

Abstract
Due to the absence of antiparistic vaccines, and the constant threat of drug resistance, the development of novel antiparasitic chemotherapies remains of major importance for disease control. A better understanding of drug transport (uptake and efflux), metabolism and the identification of drug targets, as well as potential drug resistance mechanisms would facilitate the development of more effective therapies. Here, we focus on malaria and African tyrpanosaomiasis. We review existing drugs and drug development, emphasizing high-throughput genomic and genetic approaches, which hold great promise for elucidating anti-parasitic mechanisms. We describe the approaches and technologies that have been influential for each parasite and develop some new ideas for future research directions, including strategies for target deconvolution.
Keywords: antimicrobial, chemical genomics, drug resistance, high-throughput, Plasmodium falciparum, Trypanosoma brucei
INTRODUCTION
Vaccine development against eukaryotic parasites has stalled or has progressed slowly. As a consequence, the development of novel chemotherapies against these parasites remains a priority (1). Drug resistance is also a major looming threat for both malaria and African trypanosomiasis; resistance in parasites and their vectors threatens to reverse the gains associated with decades of investment in antimicrobials and insecticides and to undermine the development of new compounds based on similar scaffolds. In short, if the drug-development pipeline is not effectively fed, some diseases may be left with no realistic therapeutic options to support control or eradication efforts.
Plasmodium falciparum and the African trypanosome, Trypanosoma brucei are parasitic protists representing the Chromalveolata and Excavata kingdoms, respectively. Malaria in humans is caused by infection by five species of Plasmodium parasites. Following transmission by mosquito vectors, the parasites enter liver cells where they multiply several thousand-fold before bursting into the blood-stream where they invade and proliferate within red blood cells (RBCs), resulting in clinical disease. The bulk of disease is caused by infection with the potentially lethal P. falciparum and the widespread P. vivax parasite. P. falciparum causes disease by proliferating to high densities within RBCs in the circulation. Severe disease can occur when infected RBCs bind to remote tissues, resulting in cerebral and placental malaria (2). P. vivax is restricted to reticulocytes and causes significant morbidity but little mortality. There are three other epidemiologically relevant Plasmodium spp., Plasmodium ovale and Plasmodium malariae, as well as the emerging zoonotic Plasmodium knowlesi.
African trypanosomes are transmitted by tsetse flies in Sub-Saharan Africa, causing sleeping sickness in humans (also known as human African trypanosomiasis or HAT) and Nagana in livestock. T. brucei circulate free in the bloodstream and tissue fluids of their mammalian hosts, migrating eventually to the central nervous system (CNS), where they cause the second, typically fatal, stage of the disease in humans. Importantly, antitrypanosomal drugs for treatment of the second stage of the disease must traverse the blood-brain barrier.
Here we focus on the opportunities afforded by genomic approaches for the development of antiparasitic chemotherapies. Studies on malaria and African trypanosomiasis reflect the broad range of genomic approaches available. Further chemical genetic approaches show great promise in this area (3) and polypharmacology, the disruption of multiple targets (4, 5), also deserves greater attention.
CURRENT STATUS OF ANTIMALARIAL THERAPY
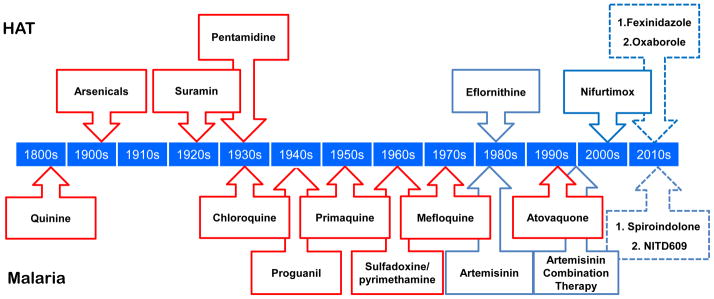
There are many antimalarial agents available (see Figure 1). Quinine, extracted from the bark of the Cinchona tree in the 1800s, was the first widely used antimalarial drug. It is a fast-acting drug that is still used in the treatment of severe malaria. The wars of the first half of the last century were a great spur for the development of many new drugs with greater efficacy and fewer side effects, resulting in many of the antimalarial agents that we still use today. The blood schizonticide chloroquine was the mainstay for antimalarial treatment through much of the last century. In response to growing antimalarial resistance, many congeners of chloroquine have been developed, including mefloquine and other arylaminoalcohols such as halofantrine, lumefantrine, piperaquine and pyronaridine, with demonstrated efficacy against chloroquine-resistant malaria. The anti-folate drug combination of sulphadoxine and pyrimethamine (Fansidar) has also demonstrated great efficacy; although widespread drug-resistance has reduced its use, it is still used in preventative therapy. More recently, the combination of atovaquone and proguanil (Malarone) has been developed, demonstrating great efficacy for prophylaxis. The introduction of artemisinin and its derivatives, particularly in combination with other antimalarials, has become central to chemotherapy, particularly due to their efficacy against chloroquine-resistant malaria, with the additional effect of reducing transmission (6). Much effort has been dedicated to the production, and recently the synthesis, of artemisinin and its congeners (7). Artemisinin is a very fast-acting antimalarial, and hence it is replacing quinine for the treatment of severe disease (8).
