

Abstract
Accurate evaluation of microbial communities is essential for understanding global biogeochemical processes and can guide bioremediation and medical treatments. Metagenomics is most commonly used to analyze microbial diversity and metabolic potential, but assemblies of the short reads generated by current sequencing platforms may fail to recover heterogeneous strain populations and rare organisms. Here we used short (150-bp) and long (multi-kb) synthetic reads to evaluate strain heterogeneity and study microorganisms at low abundance in complex microbial communities from terrestrial sediments. The long-read data revealed multiple (probably dozens of) closely related species and strains from previously undescribed Deltaproteobacteria and Aminicenantes (candidate phylum OP8). Notably, these are the most abundant organisms in the communities, yet short-read assemblies achieved only partial genome coverage, mostly in the form of short scaffolds (N50 = ∼2200 bp). Genome architecture and metabolic potential for these lineages were reconstructed using a new synteny-based method. Analysis of long-read data also revealed thousands of species whose abundances were <0.1% in all samples. Most of the organisms in this “long tail” of rare organisms belong to phyla that are also represented by abundant organisms. Genes encoding glycosyl hydrolases are significantly more abundant than expected in rare genomes, suggesting that rare species may augment the capability for carbon turnover and confer resilience to changing environmental conditions. Overall, the study showed that a diversity of closely related strains and rare organisms account for a major portion of the communities. These are probably common features of many microbial communities and can be effectively studied using a combination of long and short reads.
Metagenomics is a cultivation-independent approach for studying microbial communities. The dramatic increase in DNA sequencing throughput, accompanied by the development of new bioinformatics approaches for the assembly (Peng et al. 2012) and binning (Dick et al. 2009; Baran and Halperin 2012; Sharon et al. 2013) of metagenomic data facilitate the study of microbial communities based on the genomes of their members (Tyson et al. 2004; Goltsman et al. 2009; Wrighton et al. 2012; Brown et al. 2013; Sharon et al. 2013). One major drawback of most commonly used sequencing platforms is read length, which is typically in the range of a few hundred base pairs (bp). Short-read length is compensated by high throughput, which provides high coverage for the abundant genomes in the community, allowing assembly and sometimes even complete genome recovery (Iverson et al. 2012; Albertsen et al. 2013; Castelle et al. 2013; Di Rienzi et al. 2013; Kantor et al. 2013). Short-read assemblers sometimes fail to assemble similar repeating regions (Miller et al. 2011). For de Bruijn graph assemblers such as IDBA-UD (Peng et al. 2012) and Ray Meta (Boisvert et al. 2012), the presence of multiple similar regions should result in bubbles and short paths in the de Bruijn graph. Consequently, assembly of these regions will result in short assembled contigs or elimination of the regions altogether. Assemblers are therefore expected to perform poorly in the presence of multiple similar genomes from closely related species and strains. In addition, the assembly of rare genomes fails due to insufficient sequencing coverage. While these issues can limit understanding of the true community composition, the extent to which they do so is currently unknown.
Previously, we used short-read (150-bp) metagenomic data to study microbial communities in terrestrial sediments from a site near Rifle, Colorado (Castelle et al. 2013; Hug et al. 2013). Terrestrial sediments are major reservoirs of organic matter on Earth, and microbes play key roles in carbon turnover in these environments (Whitman et al. 1998), thus affecting the global carbon cycle. Recent studies showed that many of the microbes living in terrestrial sediments are fermenters that belong to candidate phyla or to deep branches with no close cultured representatives of other phyla. Communities in these environments are complex, with no single organism's share typically exceeding 1% (Wrighton et al. 2012; Castelle et al. 2013; Hug et al. 2013; Kantor et al. 2013). Nearly two hundred different species per community were detected in samples recovered from the same site (Wrighton et al. 2012, 2014; Castelle et al. 2013); however, the true community complexity of sediment communities is currently unknown.
Recently, a new sequencing technology (previously licensed to Moleculo, acquired by Illumina in 2012 and name changed to Illumina TruSeq Synthetic Long-Reads) that enables the sequencing of long multi-kb synthetic reads was introduced (Voskoboynik et al. 2013). Here we used the new synthetic long-read technology in tandem with the previously sequenced short (150-bp) reads to metagenomically study the sediment microbial communities described in Castelle et al. (2013) and Hug et al. (2013). The main objectives of the study were to (1) test the efficacy of assembling the reads and using them to improve the scaffolding of contigs generated by short-read assembly, (2) evaluate the accuracy of genomes reconstructed through curation of short-read assemblies, (3) provide insight into organisms present at very low abundance levels, and (4) evaluate levels of sequence variation and genomic content in populations of closely related species and strains.
Results
Sample collection and sequencing
Samples were collected from three depths (4, 5, and 6 m) in an aquifer adjacent to the Colorado River, Rifle, Colorado, USA. These samples were previously studied (Castelle et al. 2013; Hug et al. 2013) using a total of ∼200 Gbp Illumina HiSeq short-read data with median read length of 150 bp after trimming. The analysis presented here used ∼1.5 Gbp of synthetic long-read data in addition to the short-read data for the three samples. Read size distribution for the synthetic long reads is bimodal, with peaks at 1500 and ∼8000 bp and median read lengths of ∼8000 bp (Supplemental Table S1; Supplemental Fig. S1). For evaluation of one of the reconstructed genomes, we also used 40.7 Gbp of Illumina HiSeq data from a planktonic filtrate sample (GWC2) from the same site.
Assembly of the short-read data
The short-read data were assembled using IDBA-UD (Peng et al. 2012), yielding a total of 931, 1456, and 366 Mbp in scaffolds longer than 1.5 kbp for the 4-, 5-, and 6-m samples, respectively. Relatively low portions of the reads, between 18% and 33%, could be mapped to the assembled scaffolds and contigs. A low rate of read mapping is typically indicative of complex communities with a large number of low abundance genomes or with a high degree of species and strain variations. Previously, 161 different organisms were identified in the 5-m sample based on concatenated ribosomal proteins, many of which probably belong to novel phyla (Castelle et al. 2013). Organisms from at least 16 different phyla were detected, with the most abundant ones being Proteobacteria and Chloroflexi. Eighty six of the Chloroflexi members are represented by near-complete and partial genomes (Hug et al. 2013). The complete and closed genome of another organism (RBG-1) from the candidate phylum Zixibacteria was reconstructed from the 6-m sample (Castelle et al. 2013).
