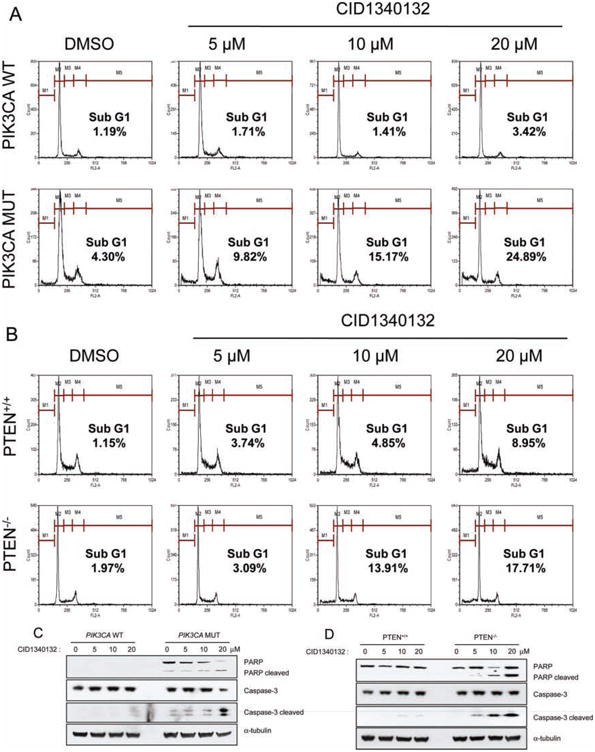
CID1340132 increases apoptosis in cells expressing mutant PIK3CA and in PTEN−/− cells. (A) isogenic HCT116 cells expressing mutant PIK3CA (PIK3CA Mut) or wild-type PI3KCA (PIK3CA WT) were treated with the indicated concentrations of CID1340132 for 48 h. cells were harvested, fixed with 70% ethanol, and labeled with propidium iodide. The sub-g1 fraction was determined by flow cytometric analysis. (B) isogenic PTEN+/+ or PTEN−/− Hec1A cells were treated with the indicated concentrations of CID1340132 for 72 h and analyzed as in a. (C) Lysates from PIK3CA Mut or PIK3CA Wt cells treated with the indicated concentrations of CID1340132 for 48 h were harvested and subjected to immunoblot analysis with the indicated antibodies. (D) Lysates from PTEN+/+ or PTEN−/− Hec1A cells were treated with the indicated concentrations of CID1340132 for 72 h and were harvested and subjected to immunoblot analysis with the indicated antibodies. PARP, poly(ADP-ribose)polymerase.
To further confirm this differential apoptosis by CID1340132 in cells with PTEN and PIK3CA mutations, we measured the extent of caspase-3 activation via Western blot for cleaved cas-pase-3 and its well-documented target, poly(ADP-ribose) polymerase (PARP). as shown in Figure 3C, CID1340132 treatment substantially increased the amount of both cleaved caspase-3 and cleaved PARP in HCT116 cells harboring mutant PIK3CA but substantially less so in cells harboring only wild-type PIK3CA. interestingly, PARP cleavage was observed in untreated HCT116 cells expressing mutant PIK3CA but not in those expressing wild-type PIK3CA, suggesting that there is more basal caspase activity in HCT116 cells expressing mutant PIK3CA. Similarly, CID1340132 treatment induced more cas-pase-3 activation in PTEN−/− than in otherwise isogenic PTEN+/+ cells (Fig. 3D). Together, these data indicate that the preferential killing of PTEN and PIK3CA mutant cells by CID1340132 is due to an increase in apoptosis.
CID1340132-induced apoptosis is accompanied by an Akt-independent decrease in cyclin D1 and increase in p27KIP1
To further investigate the molecular mechanism of the differential apoptosis, we tested whether CID1340132 treatment modified the extent of Akt phosphorylation by performing Western blots with phosphospecific antibodies. there was no inhibitory effect of CID1340132 on Akt phosphorylation; in fact, treatment with the drug seemed to increase levels of P-Akt (Fig. 4A). These data indicate that CID1340132 does not induce apoptosis via inactivation of the prosurvival factor Akt.