Figure 1.

Antiparasitic drugs. The timeline indicates the first introduction of drugs to treat HAT (top) and selected drugs to treat malaria (bottom). Red boxes indicate where resistance has been confirmed. Dashed boxes indicate drugs currently in clinical trials; only selected examples with new mechanisms of action shown for malaria.
It is assumed that drugs that are efficacious against P. falciparum will work against the other species that infect humans. Resistance to chloroquine and Fansidar have been documented in P. vivax (9, 10). Further, P. vivax parasites possess a hypnozoite form, which can remain quiescent in liver cells; this stage can be radically cured by treatment with primaquine.
There are many drugs and drug combinations in clinical development, almost all of which are based on existing antimalarial drug targets. The recently developed spiroindolone NITD609 represents the first drug in 20 years with a novel mechanism of action to enter phase IIa trials, targeting the Na+-ATPase PfATP4 (11).
CURRENT STATUS OF ANTITRYPANOSOMAL THERAPY
Antitrypanosomal drugs are detailed in Figure 1 and have been reviewed by others (12, 13). Good progress has been made recently in developing more effective therapies and this has been supported by organisations including the Drugs for Neglected Diseases initiative (DNDi, www.dndi.org), Médecins Sans Frontières (MSF, www.msf.org.uk), the Dundee Drug Discovery Unit (DDU, www.drugdiscovery.dundee.ac.uk/) and the Pan African Tsetse and Trypanosomiasis Eradication Campaign (PATTEC, sp.au.int/pattec). For example, DNDi contributed to the delivery of the nifurtimox-eflornithine combination therapy (NECT) for late-stage sleeping sickness in 2009 (14); NECT is the safest therapy for second stage HAT and the availability of these drugs has increased further in recent years (15). In terms of new drugs, pentamidine like pro-drugs (16) and other novel diamidines (17) with improved pharmacokinetic properties (18) are under development. Clinical trials are also underway for two oral drug candidates, fexinidazole (1, 19) and the benzoxaborole, SCYX-7158 (20); oxaboroles represent a class of drug not previously used against trypanosomes (1). Fexinidazole is in phase II/III testing for efficacy and safety for the treatment of either stage of HAT in the Democratic Republic of the Congo and Central African Republic. This trial is run by DNDi, with partners including MSF and the French pharmaceutical company Sanofi, and support from donors including the Bill & Melinda Gates Foundation and the British Department for International Development (DFID). An oxaborole phase I trial is also underway, run by DNDi in collaboration with Anacor Pharmaceuticals and SCYNEXIS.
CURRENT CHALLENGES AND PROBLEMS WITH ANTIPARASITE CHEMOTHERAPY
Resistance has arisen to many antiparasitic drugs (Figure 1). This perennial problem fuels a continuous effort to develop new drugs with new mechanisms to avoid cross-resistance with existing drugs. In the case of malaria, resistance to chloroquine and to the combination of pyrimethamine/sulphadoxine is widespread, while mefloquine-resistance is increasing. Of particular note, efficacy of artemisinin and its derivatives appears to be decreasing in specific foci in Southeast Asia (21). There is also some evidence for adverse side-effects of artemisinin and some of the drugs used in combination such as amodiaquine (22).