Assembly of synthetic long reads and scaffolding of short-read assemblies
We attempted to assemble the synthetic long-reads data using three programs: the Celera assembler (Myers et al. 2000), Minimus 2 from the AMOS package (Treangen et al. 2011), and a program developed for this study (named Lola) (see Supplemental Information). The Celera assembler was originally developed for the assembly of Sanger reads (typical length < 1 kbp). Minimus 2 is an overlap-based pipeline that was developed for the purpose of merging assemblies. Lola is an overlap-based program that takes a conservative approach in assembling reads that may be present in multiple genomes such as 16S rRNA genes.
The Celera assembler failed to assemble the data, most likely because of the very low coverage for any of the genomes in our samples (see below). Both Lola and Minimus 2 achieved relatively low rates of assembly on the data (8%–17% of the reads for Minimus 2%, 5%–11% for Lola), with Minimus 2 assembling more reads than Lola. More than 90% of Lola's contigs were consistent with contigs generated by Minimus 2 (see Supplemental Fig. S2). A search for potential errors in the assemblies resulted in a small number of potential misassemblies for both assemblers (<1.8% for Minimus 2 and <0.4% for Lola). Minimus 2 was significantly faster and required less computational resources to execute compared to Lola (<1 h using 1 CPU for each assembly for Minimus 2, compared to several hours on a cluster with nearly 400 CPUs for Lola due to the use of the Needleman-Wunsch derivatives).
We attribute the low levels of assembly to the extremely high species richness and community evenness in our samples, leading to relatively few reads from any single genome. For example, only 1× coverage from all samples combined was achieved by synthetic long reads for the RBG-1 genome. This genome was previously reported to be the most abundant in the 5-m sample (Castelle et al. 2013). Scaffolding of the previously assembled short-read data (Castelle et al. 2013; Hug et al. 2013) using the synthetic long reads was also limited (Supplemental Fig. S3). In part, we attribute this result to the fact that short-read assembly was effective for genomes at relatively high coverage. Thus, the synthetic long reads only rarely covered gaps in the short-read data or regions that cannot be resolved with these data, such as genome repeats. The majority of synthetic long reads (between 51% and 76%) originated from genomes with <5× coverage, roughly the coverage required for significant assembly (Fig. 1). Hence, the majority of synthetic long reads derived from organisms that were at too low abundance to be sampled using the short-read assembly approach. These findings extend the result of Castelle et al. (2013), establishing the background sediment as an environment in which almost all organisms are present at similar, very low abundance levels.
Figure 1.

(A) Overlap between short-read assembled scaffolds (orange) and synthetic long reads (blue). Numbers are in Mbp and were calculated based on all overlapping regions longer than 1000 bp aligning at 98% identity or more. (B) Coverage distribution of synthetic long reads and short-read assembled scaffolds. Coverage was computed by mapping the short reads from the same data set.
Evaluating the correctness of a genome reconstructed from short-read data
Previously, we de novo assembled the short-read data from the sediment to generate the 2.1-Mbp complete genome of a member of the Zixibacteria candidate phylum (Castelle et al. 2013). In order to evaluate the quality of the assembly, we aligned the assembled synthetic long reads from the three samples to the RBG-1 genome using BLAST (Altschul et al. 1990), allowing partial significant alignments. Next, all partial alignments were manually checked to determine whether the disagreement is the result of an error in the short-read assembly, long-read assembly, or potential strain variation. This was done by collecting short reads that mapped to the suspicious regions, reassembling them, and comparing the result to the genome. RBG-1 is the single most abundant member of its phylum in our samples, thus alignment was either highly significant or did not occur. Overall, 87 assembled synthetic long-read contigs and 99 unassembled synthetic long reads aligned to the Zixibacteria (RBG-1) genome covering 75% of its length. One hundred sixty-two of the 186 long-read sequences aligned at >99% over their entire length to the genome. Twenty-two other sequences only partially aligned to the RBG-1 genome with no errors detected in the assembly of the genome. These discrepancies may have derived from other Zixibacteria genotypes, which occur in the sediment at low abundance levels (Castelle et al. 2013), or from errors in the assemblies of the synthetic long reads. The remaining two sequences revealed two local misassemblies, not of the type involving chimeras that are a concern in metagenomic assemblies (Supplemental Fig. S4; Supplemental Information). Alignment of the assembled 87 long-read contigs did not reveal any misassemblies.
Community composition as revealed by the short-read assemblies and long-read data
Microbial community composition is often evaluated using the 16S rRNA gene. Assemblers often fail to assemble sequences for this gene from metagenomic data sets (Miller et al. 2011), but taxonomically informative ribosomal protein sequences are typically reconstructed (Hug et al. 2013). Thus, we used the phylogenetic informative ribosomal protein S3 (rpS3) for evaluating community composition in the studied samples. We developed a statistical model for calibrating the rpS3 tree to the 16S rRNA tree to determine equivalent divergence levels between rpS3 and 16S rRNA genes. Parameters for the model were calculated based on data from this study as well as 1976 published genomes. This calibration should be generally applicable for other studies as well (Supplemental Information).
More 16S rRNA than rpS3 genes were recovered from the long-read data in all three samples (Supplemental Fig. S15). This is expected for unbiased data sets both because the rpS3 gene is a single-copy gene while the 16S rRNA gene may appear in multiple copies per genome and also because the 16S rRNA gene is longer. For the short-read data, however, significantly more rpS3 genes were recovered compared to the 16S rRNA genes. Most likely this reflects relatively low levels of recovery of 16S rRNA genes from short-read assemblies due to a high degree of sequence similarity. Clustering of the 16S rRNA genes based on 99% identity (species level) shows that roughly one-third of the 16S rRNA genes recovered from the long-read data originated from closely related genomes. This highlights the fact that inventories of 16S rRNA genes recovered from short-read metagenomic data can provide an unreliable view of community structure (Miller et al. 2011), biased against species presenting high strain variability.