Fig. 4.

CID1340132 inhibits proliferation of isogenic HCT116 cells expressing mutant PIK3CA by decreasing cyclin D1 and increasing p27 independently of Akt phosphorylation. (A) Lysates from PIK3CA Mut or PIK3CA WT cells were treated with the indicated concentrations of CID1340132 for 48 h and were harvested and subjected to immunoblot analysis with the indicated antibodies. Results from two individual experiments are shown. (B) Cell lysates prepared as in A were subjected to immunoblot analysis with the indicated antibodies. results from three individual experiments are shown. (C) The fold induction of the normalized protein amount of cyclin D1 or p27KIP1 in cells treated with CID1340132 at the indicated concentrations over that without treatment was quantified from three experiments. Results represent mean ± SEM. *p < 0.05. **p < 0.01 (PIK3CA Wt vs. PIK3CA Mut). (D) PIK3CA Mut or PIK3CA Wt cells were treated with DMSO or 20 μM CID1340132 for 48 h, and DNA synthesis was detected by the BrdU incorporation assay as described in Materials and Methods.
We next measured the amount of cyclin D1 and p27KIP1 in cells treated with CID1340132. We found that CID1340132 preferentially decreased cyclin D1 and increased p27KIP1 levels in cells expressing mutant PIK3CA (Fig. 4B,C). Similar results were observed in HECLA PTEN−/− cells treated with CID1340132 (data not shown). Because the reduction of cyclin D1 and accumulation of p27KIP1 indicates inhibition of cellular proliferation, we evaluated DNA synthesis using a BrdU incorporation assay in cells treated with CID1340132. Incubation with 20 μM CID1340132 for 48 h suppressed DNA synthesis to a greater degree in PIK3CA mutant cells than in PIK3CA wild-type cells (Fig. 4D). These studies provide evidence to suggest that CID1340132 preferentially inhibits DNA synthesis in cells with mutant PTEN and PIK3CA genes by decreasing cyclin D1 and increasing p27KIP1.
CID1340132 induces DNA damage in PIK3CA mutant cells
We and others have previously demonstrated that activation of the Pi3K signaling pathway can activate p53.17,22 Therefore, we wanted to elucidate the role of p53 activation in the preferential cytotoxicity of CID1340132 toward PTEN and PIK3CA mutant cells. to test this, steady-state levels of p53 and its effector p21WAF1/CIP1 were measured by Western blot in CID1340132-treated HCT116 PIK3CA gene-targeted cells. This experiment demonstrated that, in fact, CID1340132 induced upregulation of p53 and p21. Furthermore, CID1340132 induced greater p53 accumulation in cells harboring mutant PIK3CA than in otherwise isogenic cells harboring wild-type PIK3CA (Fig. 5A). These data suggest that the preferential apoptosis could be due to enhanced activation of p53 in PTEN and PIK3CA mutant cells.
Fig. 5.

CID1340132 induces DNA damage in isogenic PIK3CA mutant HCT116 cells. (A) Lysates from PIK3CA Mut or PIK3CA WT cells treated with the indicated concentrations of CID1340132 for 48 h were harvested and subjected to immunoblot analysis with the indicated antibodies. Results from three independent experiments are shown. (B) PIK3CA Mut or PIK3CA WT cells growing on cover glass were treated with DMSO or 20 μM CID1340132 for 30 h and were fixed with cold methanol and immunostained with antibody specific for γ-H2aX. PIK3CA WT cells treated with 10 μg/mL etoposide for 8 h were used as positive control. γ-H2aX foci were observed by confocal microscopy. representative images from three independent experiments are shown. (C) PIK3CA Mut or PIK3CA WT cells were treated with DMSO or CID1340132 at the indicated concentration. After incubation for 30 h, cultures were washed with phosphate-buffered saline, and attached cells were harvested and analyzed by the Oxiselect comet assay Kit as described in Materials and Methods. Representative electrophoresis images from three independent experiments are shown. Arrows indicate damaged nuclei. **p < 0.01. ***p < 0.001.