The concept of the ideal drug - Single Encounter Radical Cure and Prophylaxis (SERCaP) - has emerged (23). This presents a major challenge for drug development. In addition, eradication of P. vivax will be more difficult unless an efficacious anti-hynozoite drug can be developed which can also be used for mass drug administration. The mechanism of action of primaquine, the only drug available for radical cure, is unknown. Indeed, the mechanism by which primaquine treatment results in life-threatening hemolysis in patients with G6PD-deficieny, which is widespread in malaria-endemic regions (24), is also poorly understood (25). The use of antimalarials for mass drug administration and intermittent preventative treatment requires an extremely high safety profile. It would also be desirable for the drugs to be active against all Plasmodium spp., as well as being fast-acting so that they can be used in treating severe malaria. Additionally, the ability to prevent transmission by targeting the sexual forms and hypnozoite forms of the parasite will become increasingly important as we approach eradication.
Only one antitrypanosomal drug, eflornithine, has a known enzymatic target, meaning that there are typically major gaps in our understanding of toxicity or resistance where these are observed. In addition, eflornithine lacks efficacy against T. b. rhodesiense due to the shorter half-life of the target, ornithine decarboxylase (26). Another drug, melarsoprol is a melaminophenyl arsenical that emerged after even more toxic arsenicals were introduced for use against sleeping sickness in the early 1900’s. High levels of toxicity are still displayed by melarsoprol, which causes an often fatal reactive encephalopathy in approximately 10% of patients (27). This drug is still used to treat HAT in East Africa caused by T. b. rhodesiense because eflornithine lacks efficacy there (26). Drug resistance is a major current problem or a looming threat (Figure 1). The most prominent example of drug resistance is cross-resistance between arsenicals and diamidines (28–30), first reported over 60 years ago (31). This is most commonly observed in the laboratory for melarsoprol and pentamidine, an aromatic diamidine first introduced for use against the first stage of African trypanosomiasis in the 1930’s. Thus, limited efficacy and potency, problems with toxicity and/or resistance and major gaps in our understanding of the mechanisms underlying some of these properties are among the problems associated with current antiparasitic therapies.
UNDERSTANDING DRUG ACTION – BEFORE GENOMICS
An understanding of drug action and interaction with the target parasite should facilitate the development of new and improved therapies for the treatment of parasitic diseases. Below, we consider three aspects of drug action and interaction that impact drug efficacy (Figure 2); transport into, within and out of the parasite; metabolic modification by the parasite which alters potency and, finally; the drug target or targets themselves. In each case for the malaria parasite, we must also consider the host infected RBC environment and its modification by the parasite (Figure 2).
Figure 2.
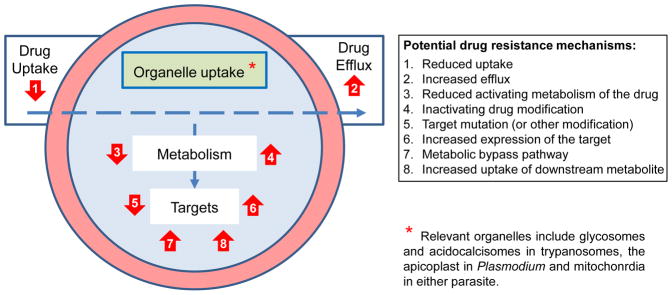
How drugs interact with parasites. The blue circle represents the parasite. The outer red circle represents the infected RBC in the case of Plasmodium spp. When considering mechanism of action, we need to take 1. Transport, 2. Metabolism and 3. Target(s) into account, as illustrated. Active transport typically involves passage through membrane channels, into the cell, into organelles and out of those organelles/cells; not relevant for extracellular targets but these are not common. Metabolism within the parasite may activate or detoxify the (pro)drug. Drug targets can be enzymes, complexes (protein-protein interaction blockers) or other macromolecules and metabolites.
Antiparasitic drug transport
Drugs must effectively access their targets. This may be a passive process in some cases but membrane channels and transporters are often important determinants of drug uptake or efflux and can display a high degree of specificity. A deeper understanding of the interplay between various drug transporters, their expression and genetic variation, will improve strategies to optimise drug design and delivery.
Many of the antimalarials, including the quinolines, chloroquine and mefloquine, accumulate to high concentrations within the parasite. The blood schizonticides appear to target the biochemical process of heme polymerization that is required for the detoxification of the heme moiety within the parasite. Significant digestion of host hemoglobin occurs within the digestive vacuole of the parasite during the intracellular growth phase. Resistance against these drugs involve reductions in the accumulation of the drug within the parasite through the mutation and amplification of the Pfcrt and Pfmdr1 transporters (32, 33).