We clustered rpS3 genes from both the long-read data and short-read assemblies at the 99% nucleic acid identity level to form species-level groups. This threshold was found by our model to be equivalent to the 99% similarity threshold often used for determining species relations based on 16S rRNA sequences (Janda and Abbott 2007). Overall, we recovered 401, 506, and 244 rpS3 genes from the 4-, 5-, and 6-m samples, respectively. We identified 376 distinct species in the sediments collected at the 4-m depth, 457 in the 5-m sample, and 233 in the 6-m sample. The lineages with the largest number of identified species were, in decreasing order, Chloroflexi, Deltaproteobacteria, and Microgenomates (candidate phylum OP11).
Nearly all rpS3 genes with coverage ≤2× from all samples were recovered from the long-read data. These genes represent the least abundant organisms in the samples and were not detected by the assembled short reads. Many of these genes have zero coverage by the short-read data, suggesting that our short-read data sets are far from being exhaustive. The majority of rpS3 genes with sufficient coverage for short-read assembly were recovered from the short-read assemblies alone. This is probably the result of better coverage of these genomes by the short-read data compared to the long-read data. Notably, some of the most abundant species were typically represented on synthetic long reads but not in the short-read assemblies (Fig. 2; Supplemental Figs. S5,S6). For example, the most abundant species in the 4-m sample is a member of candidate phylum Aminicenantes (OP8) for which four copies of rpS3 (and five 16S rRNA) genes were recovered from the long-read data. None were identified in the short-read assembly (Supplemental Fig. S5). Even more significantly, for both the 5-m and 6-m samples, the most abundant and numerous other abundant species (Supplemental Information) were not detected by the short-read assembly. These all belong to the same lineage, a new genus within the Deltaproteobacteria. The most abundant Deltaproteobacteria species has an estimated short-read coverage of 418×, higher than the coverage of the RBG-1 genome (294×), previously thought to dominate the 5-m sediment. We attribute failure to detect these species in standard short-read assemblies to strain variation that fragmented assemblies (Supplemental Information).
Figure 2.
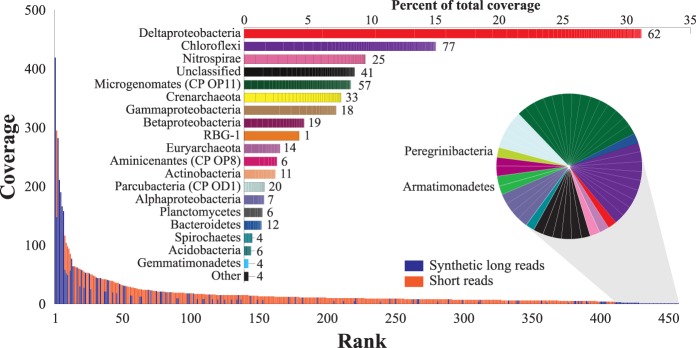
Rank abundance curve for the 5-m community including all species for which the rpS3 gene could be recovered. (Bottom) While most of the rpS3 genes were recovered from the short-read assembly (orange), least and most abundant species were represented almost exclusively by the long-read data (blue). (Top) Stacked bar graph shows abundance of phyla and Proteobacteria classes; stacked boxes indicate abundance of individual species (number of species indicated). Deltaproteobacteria is the most abundant lineage in the sample, with five of the seven most abundant species being closely related. (Pie chart) Species with zero short-read coverage in the short-read data, detected in the synthetic long reads only.
The availability of both short- and long-read data allowed us to explore patterns of population diversity, taxonomic diversity, and organism abundance levels using genome sequence information for rare as well as more abundant organisms. We considered the possibility that some phyla may be only represented by low-abundance organisms but found no evidence for this. Specifically, almost all rare genotypes in the three samples belong to phyla also represented by more abundant genotypes. Two exceptions, members of the Saccharibacteria (TM7) and Peregrinibacteria (PER) phyla, were not found in high abundance in our samples, but have been found at relatively high abundance in acetate-stimulated sediment and groundwater from the same site (Wrighton et al. 2012; Kantor et al. 2013). Interestingly, members of the Microgenomates (OP11) and Parcubacteria (OD1) candidate phyla were common in the 5-and 6-m samples, but very few genotypes were abundant. Chloroflexi and Deltaproteobacteria were represented by a large number of genotypes at varying abundance levels (Fig. 2; Supplemental Figs. S5, S6). Examples of abundant genotypes from phyla with a single or few representatives in the sediment are Aminicenantes (all samples), Gammaproteobacteria and Nirospirae (5 m), and Zixibacteria RBG-1 (5 and 6 m). Most notable of this group is RBG-1, which was represented by a single abundant strain (Fig. 2). This may reflect strong selection for this specific RBG-1 strain under the current conditions.
Reconstruction of genome architecture and metabolic potential for the most abundant populations of species and strains
In order to construct an approximate genomic and metabolic representation of the dominant populations, we collected clusters of synthetic long reads and sequences that aligned at 90% identity or higher over at least 500 bp (Supplemental Information). For the 5-m sample, we identified four clusters with 100 reads or more (3711 reads across the four clusters). All clusters had similar phylogenetic profiles dominated by Deltaproteobacteria hits. (Supplemental Fig. S7; additional files: concatenated_rp_tree.pdf and 5m.16S.bacteria.pdf). The closest published genome is Desulfobacca acetoxidans DSM 11109, sharing only 90% identity with the 16S rRNA gene sequence of the cluster. For the 4-m sample, we identified 3023 synthetic long reads in clusters with 100 reads or more. A similar phylogenetic profile with a significant number of Aminicenantes hits was found for all but one cluster, with a total of 2886 reads (Supplemental Fig. S8; additional files: concatenated_rp_tree.pdf and 4m.16S.bacteria.pdf). Clustering of the sequences was confirmed using 3-mer-based emergent self-organizing maps (ESOM) maps of synthetic long reads, generated for each sample separately (Supplemental Fig. S9).