Because p53 accumulation can be an indicator of DNA damage, we next tested whether CID1340132 could induce DNA damage. First, DNA damage was examined by immunostaining for γ-H2AX, a marker for DNA double-strand breaks (DSBs). Cell staining was performed 24 h after treatment with vehicle or with CID1340132 to eliminate the effect of apoptosis-related DNA damage. Treatment with etoposide, a known DNA-damaging reagent, was used as a positive control (data not shown). Representative images are shown in Figure 5B (left panel), and quantification of results is shown in Figure 5B (right panel). We found that 20 μM CID1340132 significantly induced γ-H2aX foci in PIK3CA mutant cells but less so in isogenic cells expressing wild-type PIK3CA (Fig. 5B).
DNA damage was next evaluated by a comet assay (also known as single-cell electrophoresis) in cells treated with various concentrations of CID1340132 for 30 h. Representative images and quantification of results are shown in Figure 5C. Treatment with CID1340132 at 30 μM and 50 μM significantly increased tail moments in PIK3CA mutant cells but not in PIK3CA wild-type cells. Likewise, tail moments in HCT116 cells expressing mutant PIK3CA were significantly increased over that of wild-type PIK3CA cells under all conditions, even in the absence of CID1340132. These results are consistent with those described for γ-H2aX staining and indicate that CID1340132 preferentially induces DNA damage in cells with increased PI3K pathway activity.
Discussion
As one of the most commonly mutated genes in human cancer, PTEN is an appealing target for the development of anti-cancer agents. In this study, we developed and implemented a high-throughput screen for the identification of small molecules that were preferentially growth inhibitory toward cells with PTEN mutations. Several hits were identified, and one, named CID1340132, was further characterized in a variety of cancer cell systems. CID1340132 was generalizably cytotoxic toward cells with mutations in PTEN. As such, it represents a promising small-molecule lead compound for further study, derivatization, and development.
Evaluation of CID1340132 showed that its preferential cytotoxicity for PTEN-deficient cells was at least partly due to enhanced apoptosis. We also found that CID1340132 was preferentially cytotoxic for cells harboring mutant PIK3CA genes. Importantly, however, CID1340132 does not inhibit Akt phosphorylation and is therefore neither a PI3K inhibitor such as LY294002 nor an allosteric Akt inhibitor such as MK2206.
Instead, our data suggest that CID1340132 may have properties of a DNA-damaging agent and that these properties are enhanced in cells with mutant PTEN or PIK3CA. We should note, however, that CID1340132 has no structural properties in common with known DNA-damaging chemotherapeutics. Of note, other DNA-damaging reagents have been previously shown to selectively inhibit the growth of isogenic cells with mutations of PTEN. For example, we and others have previously shown that PTEN−/− cells are sensitized to ionizing radiation, doxorubicin, and cisplatin.14–16 it has also been reported that PARP inhibitors selectively inhibit the survival of PTEN−/− isogenic cells.15 Further studies will be required to determine the extent to which CID1340132-mediated cytotoxicity is due to the induction of DNA damage or other, as yet unidentified mechanisms.
In addition to the discovery of a novel, PTEN-specific lead compound, this study provides proof of principle for a relatively novel approach for the discovery of small molecules that preferentially kill cells with loss-of-function mutations in tumor suppressor genes. Despite the fact that tumor suppressor gene mutations are the most common mutations driving malignancy, there are no anticancer drugs specifically designed to kill cells with tumor suppressor gene inactivation, and few (if any) such compounds are in the pipeline. This paucity of compounds is due to the fundamental difficulty in targeting “loss of functions” with therapeutics.