Past approaches to the identification of T. brucei genes associated with drug transport include genetic complementation screening in transport-defective yeast (34) or the study of resistant mutants generated in the laboratory. Using the former approach, melarsoprol uptake was shown to involve an adenosine transporter [36]. In some cases, accumulation in organelles, such as the mitochondrion, is also likely to be important. Several laboratory-selected resistant strains or genetically modified strains have been used to characterize gene function, and radiolabelled or fluorescent drug-derivatives have been particularly useful for this purpose (35).
Antiparasitic drug metabolism
Drug metabolism, that may be specific to the parasite, can lead to the production of more potent (or more toxic) metabolites (Figure 2). Thus, an improved understanding of drug metabolism, specifically in those cases where it increases potency, may present new strategies for pro-drug design. The mechanism of action of the artemisinin-based drugs is still not clearly understood. It appears that antimalarial activity requires the formation of a free radical form. The endoperoxide bond is essential for its activation following interaction with heme and other pools of iron (36). Once activated, the drug may directly alkylate heme, resulting in the inhibition of heme polymerization and the generation of free radicals. Alternatively or additionally, the activated drug could directly bind to other critical essential parasite proteins, such as the parasite PfATP6 (a SERCA type Ca2+-ATPase) or iron-sulphur proteins, or could oxidize parasite membranes (37).
Nifurtimox, and other nitro pro-drugs, used against African and South American trypanosomes, are ‘activated’ by a trypanosomal, mitochondrial nitroreductase (38, 39). As above for drug transport, the generation of resistant mutants in the laboratory, followed by a focus on mutations in candidate genes, lead to the identification of this activation mechanism (38). Notably, genome-wide analysis of nitroimidazole-resistant Mycobacterium tuberculosis also revealed mutations in a nitroreductase responsible for drug activation (40). There is now a real concern that cross-resistance with nifurtimox will reduce the efficacy of fexinidazole, the nitro pro-drug currently in clinical trials for the treatment of HAT (41). As in the case of drug transporters, pro-drug activators present the possibility of selective action against the parasite but also present opportunities for the emergence of resistance.
Antiparasitic drug targets
When considering drug targets, we must include proteins (often enzymes), other macromolecules and their interactions. Despite the long list of potential targets, whole parasite-based approaches yielded most of the current and emerging antiparasitic drugs. Folate metabolism has been targeted, as Plasmodium parasites cannot salvage this factor that is essential for proliferation. The antimalarial combination of pyrimethamine and sulphadoxine target the enzymes dihydrofolate reductase (dhfr) and dihydropteroate synthase (dhps) respectively, which catalyze different steps in the folate biosynthetic pathway. When administered in combination, the drugs act synergistically. Mutations associated with Fansidar-resistance have been identified and validated in dhfr and dhps (42–44). Atovaquone and proguanil, the components of Malarone, target mitochondrial electron transport, with atovaquone being specific for cytochrome b while proguanil, when administered with atovaquone, appears to function by collapsing the electron transport chain (45).
Eflornithine is known to be a specific inhibitor of ornithine decarboxylase but, otherwise, insight into the targets of the antitrypanosomal drugs is more limited. For example, the diamidines, including pentamidine, are nucleic acid binding drugs (46) that typically become concentrated within, and destroy, the kinetoplast (47), the complex mitochondrial genome. One concern here is that some parasitic trypanosomes lack a kinetoplast (48) suggesting that DNA-binding drugs that specifically target this structure could be prone to the emergence of resistance. Perhaps fortunately then, pentamidine appears to also disrupt mitochondrial membrane potential (49, 50) and other related compounds are seen in the nucleus and in acidocalcisomes (51, 52). Melarsoprol is thought to act primarily as a trypanothione adduct, Mel T (53). Other potential targets with trypanosomatid-specific features include compartmentalized glycolysis, glycosylphoshatidylinositol anchoring (on variant surface glycoproteins) and RNA editing in trypanosomatid mitochondria (54). N-myristoyltransferase is another promising and validated target (55).