Clustering of predicted protein sequences identified 2960 and 2949 protein families with at least two members for the Deltaproteobacteria and Aminicenantes, respectively. Given that closely related genomes are largely syntenic, especially within functional modules (Yelton et al. 2011), we used shared synteny of protein-coding genes to define genome architecture (Supplemental Information). The analysis used a new algorithm that reconstructs the long-range order of blocks of genes based on shared gene order and gene content and resolves complications that arise when genes occur in multiple contexts. One hundred and eight syntenic regions longer than 10 kbp were reconstructed for the Deltaproteobacteria population (5 m), covering ∼1.9 Mbp, with the longest region spanning 111 kbp. Seventy syntenic regions larger than 10 kbp, covering a total of 2.3 Mbp, were reconstructed for the Aminicenantes population (4-m sample), four of which were longer than 100 kbp (Supplemental Information). These syntenic regions represent sequences that are common to some or all of the genomes. We estimate that these regions cover the majority of the genomes, based on the presence of 51 and 48 protein families out of 51 single-copy marker genes for the Deltaproteobacteria and Aminicenantes, respectively. Within each group, we found that families of predicted proteins for single-copy genes shared high sequence similarity (typically 95% identity or higher) (Supplemental Tables S3, S4), demonstrating the presence of many closely related members of both Deltaproteobacteria and Aminicenantes in the two sediment samples.
The sequences for the Aminicenantes and Deltaproteobacteria do not represent genomes in the strict sense because the data sets incorporate information from multiple strains and species. To test the overall reliability of the approach, we leveraged the availability of a manually curated, near-complete genome of a fairly closely related organism from a simpler sample (GWC2 groundwater filtrate) (genome is available in Supplemental File GWC2_DPX2_65_14.tar.gz). Gene order for the GWC2 genome was similar to that in the Deltaproteobacteria data set (with some insertions/deletions), supporting the validity of the approach (Fig. 3). In the case of Aminicenantes, our genomic data set comprises 2.7 Mbp of sequence and augments the current genomic sampling of the Aminicenantes. Currently the Aminicenantes are represented by 36 partial single-cell genomes from organisms with a maximum of 88% 16S rRNA gene sequence identity to the Aminicenantes group from this study. Notably, the estimated completeness of the OP8 single-cell genomes ranged from 4% to 67%, and the largest single-cell genome was only 1.92 Mbp in length (Rinke et al. 2013).
Figure 3.
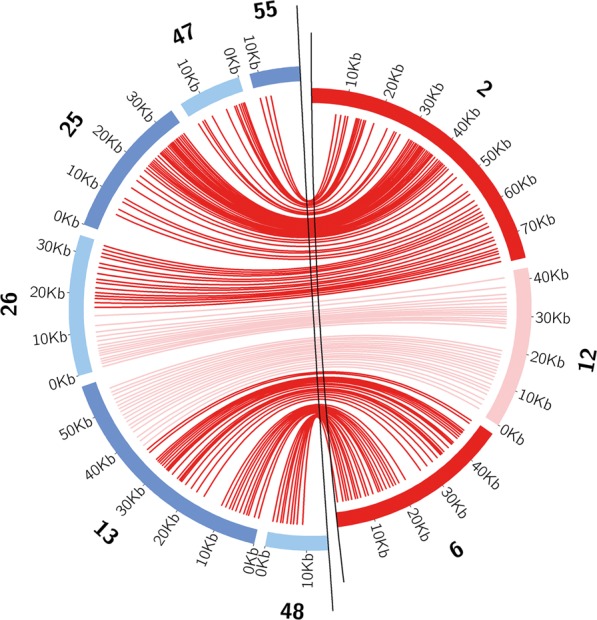
Alignment of three Deltaproteobacteria reconstructed syntenic regions from the 5-m sample to a closely related genome reconstructed from the planktonic filtrate sample GWC2. Lines connect homologous genes.
The genome data sets provide insight into the metabolic potential of important sediment-associated organisms (Fig. 4). For Aminicenantes, whose metabolic potential has not been described previously, we predict the capacity for a fermentative, saccharolytic, and aerobic lifestyle. The presence of putative extracellular and cytoplasmic glycosyl hydrolases indicates the potential for degradation and utilization of complex sediment-associated organic carbon compounds. Carbon degradation might occur via fermentation processes (including hydrogen metabolism) or respiration, based on the capacity for aerobic respiration (i.e., oxygen reductase). The Deltaproteobacteria are predicted to be capable of aerobic/anaerobic heterotrophic growth and fermentation, including the potential for nitrate reduction and oxygen reduction.
Figure 4.
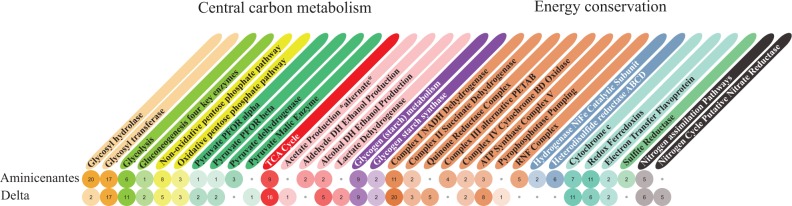
Summary of metabolic potential for the Deltaproteobacteria (5-m) and Aminicenantes (4-m) strains.
Clustering of predicted proteins from synthetic long reads
Genes and their proteins were predicted on synthetic long reads longer than 5 kbp. We clustered complete proteins predicted from the synthetic long reads in order to estimate the number of species in our samples and to identify families that are significantly more abundant among rare organisms. We used the MCL algorithm (Van Dongen 2000; Enright et al. 2002) for the clustering. Unlike short reads, synthetic long reads are sufficiently long for effective gene prediction, regardless of the coverage of the genome to which they belong. Short reads, on the other hand, are too short for reliable recognition of genes and their assembly depends on sufficient coverage. As a result, any analysis that is based on short reads is expected to be biased and to miss rare genomes, which may account for a significant portion of the community.
For each family, we compared the number of representatives from high- and low-coverage sequences using 2× coverage as a threshold. This threshold was chosen because it is slightly lower than the coverage for the least abundant genomes we were able to partially reconstruct (Fig. 1). For each sample, the expected frequency of proteins from low-coverage genomes was calculated based on the fraction of bp in reads with ≤2× coverage. Significance (P-value) of the observed frequency was computed based on the binomial distribution (see Methods).
Overall, the clustering resulted with 2907, 2833, and 2432 families with 20 members or more for the 4-, 5-, and 6-m samples, respectively. Clusters with more than 1000 members in our data sets (three from each sample) (Supplemental Tables S13–S15) represented ABC transporter ATP-binding protein and dehydrogenases. Both types of proteins were highly abundant among bacteria, and both annotations were too general to draw meaningful conclusions. A few of these clusters were significantly more prevalent in abundant species compared to rare species (see below).