Solving the loss-of-function problem that results from tumor suppressor gene mutations could dramatically accelerate anti-cancer drug discovery efforts because virtually all current efforts are focused on identifying/designing pharmacological inhibitors of activated oncogenes. Although targeting oncogenes has already resulted in the discovery of novel, effective anticancer compounds (e.g., imatinib, sunitinib), the strategy is, by definition, limited to the small minority of cancer-causing mutations that lead to a gain of function. We believe that if we can perfectly recapitulate a mutant tumor suppressor gene in cultured human cancer cells, we can employ paired cell screens using otherwise isogenic cells to identify compounds that preferentially kill cells with mutant tumor suppressor genes. This strategy is quite distinct from the nci 60-cell screen because the cell lines composing the 60-cell screen were genetically unrelated, and therefore it was not a target-based effort.
Of note, although paired cell-based screens based on gene-targeted cells are not strictly mechanism based, we believe this to be an advantage because the screening campaign is not based around potentially incorrect and always limiting notions about the function of the tumor suppressor gene. Instead, it relies on the simple notion that if we can perfectly recapitulate a genetic difference between cancer cells and normal cells, we can exploit it to identify novel lead compounds that target the loss of function.
Although we are optimistic about the potential value of such paired, cell-based phenotypic screens, there are several potential challenges involved in their execution and hit validation. First, many of the compounds identified in such screens display cytotoxicity not only toward the cells with deletion of the tumor suppressor gene but also toward the control cells (albeit less so). Therefore, it remains a possibility that such compounds could display toxicity in an in vivo setting. Second, these screens require that any hits be cell-permeable compounds. Third, performing SAR in the context of a cell-based screen with an unknown biochemical target of action is substantially more challenging than performing SAR with a purified biochemical target.
In conclusion, we have identified a novel compound with preferential specificity for killing PTEN−/− cells. The strategy employed validates and provides proof of principle for the use of gene-targeted human cancer cells to identify compounds that target loss-of-function mutations. Furthermore, the compound identified may be useful as a lead compound for the generation of a potential PTEN and PIK3CA-specific anticancer agent.
Acknowledgments
We thank David Solomon for comments on the manuscript. The complete data for the high-throughput screens are available on PubChem at AIDs 824, 827, 999, and 1047.
This work was supported by NIH grants R01 ca115699 and R03 NS050857 to T.W., American Cancer Society RPG MGO-112078 to T.W., and NIH U54 HG003917 to G.A.P.
Footnotes
The online version of this article can be found at: http://jbx.sagepub.com/content/16/4/383
References
- 1.Kamb A, Wee S, Lengauer C. Why is Cancer Drug Discovery So Difficult? Nat Rev Drug Discov. 2007;6:115–120. doi: 10.1038/nrd2155. [DOI] [PubMed] [Google Scholar]
- 2.Benson JD, Chen YN, Cornell-Kennon SA, Dorsch M, Kim S, Leszczyniecka M, Sellers WR, Lengauer C. Validating Cancer Drug Targets. Nature. 2006;441:451–456. doi: 10.1038/nature04873. [DOI] [PubMed] [Google Scholar]
- 3.Kaelin WG. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat Rev Can. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 4.Powell D, Gopalsamy A, Wang YD, Zhang N, Miranda M, McGinnis JP, Rabindran SK. Pyrazolo[1,5-a]pyrimidin-7-yl Phenyl Amides as Novel Antiproliferative Agents: Exploration of Core and Headpiece Structure-Activity Relationships. Bioorg Med Chem Lett. 2007;17:1641–1645. doi: 10.1016/j.bmcl.2006.12.116. [DOI] [PubMed] [Google Scholar]