THE IMPACT OF GENOME SEQUENCES ON TARGET-BASED DRUG DISCOVERY
The acquisition of full reference genome sequences for the different Plasmodium species (56) and trypanosomatids, including T. brucei (57), provide blueprints for exploitation, immediately revealing multiple candidates to pursue for the development of antiparasitic interventions. Clearly, specific targets within probable druggable pathways were identified through genome sequencing and bioinformatics, and also many kinases (58, 59), phosphodiesterases (60), protein deacetylases (61), proteases (62, 63) and tRNA synthetases, to name a few. Indeed, the full complement of each druggable genome became accessible and the TDR-targets database was constructed to provide a chemogenomics resource for the genome-scale prioritization of drug targets (64). Thus, genome sequences had a major impact on research on, and drug development for, these parasites.
A key question is how does one prioritize targets for antiparasitic drug development? Drug repurposing can leverage past investments and a wealth of information from successful drug development programmes and can also facilitate a focus on one or a few defined protein targets. However, this largely ignores parasite genes annotated ‘hypothetical, conserved’ and parasite-specific biology. For example, 50% of genes associated with loss-of-fitness in T. brucei are of unknown function (65). Whether repurposing is the goal or not, genetic validation is widely used to indicate that a gene is essential. Regulated or conditional expression (66), particularly through RNA interference (RNAi), has been widely used for this genetic validation in T. brucei. Challenges remain however, since the genome sequences, taking only protein coding genes into account, present approximately 7,500 potential targets. Genome-scale genetic validation efforts can be used to prioritise (65) and these data can be combined with other sources of information through the TDR-targets database (67), but this still leaves hundreds of potential targets. The reader is referred to (68) to see the challenges that remain, even with the resources of GlaxoSmithKline, when following up large numbers of post-genomic potential drug targets.
Unfortunately, RNAi is not functional in Plasmodium parasites (69). Instead, systems for conditional transcriptional expression are now being developed (70). Additionally, conditional protein expression systems have been useful to demonstrate essentiality of the PfCDPK5 kinase (71), while also demonstrating its functional requirement for egress from RBCs.
ELUCIDATING NEW MECHANISMS OF DRUG ACTION USING GENOMIC APPROACHES
Phenotypic screening against whole pathogens is now being more widely adopted as an alternative to target-based approaches, allowing access to the portion of the potentially druggable genome that is not otherwise readily accessible. Another approach which has yielded several successes has been to screen FDA-approved drugs to identify those with antimalarial activity (72, 73). Indeed, many high-throughput drug screens using cell-based assays have been conducted by Novartis-GNF and GlaxoSmithKline recently to identify inhibitors of the asexual stage of Plasmodium growth (74–77). It is true that many of these molecules are likely to hit known targets and processes, and previously developed assays can be used to confirm specificity through reverse chemical genetic approaches where the small molecules were tested against validated targets (74). Chemoinformatics and in silico activity profiling was able to suggest the antimalarial targets of many of the active compounds, for example (76, 78) and phylochemogenetics can be carried out through comparison with drug sensitivity in other organisms (79). However, these high throughput screens, also carried out for T. brucei (80), typically produce many promising compounds with no known mechanism of action.
Classical genetic approaches are now being scaled to a genomic level. Together these approaches can close major gaps in knowledge. They can provide insights into drug transport and metabolism, they can facilitate the prioritisation of drug targets and can achieve target validation. They can also achieve drug target deconvolution, the identification of the molecular targets and elucidation of mechanism of action (81, 82). Below, we present a range of relevant genetic approaches to these problems, often focusing on the parasite where most work has been done to illustrate the key points. We also briefly describe some other complementary approaches.
MATCHING GENETIC CHANGE TO PHENOTYPE
Genetic cross mapping
The first three approaches we describe below involve using drug resistant strains to find the genotypic changes underlying a particular phenotype. High-throughput sequencing can facilitate the process in each case and reverse genetics is typically employed to confirm the contribution of the genetic change, either single-nucleotide polymorphism (SNP) or copy-number variation (CNV), to drug-resistance. Classical forward genetic mapping involves the generation of progeny from a genetic cross of two parental lines which exhibit different traits. This was applied to P. falciparum and chloroquine drug susceptibility (83), and eventually resulted in the identification of the PfCRT gene that encodes a membrane transporter and a determinant of chloroquine-resistance (84, 85). This approach has also been used to uncover the genetic basis of quinine-resistance (86) and nutrient uptake, revealing the clag genes which encode integral membrane proteins localized to the surface of infected RBCs (87). Similar approaches also have the potential to uncover the genetic basis of a range of phenotypes in African trypanosomes (88).