Estimating the number of species in each of the samples
In order to evaluate the fraction of microbial cells represented by low-coverage, long-read sequences, we examined the abundance of families of single-copy genes in the three samples. Single-copy genes were found to be significantly more abundant in high-coverage sequences (Supplemental Tables S7–S9; Supplemental Fig. S18). Several reasons related to methodological limitations and the nature of the samples could explain this result. First, it is possible that a significant amount of reads from nonmicrobial genomes such as phages and plasmids are present in our samples. While we were able to identify only a handful of such sequences, a reliable identification of all these sequences is difficult since many reads do not contain potential marker genes and do not have close hits in public databases. Second, it is possible that the average genome size for abundant organisms in our samples is smaller than the average size of the genome for rare organisms. Note that since the number of abundant species is relatively small, the average size for these genomes may be misleading. Finally, it is possible that our tools failed to identify single-copy genes from novel lineages in the bacterial tree and from domains other than bacteria. This possibility is supported by the high fraction of sequences with no phylogenetic affiliation (Supplemental Fig. S17).
We estimated the number of species in each sample using the fraction of single-copy genes on reads with ≤2× coverage as proxy for the portion of cells in these fractions. These numbers were used in conjunction with coverage values computed for rpS3 genes from both long reads and long-read assemblies (see Methods). Our results suggest that 336, 415, and 189 species with frequencies of ≥0.1% are present in the 4-, 5-, and 6-m samples, respectively. Extending the analysis further to species with a lower abundance suggests that at least 1200 (4-, 6-m) and 2100 (5-m) different species are present in the different samples. These values were computed as strict lower bounds. The true number of species is therefore expected to be much higher—probably in the range of several thousand or tens of thousands of different species.
Analysis of abundant protein families among rare organisms
Rare organisms may increase the metabolic potential of their community by carrying genes that will facilitate their adjustment to changing environmental conditions. In order to evaluate whether this is the case in our communities, we considered two automated strategies for identifying significant functions. The first approach is based on the identification of protein families, determined through sequence similarity-based clustering, that are significantly more abundant than expected in rare genomes. Overall we found 15, 8, and 20 such protein families in the 4-, 5-, and 6-m samples (Supplemental Tables S10–S12). Some of these families represent functions that could be related to adjustment to the environment, including ABC transporter-related proteins, glycoside hydrolase family 4, sulfatases, and TonB-dependent receptors. Other families could not be directly linked to adjustment for changing conditions or have annotations that are too general (e.g., dehydrogenases and oxidoreductases) or have no specific annotation (e.g., hypothetical proteins and tetratricopeptide repeat protein). The second automated approach is based on the grouping of multiple clusters that represent the same function based on KEGG terms function assignments (see Methods). This approach yielded a few functions in each sample that were significantly more abundant in the rare genomes, but did not improve significantly the information acquired through the first approach (Supplemental Tables S16–S18).
We hypothesized that glycosyl hydrolases (EC:3.2.1.-) in rare organisms could extend the community metabolic repertoire and resilience to changing environmental conditions and chose to focus on them. These enzymes catalyze the cleavage of glycosidic bonds and serve multiple purposes in bacteria and archaea. One of the major roles of glycosyl hydrolases is energy uptake (Davies and Henrissat 1995). Glycosyl hydrolases are also well characterized and studied due to their central role in biofuel production and disease research (Vuong and Wilson 2010). There are currently 113 families of glycosyl hydrolases that were compiled based on sequence similarity and function (Cantarel et al. 2009). We hypothesize that a greater repertoire of glycosyl hydrolases may allow the community to take advantage of a wider range of nutrients, which may become available under changing conditions.
Overall, we found 2826, 2138, and 2385 proteins annotated as glycosyl hydrolases in the 4-, 5-, and 6-m samples, respectively. In all cases, the majority of proteins were clustered in families with representatives in both rare and abundant genomes (Supplemental Fig. S19), but the number of proteins in the rare organisms' fraction was significantly higher than expected (P-values of 0, 2.7 × 10−14, and 5.2 × 10−15, respectively). The number of different protein families may be a better indicator for functional diversity: We found that in all cases the number of families represented only in the rare organisms was higher than the number of families represented solely in the abundant genomes. The majority of these families were singletons, and for all three samples, the number of these families in the rare organisms' fraction is significantly higher than expected.
Discussion
Previously, short-read data from aquifer sediments enabled the recovery of one complete genome (RBG-1), many partial Chloroflexi genomes, and many long fragments that are currently being binned to genomes that will be reported separately (K Anantharaman, CT Brown, I Sharon, BC Thomas, A Singh, LA Hug, CJ Castelle, KH Williams, EL Brodie, JF Banfield, et al., unpubl.). Alignment of synthetic long reads to the manually curated RBG-1 genome confirmed the assembly of the genome and revealed two local misassemblies. This result confirms that high-quality genomes can be recovered from short-read metagenomic data when the proper quality control steps are taken. The short-read assembly fails to reconstruct rare genomes and genomes from groups of closely related organisms. Notably, the most abundant organisms in all the samples were missed in short-read data assemblies. This outcome demonstrates one of the major limitations of de Bruijn graph assemblers for metagenomics data. These assemblers require a high degree of sequence conservation for the assembled genomes, and therefore may fail in the presence of closely related heterogeneous genomes. The majority of short reads in all our samples remained unassembled, suggesting that rare and closely related genomes account for significant portions of the studied communities. However, given the high number of synthetic long reads with low coverage, we estimate that the majority of unassembled reads in the short-read data were left unassembled because of low coverage. Similar or lower levels of assemblies were also reported in other metagenomic studies of complex environments such as the ocean (Iverson et al. 2012) and soil (Howe et al. 2014). Mapping of short reads to the synthetic long reads shows that the coverage for 8%–17% of the synthetic long reads in the three samples is lower than 0.1×. We therefore estimate that ∼100 times more short-read sequencing data than available for this study will be required for the recovery of the genomes for 80%–90% of community members in the three samples. The increase in sequencing throughput is expected to improve the recovery of rare genomes from short-read data, but is not expected to improve the assembly of closely related genomes.