- 5.Wang YD, Honores E, Wu B, Johnson S, Powell D, Miranda M, Mcginnis JP, Discafani C, Rabindran SK, Cheng W, et al. Synthesis, SAR Study and Biological Evaluation of Novel Pyrazolo[1,5-a] pyrimidin-7-yl Phenyl Amides as Anti-Proliferative Agents. Bioorg Med Chem Lett. 2009;17:2091–2100. doi: 10.1016/j.bmc.2008.12.046. [DOI] [PubMed] [Google Scholar]
- 6.Jennings LD, Kincaid SL, Wang YD, Krishnamurthy G, Beyer CF, Mcginnis JP, Miranda M, Discafani CM, Rabindran SK. Parallel Synthesis and Biological Evaluation of 5,6,7,8-tetrahydrobenzothieno[2,3-d] pyrimidin-4(3H)-one Cytotoxic Agents Selective for p21-Deficient Cells. Bioorg Med Chem Lett. 2005;15:473–475. doi: 10.1016/j.bmcl.2005.07.072. [DOI] [PubMed] [Google Scholar]
- 7.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Small Molecule RITA binds to p53, Blocks p53-HDM-2 Interaction and Activates p53 Function in Tumors. Nat Med. 2004;10:1321–1328. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 8.Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. Cancer Cell. 2003;3:285–290. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 9.Stockwell BR, Haggarty SJ, Schreiber SL. High-Throughput Screening of Small Molecules in Miniaturized Mammalian Cell-Based Assays Involving Post-Translational Modifications. Chem Biol. 1999;6:71–8. doi: 10.1016/S1074-5521(99)80004-0. [DOI] [PubMed] [Google Scholar]
- 10.Rago C, Vogelstein B, Bunz F. Genetic Knockouts and Knockins in Human Somatic Cells. Nat Protoc. 2007;2:2734–2746. doi: 10.1038/nprot.2007.408. [DOI] [PubMed] [Google Scholar]
- 11.Waldman T, Lee C, Nishanian TG, Kim JS. Human somatic cell Gene Targeting. Curr Protoc Mol Biol. 2003;Chapter 9 doi: 10.1002/0471142727.mb0915s62. Unit 9.15. [DOI] [PubMed] [Google Scholar]
- 12.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN Tumor Suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 13.Parsons R. Human Cancer, PTEN and the Pi-3 Kinase Pathway. Semin Cell Dev Biol. 2004;15:171–176. doi: 10.1016/j.semcdb.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 14.Lee C, Kim JS, Waldman T. PTEN Gene Targeting Reveals a Radiation-Induced Size Checkpoint In Human Cancer Cells. Cancer Res. 2004;64:6906–6914. doi: 10.1158/0008-5472.CAN-04-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendes-Pereira AM, Martin SA, Brough R, Mccarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ, Ashworth A. Synthetic Lethal Targeting of PTEN Mutant Cells with PARP Inhibitors. EMBO Mol Med. 2009;1:315–322. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vitolo MI, Weiss MB, Szmacinski M, Tahir K, Waldman T, Park BH, Martin SS, Weber DJ, Bachman KE. Deletion of PTEN Promotes Tumorigenic Signaling, Resistance to Anoikis, and Altered Response to Chemotherapeutic Agents in Human Mammary Epithelial Cells. Cancer Res. 2009;69:8275–8283. doi: 10.1158/0008-5472.CAN-09-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-Dependent Growth Suppression in Human Cells by Mutations in PTEN or PIK3CA. Mol Cell Biol. 2007;27:662–677. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sui Y, Wu Z. Alternative Statistical Parameter for High-throughput Screening Assay Quality Assessment. J Biomol Screen. 2007;12:229–234. doi: 10.1177/1087057106296498. [DOI] [PubMed] [Google Scholar]
- 19.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 20.Samuels Y, Waldman T. Mutations of PIK3CA in Human Cancer. New York: Springer Verlag; in press. [Google Scholar]
- 21.Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, et al. Mutant PIK3CA Promotes Cell Growth and Invasion of Human Cancer Cells. Cancer Cell. 2005;7:561–573. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 22.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial Role of p53-dependent Cellular Senescence in Suppression of PTEN-Deficient Tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]