Genome-wide association studies (GWAS)
GWAS is similar to genetic mapping but involves epidemiology, looking at natural trait variation and the identification of resistance-associated mutations in clinical (or veterinary) samples. Several studies have looked for associations between sensitivity to antimalarial drugs and genetic variation in a series of diverse parasite isolates (89–91). Recently, loci associated with delayed clearance of artemisinin were mapped (92). Further mapping and functional analysis will hopefully identify the causative gene(s). In another study, a forward genetic screen was carried out with 32 antimalarial compounds, and several genetic loci associated with differential sensitivity were identified (93). One limitation of this approach is that candidate loci can be in regions with long-range haplotypes, presumably due to recent selection, and it can be difficult to identify the etiological variant.
Laboratory selection of resistant mutants
A powerful and relatively rapid approach that has been employed with great success recently involves the in vitro selection of resistant mutants using small molecule inhibitors (94). Once resistance has been confirmed, parasite clones are derived and analyzed to identify novel genetic changes relative to the parental genome sequence that could be associated with drug-resistance. It is possible to focus on candidate genes or to search more widely by either hybridizing DNA to microarrays or, more commonly now, by genome sequencing. This methodology has been used to identify the genetic basis of resistance to fosmidomycin, an inhibitor of isoprenoid biosynthesis, as a CNV in the Pfdxr gene, which is the likely drug target (95). The novel antimalarial piperaquine is becoming more widely used in Southeast Asia in combination with dihydroartemisinin. Piperaquine-resistance lines were obtained and whole genome profiling revealed the resistance was associated with a CNV event on chromosome 5 (96). Similarly, thioisoleucine and decoquinate possess potent activity against P. falciparum, and lines selected for resistance resulted in mutations in the cytoplasmic isoleucyl-tRNA synthetase and the cytochrome b gene respectively (97).
The spiroindolones were identified in a large screen for new antimalarials. Resistant lines were obtained and the resistance mapped to the ATPase4 gene (98, 99). Similarly, this approach has been used to validate signal peptide peptidase as a valid target within the ERAD pathway for highly selective SPP inhibitors at a nanomolar level (99). Interestingly, liver-stage parasites are also sensitive to these compounds.
Nitroreductase was identified as the trypanosome enzyme that activates nifurtimox using this approach. Indeed the entire NTR-encoding chromosome was lost from resistant Trypanosoma cruzi (38). Since eflornithine is an amino-acid analogue, resistant mutants, generated in the laboratory, were screened for mutations in amino-acid transporter genes and the AAT6 gene was identified as the gene responsible for drug uptake; a finding confirmed by RNA interference (RNAi) library screening (100–102). Finally, melarsoprol-pentamidine cross-resistance was confirmed to be dependent upon the aquaglyceroporin gene AQP2, found to be mutated in a laboratory selected resistant strain (103). The success of these approaches depends upon the ability to identify the key SNPs, CNVs and indels, which is more challenging in a diploid organism such as T. brucei.
Perturbing gene expression
Gene disruption and conditional gene expression, as well as mutagenesis with transposons, are scalable approaches. Complex populations can be monitored in parallel using microarrays or by deep DNA sequencing. Indeed, relative abundance within a pool of mutants under different conditions can be used to probe a range of phenotypes. There has been good progress in this area but much still remains to be done to realise the potential (see Figure 3).
Figure 3.