The TrueSeq Synthetic Long-Read technology currently offers ∼8 kbp reads at almost two orders of magnitude less throughput (bp per lane), compared to the Illumina HiSeq platform. The study of rare genomes is facilitated by the synthetic long-read technology due to the read size, which is usually sufficient for reliable gene prediction of unassembled reads. In addition, the presence of multiple genes on each synthetic long read enabled the development of a synteny-based approach for the recovery of genome architecture for groups of closely related genomes. The outcome of this process is a gene-centric description of the regions shared in the genomes of organisms in the population. This approach allowed us to recover metabolic features that are common to all related genomes despite not recovering any specific genome. On the other hand, no significant assembly of synthetic long reads was achieved for any single genome in our samples due to the relatively low throughput of the technology and the high complexity of the samples. Scaffolding of short-read assemblies using the long reads was also limited for the same reason: Abundant genomes in the samples typically had sufficient coverage by the short-read data for extensive assembly, and the numerous synthetic long reads from each genome rarely matched a region that was required for scaffolding. Overall, short- and long-read data provide complementary advantages for metagenomics studies, thus making the use of both technologies together more powerful than use of one alone.
Analysis of the long-read data revealed that the most abundant species in all three samples belong to populations of multiple (possibly dozens of) different closely related strains and species. As previously described for sediment-associated RBG-1 populations, the abundance of the Aminicenantes (4-m) and Deltaproteobacteria (5- and 6-m) populations in sediment close to and below the water table may be attributed to metabolic flexibility. The Aminicenantes and Deltaproteobacteria populations are numerous and fairly closely related. In addition to their detection in the acetate-amended groundwater, organisms closely related to the Deltaproteobacteria identified here were also recovered from other locations in the same aquifer (LA Hug, BC Thomas, I Sharon, CT Brown, MJ Wilkins, KH Williams, A Singh, and JF Banfield, in prep.), suggesting that this lineage is a key player in the Rifle sediment environment. The presence of many strains from this lineage may increase the niches occupied by these organisms. It may also improve the overall resistance of the population to threats such as phages (Sharon et al. 2013). RBG-1, on the other hand, was found to be abundant only in the samples studied here, which may suggest that specific environmental conditions at the time of sampling contributed to its high abundance.
Our results show that microbial communities in sediment consist of a few abundant species and a “long tail” of thousands of rare species whose abundance is <0.1%. The majority of these rare organisms belong to phyla also represented by abundant organisms. Our results show that significantly more glycosyl hydrolases than expected are found in the rare organisms' fraction and that these glycosyl hydrolases cluster into more families than glycosyl hydrolases detected in abundant organisms. Theses enzymes can, therefore, increase the repertoire of nutrients degraded by community members, thus increasing the community's ability to adjust to changing environmental conditions.
Methods
Sample collection
Refer to Castelle et al. (2013) and Hug et al. (2013) for a complete description of sample collection and DNA extraction.
Sequencing of the synthetic long-read data
Three sequencing libraries were prepared by Moleculo, Inc. as described in Voskoboynik et al. (2013), Kuleshov et al. (2014), and McCoy et al. (2014) and sequenced on three Illumina HiSeq lanes by the Joint Genome Institute. The synthetic long-read sequencing process consisted of the following steps. First, the DNA was mechanically sheared into ∼8-kbp fragments. Next, the DNA fragments went through end repair and ligation of amplification adapters and then were diluted into 384-well plates, resulting in ∼200 molecules per well. Following this step, the molecules were amplified in the wells using PCR and then went through parallel Nextera-based fragmentation and barcoding. DNA from all 384 wells was then sequenced on one lane (per sample) of an Illumina HiSeq 2000 sequencer. Finally, the sequenced DNA was assembled using an assembly pipeline developed for this purpose. Supplemental Table S1 summarizes the amount of sequencing and the N50 length for the reads in the different samples. Supplemental Figure S1 shows read-size distributions for the different samples.
Sequencing and assembly of short-read data
The three samples were sequenced (paired-end) on four lanes of Illumina HiSeq at the Joint Genome Institute. Sequencing yield was between 140 and 498 million 150-bp reads for the three samples (Supplemental Table S1). The data sets were previously deposited at the NCBI Sequence Read Archive (SRA) database with project number BioProject ID# PRJNA167727, under the accession code SRP013381. Average insert size ranged between 220 and 280 bp for the three samples. Reads were trimmed using Sickle (https://github.com/najoshi/sickle) with default parameters. Data for the different samples were assembled separately using IDBA-UD (Peng et al. 2012) with default parameters. Due to low levels of assembly in the first round of assembly for the 6-m sample, we applied a second round of assembly for this sample with reads that were not assembled in the first round.
Assembly of long-read data
We assembled the data using Minimus 2 from the AMOS package (parameters -D OVERLAP = 500 -D MINID = 99) and also using a new program (Lola) we developed that implements an overlap strategy for assembly. The program is divided into two modules. First, it uses BLAST and variants of the Needleman-Wunsch global alignment algorithm (Needleman and Wunsch 1970) to identify overlaps between reads and determine how they should be used for the assembly of the sequences. Next, this information is used to assemble sequences with overlapping ends and no ambiguities with other sequences. Parameters for the program were set to consider overlaps of 500 bp or more with ≥99% identity. Refer to the Supplemental Information for a complete description of the program; the code is available in Supplemental File Lola-1.02.tar.gz and is maintained under https://github.com/CK7/Lola.
Scaffolding of short-read assemblies
This step was performed using Minimus 2 with the same parameters as above, using the synthetic long reads and the short-read assemblies as two separate input data sets. For computing scaffolding statistics, only sequences that include short-read scaffolds and contigs were considered.
Read mapping
All read mappings were performed using Bowtie 2 (Langmead and Salzberg 2012) with parameter –sensitive.