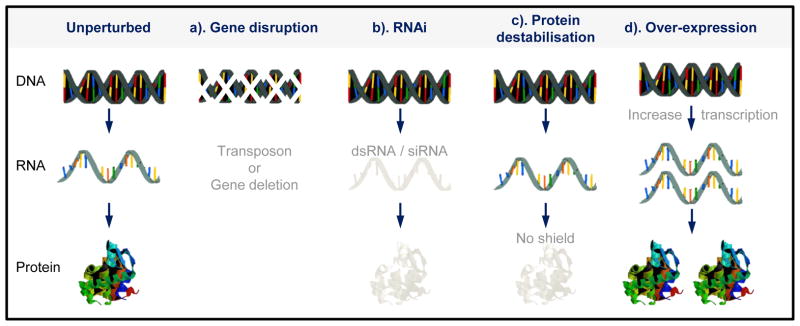

Chemical genetic screens. Genome profiling using knockdown or over-expression of every reading frame to probe mechanism of action (MOA) and interaction, most of what is shown is yet to be implemented for parasites. Open arrows indicate high and low-dose chemical exposure. The schematic illustrates highly parallel co-culture fitness tests following conditional gene expression changes, either loss-of-function (LOF) or gain-of-function (GOF), and chemical exposure. Complex populations with protein expression down or upregulated are grown competitively and then quantitatively assessed using a read-out, typically generated using DNA sequencing and mapping to a reference genome. In the case of green genes, loss of fitness in map 1 means that it is more likely that this gene is a target. A more moderate knockdown could be used to increase the differential read-out, due to drug-RNAi synergy, in maps 1 and 3. Genes indicated in red and blue may also be associated with a fitness cost when knocked down (or occasionally when over-expressed, neither indicated). In these cases, differential representation may still reveal the chemical interaction indicated for red genes in maps 1 to 3. Conversely, genes indicated in green may not be associated with a fitness cost when knocked down if they are not targets; these could be dispensable pumps involved in drug export for example. Trypanosomes are shown but the principles are similar for other cell types. ‘Functional variomics’ screens, combining both mutations and over-expression, have recently been applied to budding yeast (156).
The fungal secondary metabolite cladosporin, shown to possess antimalarial activity, was used in a haploinsufficiency screen with a pool of yeast mutants. This revealed the KRS1 gene encoding a lysyl-tRNA synthetase as the target of the drug (104). Recently both protein and transcription inducible systems have been described for P. falciparum, suggesting that these approaches could form the basis for systematic screens. Several groups have also attempted to knock out large sets of genes, including genes involved in cytoadherence (105), and the various members of the parasite kinomes (106, 107).
Transposon (signature-tagged) mutagenesis, used initially in bacterial pathogens (108, 109), has now been utilized to produce mutant clones in P. falciparum, which is haploid, at a frequency that opens the door to high throughput (110–112), but saturation mutagenesis to identify the full complement of essential genes has yet to be realized. Transposon mutagenesis has also been applied to insect-stage T. brucei, revealing haploinsufficiency in one case (113) and loss-of-heterozygocity in another; T. brucei is diploid (114). Transposons can also be used in a gain-of-function format if associated with outward-facing promoters (115).
RNAi Target Sequencing (RIT-seq) screening, for gene knockdowns associated with significant fitness costs, has also been applied at a genomic scale (65) and the data have been incorporated into the TDRtargets Database (67) facilitating drug–target prioritization efforts. Indeed, these data represent the largest gene function dataset in this database. In total, 750 T. brucei genes (10% of all genes) were associated with a significant loss-of-fitness in all conditions tested, representing a wide range of potential targets. For example, the majority of proteasome components were associated with loss-of-fitness, notable since it has been estimated that 5–10% of all pharmaceutical targets being pursued are proteases (116). The eight subunit TCP-1 chaperonin complex, known to be inhibited by trivalent arsenic in yeast (117), was also associated with a substantial loss-of-fitness in T. brucei.
RIT-seq screens using antitrypanosomal drugs have also been highly informative (118), revealing drug transporters and mechanisms of pro-drug activation (118). Importantly, the drug-selection approach to RIT-seq reveals genes with knockdowns associated with no or little detectable concomitant fitness cost, but also genes associated with a concomitant fitness cost. The former group may be more likely to be disrupted in resistant parasites while the latter group is probably less likely to allow propagation and spread when mutated in patients.
In terms of drug transport, aquaglyceroporin 2 (AQP2) was linked to melarsoprol–pentamidine uptake, which may require a proton-motive force (103, 119). In addition, receptor-mediated endocytosis was linked to suramin resistance. These findings go some way to explaining the selective toxicity of these compounds and possibly also revealing the mechanisms underlying loss of efficacy. The findings have reinforced the importance of drug transport and the possibility of exploiting parasite transporters for selective drug delivery. Receptor-mediated endocytosis could be exploited for toxin delivery, for example (120), since suramin and human trypanolytic factor are known to be delivered via this route.