Reconstruction of syntenic regions for Deltaproteobacteria and Aminicenantes populations
As a first step we clustered synthetic long reads and Lola contigs with significant overlaps (at least 90% identity over 500 bp). Next, we predicted genes for reads longer than 5 kbp in clusters with at least 100 reads using prodigal (Hyatt et al. 2010), allowing closed-ends genes only (-c). Predicted proteins were then clustered using UCLUST from the USEARCH suite (Edgar 2010) with parameter --id 0.75. More proteins from reads shorter than 5 kbp as well as incomplete proteins were added to the different clusters through an iterative BLAST-based procedure for predicting genes based on genes already predicted by prodigal. In each iteration, the remaining regions without predicted genes on reads are aligned (BLAST with parameters -p blastx –F F –b 1 –v 1 –e 1e-30) against the proteins predicted by prodigal. Regions aligned to hits at ≥75% identity over ≥60% of the hit's length are marked as new genes and added to their hit's cluster. This procedure ends when no more new genes are detected. Finally, we used an algorithm we developed for identifying gene architecture that is based on graph representation of gene order on the reads. The algorithm looks for paths in a weighted gene/protein graph that is constructed based on the predicted proteins and their genes. Nodes represent protein families, and edges connect nodes whose corresponding genes can be found next to each other on at least one read (number of reads is the weight). The algorithm searches for both linear paths as well as paths with “bubbles” (splitting and then rejoining of the path). Code for the software implementing the algorithm is available in Supplemental File synteny-1.02.tar.gz and is maintained under https://github.com/CK7/synteny. Refer to the Supplemental Material for a complete description of the algorithm.
Verification of overlap-based clusters through ESOM
Reads from the 4- and 5-m samples were clustered separately using the Databionic implementation of ESOM (Ultsch and Moerchen 2005; http://databionic-esom.sourceforge.net/) with 3-mer frequencies (Dick et al. 2009). Reads ≥5 kbp were considered. Each read was represented by a single data point. A robust ZT transform was applied on the data prior to training. Nondefault parameters used for training were: training algorithm = K-batch training, number or rows in map = 400, number of columns in map = 700, start value for radius = 50. The perl script prepare_esom_files.pl (available as a Supplemental File and maintained under https://github.com/CK7/esom/blob/master/prepare_esom_files.pl) was used for preparing the input files for ESOM with parameters –k 3 –m 5000 –w 100000.
Verification of the RBG-1 genome
Assembled (Lola) and unassembled synthetic long reads that belong to the RBG-1 genome were identified through alignment of the RBG-1 genome against the three long-read data sets using BLAST (parameters -r 2 -q -3 -F F -e 1e-100). All sequences that aligned at 93% identity or more over at least 1500 bp were further checked. Since RBG-1 was represented by a single abundant strain in our samples (see below), all sequences collected based on these criteria typically aligned at 99% identity or more over most of their length, indicating that they are true RBG-1 sequences. All regions on the RBG-1 genome to which long-read sequences aligned at <99% or did not align throughout their entire length were examined manually. This included examination of read mapping to the regions and re-assembly of the suspected region using a local assembly process described in Sharon et al. (2013). Inconsistencies between the local assembly and the RBG-1 genome were recognized as misassemblies.
Clustering of protein families
The proteins were clustered in families based on similarity networks as follows. An all-against-all protein similarity search was performed using UBLAST from the USEARCH suite (Edgar 2010). A weighted network was constructed by connecting pairs of proteins that shared a similarity of e-value lower than 0.0001 that covers at least 80% of both proteins. The weight of the edge connecting the proteins was the bit score of the similarity between them. Clusters of protein families were extracted from the similarity network using the MCL algorithm with an inflation parameter of 2.0 (Enright et al. 2002).
Identification of single-copy genes
We considered 51 single-copy genes (Supplemental Tables S7–S9) and used a two-step procedure for assigning MCL protein families to each of these genes. Each sample was analyzed separately. In the first step, we identified 51 preliminary sets of genes (regardless of their families) from each sample using a reciprocal best BLAST hits procedure (see below). Next, we used these preliminary sets to identify MCL protein clusters enriched with single-copy genes. Each cluster with >50% of its members belonging to one of the preliminary sets was assigned to the gene of the enriched set. This procedure aimed at reducing the number of false positives and also at improving the identification of proteins with low similarity to the proteins in our reference set.
Identification of the preliminary set was carried out using a reciprocal best BLAST hit procedure as follows. First, we identified a set of potential single-copy genes by running BLAST (-F F –e 1e-5) of proteins in each sample against a set of single-copy genes from 30 reference genomes covering most known bacterial phyla with sequenced genomes. Next, we BLASTed all potential single-copy genes against all proteins from the reference genomes and kept those with a single-copy gene as their best hit. Candidates with single-copy genes as their best hits were added to the set of their best hit.
Identification of glycosyl hydrolases
Glycosyl hydrolase MCL clusters were identified in each sample using a two-step procedure similar to the one used for the single-copy genes (see above). A preliminary set of proteins annotated as glycosyl hydrolases was identified using the ggKBase platform (http://ggkbase.berkeley.edu/) with a list of glycosyl hydrolase-related terms (Supplemental Table S19; http://ggkbase.berkeley.edu/custom_lists/5572-Glycosyl_hydrolase). Glycosyl hydrolase MCL clusters were identified as described above for the single-copy genes.
Determining sets for KEGG terms
Similarly to the above, this analysis involved the identification of a preliminary set for each KEGG term and assignment of MCL cluster to KEGG terms based on these sets. Preliminary sets were determined based on KEGG annotations assigned to the proteins by the ggKBase annotation pipeline.
Data access
Synthetic long reads for the Rifle sediment metagenome have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) and Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under BioProject ID# and SRA accession numbers 4-m: PRJNA263279/SRX727132, 5-m: PRJNA263282/SRX727164, and 6-m: PRJNA263284/SRX727169. Information for genome architectures described in the paper as well as synthetic long-read data for the three samples and the GWC2 Deltaproteobacteria genome is available in Supplemental Material and at http://ggkbase.berkeley.edu.
Competing interest statement
D.P., T.B., and M.K. performed the research at Moleculo, Inc. (acquired by Illumina, Inc.). T.B. is employed by Illumina, Inc. The library preparation protocol is covered by U.S. and international patents with numbers 61/532,882 and 13/608,778 on which D.P. and M.K. are listed as inventors. The TruSeq Synthetic Long-Read technology is offered commercially by Illumina, Inc.
Supplementary Material
Acknowledgments
J.F.B., I.S., L.A.H., B.C.T., and C.J.C. were supported as part of the Sustainable Systems Scientific Focus Area funded by the US Department of Energy, Office of Science, Office of Biological and Environmental Research under award number DE-AC02-05CH11231 and DOE Kbase grant DE-SC0004918. Sequencing was performed at the DOE Joint Genome Institute under the CSP Program. The work was conducted in part by the US Department of Energy Joint Genome Institute.
Author contributions: J.F.B., I.S., and M.K. conceived the idea for the study. M.K., D.P., and T.B. developed the synthetic long-read laboratory preparation protocol and prepared the libraries. S.G.T. and M.A. prepared the short-read libraries and sequenced both the short- and long-read libraries. I.S. performed computational analysis for the long-read data. I.S. and B.C.T. analyzed the short-read data. I.S., J.F.B., D.B., and L.A.H. performed community and protein family analyses, B.C.T. assembled and J.F.B. manually curated the near-complete Deltaproteobacteria genome, and C.J.C. and J.F.B. performed metabolic analysis for the Deltaproteobacteria and Aminicenantes genotypes. I.S. and J.F.B. wrote the manuscript. All authors reviewed and revised the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.183012.114.
References
- Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Baran Y, Halperin E. 2012. Joint analysis of multiple metagenomic samples. PLoS Comput Biol 8: e1002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert S, Raymond F, Godzaridis E, Laviolette F, Corbeil J. 2012. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol 13: R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CT, Sharon I, Thomas BC, Castelle CJ, Morowitz MJ, Banfield JF. 2013. Genome resolved analysis of a premature infant gut microbial community reveals a Varibaculum cambriense genome and a shift towards fermentation-based metabolism during the third week of life. Microbiome 1: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37: D233–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelle CJ, Hug LA, Wrighton KC, Thomas BC, Williams KH, Wu D, Tringe SG, Singer SW, Eisen JA, Banfield JF. 2013. Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat Commun 4: 2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Henrissat B. 1995. Structures and mechanisms of glycosyl hydrolases. Structure 3: 853–859. [DOI] [PubMed] [Google Scholar]
- Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP, Banfield JF. 2009. Community-wide analysis of microbial genome sequence signatures. Genome Biol 10: R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzi SC, Sharon I, Wrighton KC, Koren O, Hug LA, Thomas BC, Goodrich JK, Bell JT, Spector TD, Banfield JF, et al. 2013. The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. eLife 2: e01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. [DOI] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30: 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltsman DS, Denef VJ, Singer SW, VerBerkmoes NC, Lefsrud M, Mueller RS, Dick GJ, Sun CL, Wheeler KE, Zemla A, et al. 2009. Community genomic and proteomic analyses of chemoautotrophic iron-oxidizing “Leptospirillum rubarum” (Group II) and “Leptospirillum ferrodiazotrophum” (Group III) bacteria in acid mine drainage biofilms. Appl Environ Microbiol 75: 4599–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe AC, Jansson JK, Malfatti SA, Tringe SG, Tiedje JM, Brown CT. 2014. Tackling soil diversity with the assembly of large, complex metagenomes. Proc Natl Acad Sci 111: 4904–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug LA, Castelle CJ, Wrighton KC, Thomas BC, Sharon I, Frischkorn KR, Williams KH, Tringe SG, Banfield JF. 2013. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. 2012. Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335: 587–590. [DOI] [PubMed] [Google Scholar]
- Janda JM, Abbott SL. 2007. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol 45: 2761–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor RS, Wrighton KC, Handley KM, Sharon I, Hug LA, Castelle CJ, Thomas BC, Banfield JF. 2013. Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. mBio 4: e00708–e00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov V, Xie D, Chen R, Pushkarev D, Ma Z, Blauwkamp T, Kertesz M, Snyder M. 2014. Whole-genome haplotyping using long reads and statistical methods. Nat Biotechnol 32: 261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy RC, Taylor RW, Blauwkamp TA, Kelley JL, Kertesz M, Pushkarev D, Petrov DA, Fiston-Lavier A. 2014. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly repetitive transposable elements. PLoS ONE 9: e106689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CS, Baker BJ, Thomas BC, Singer SW, Banfield JF. 2011. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol 12: R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al. 2000. A whole-genome assembly of Drosophila. Science 287: 2196–2204. [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443–453. [DOI] [PubMed] [Google Scholar]
- Peng Y, Leung HC, Yiu SM, Chin FY. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28: 1420–1428. [DOI] [PubMed] [Google Scholar]
- Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, et al. 2013. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437. [DOI] [PubMed] [Google Scholar]
- Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. 2013. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res 23: 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Sommer DD, Angly FE, Koren S, Pop M. 2011. Next generation sequence assembly with AMOS. Curr Protoc Bioinformatics 33: 11.8.1–11.8.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson G, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. 2004. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43. [DOI] [PubMed] [Google Scholar]
- Ultsch A, Moerchen F. 2005. ESOM-Maps: tools for clustering, visualization, and classification with Emergent SOM. Technical Report No. 46, Department of Mathematics and Computer Science, University of Marburg, Germany. [Google Scholar]
- Van Dongen S.2000. “Graph clustering by flow simulation.” PhD thesis, University of Utrecht. [Google Scholar]
- Voskoboynik A, Neff NF, Sahoo D, Newman AM, Pushkarev D, Koh W, Passarelli B, Fan HC, Mantalas GL, Palmeri KJ, et al. 2013. The genome sequence of the colonial chordate, Botryllus schlosseri. eLife 2: e00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong TV, Wilson DB. 2010. Glycoside hydrolases: catalytic base/nucleophile diversity. Biotechnol Bioeng 107: 195–205. [DOI] [PubMed] [Google Scholar]
- Whitman WB, Coleman DC, Wiebe WJ. 1998. Prokaryotes: the unseen majority. Proc Natl Acad Sci 95: 6578–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, et al. 2012. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337: 1661–1665. [DOI] [PubMed] [Google Scholar]
- Wrighton KC, Castelle CJ, Wilkins MJ, Hug LA, Sharon I, Thomas BC, Handley KM, Mullin SW, Nicora CD, Singh A, et al. 2014. Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer. ISME J 8: 1452–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelton AP, Thomas BC, Simmons SL, Wilmes P, Zemla A, Thelen MP, Justice N, Banfield JF. 2011. A semi-quantitative, synteny-based method to improve functional predictions for hypothetical and poorly annotated bacterial and archaeal genes. PLoS Comput Biol 7: e1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.