In terms of drug metabolism, RIT-seq screens confirmed the role of nitroreductase in nitro pro-drug activation (100, 118) and also revealed other metabolic drug interactions (118). The previously reported melarsoprol trypanothione adduct, Mel T (53) was highlighted, suramin action was shown to be enhanced by the spermidine biosynthetic pathway, as well as a glycosylation pathway and a pair of deubiquitinases, and nifurtimox action was shown to be enhanced by the ubiquinone biosynthetic pathway. In the case of suramin, glycosylation and ubiquitin transfer are likely to be required for effective trafficking of the suramin-binding receptor, ISG75. In contrast, ubiquinone is the likely natural substrate for nitroreductase, the activity of which is, therefore, probably dependent upon the presence of the substrate.
OTHER APPROACHES
Expression profiling
Changes in expression profiles, measured using DNA microarrays, RNA-seq or proteomics, following the administration of compounds, can be used to generate ‘connectivity maps’ and can reveal mechanism of action. To understand artemisinin resistance, transcriptional profiling of artemsinin-resistant lines was carried out. Metabolic activities were found to be reduced in the early ring stage, while protein synthesis was enhanced in the schizont stage to counteract the oxidative stress caused by the drug (121). Transcriptional changes resulting from a given compound can also be compared with a compendium of small molecule perturbations to identify mechanisms. Transcriptional profiles were obtained following the administration of twenty compounds that inhibit P. falciparum schizont-stage growth. Together with yeast two-hybrid data, an interaction network was constructed to predict novel antimalarial mechanisms (122).
RNA-seq has been used quite extensively in T. brucei (123) but application to drug resistant cells has yet to be reported. RNA profiling applied to another trypanosomatid, Leishmania, revealed changes caused by gene amplification, deletion and chromosome aneuploidy in association with methotrexate resistance (124).
Chemoproteomics
A powerful approach that has been developed for target identification involves making a derivative of the small molecule of interest to produce an affinity probe (125). Affinity matrices can be made and RBC and parasite lysate passed over these to identify molecules that bind specifically. This has been done with quinoline antimalarials and the RBC proteins aldehyde dehydrogenase 1 and quinine reductase 2 were recovered (112). With artemsinin-based endoperoxide probes, the P. falciparum endoplasmic reticulum resident binding protein was identified as a potential target (126, 127). However, follow-up to define specificity is a major problem with chemical proteomics approaches.
A development of this approach which has proven to be very successful is the use of activity-based probes. Affinity probes are made from antimalarial drugs for proteomic profiling as described above, while at the same time inactive derivatives of the drugs are also produced by introducing small chemical changes. The latter serves to identify the active protein within the proteome of the cell by increasing the specificity of the profiling. In this way the P. falciparum protease falcipain 1 was identified as playing a role in RBC invasion (128). Recently, a natural product, symplostatin 4, demonstrating nanomolar inhibition of parasite vacuolar falcipains has been identified by this approach (129). Similarly, activity-based probes designed on a bestatin scaffold were used to identify specific roles for the P. falciparum M1 aminopeptidase in hemoglobin digestion and the M17 aminopeptidase earlier in the life-cycle (130).
Chemoproteomic approaches have also been applied to T. brucei and identify several kinases as potential targets of novel compounds (131–134). Other affinity-based approaches that exploit drug-target interactions to enrich targets, include the yeast three hybrid system, mRNA or phage display, protein microarrays and ‘reverse transfected’ cell microarrays.
Metabolomics
The goal of metabolomics is to measure all the low molecular weight chemicals in a system. Metabolomic analysis in trypanosomes (135) has recently been used to probe drug action and reveals a lack of synergy between nifurtimox and eflornithine (136). Another study revealed a potential problem with matching drug efficacy in vitro to drug efficacy in vivo; the growth medium was found to contain unnecessarily high concentrations of nutrients that decreased sensitivity to pentamidine and methotrexate by 400-fold (137). Metabolomics approaches have also been developed to interrogate P. falciparum (138) and should yield new insights in the near future.
NEW GENOMIC APPROACHES TO DRUG DEVELOPMENT
Genetic screens have proven to be particularly powerful, since they take a lot of the guess-work out of hypothesis-generation. This can be even more powerful when investigating molecular mechanisms underlying phenotypes lacking prior mechanistic insight and when applied in high-throughput or genome-scale formats (Figure 3). The tremendous potential of these screens justifies the design and development of new tools and approaches in order to access the genetic basis of parasite biology, including druggable biology. Such studies have begun to close some major gaps in our understanding of the mechanisms underlying drug efficacy and potential resistance and this should have a substantial impact on the drug development process in the future.
Target deconvolution strategies
Genome sequencing in drug-resistant strains may have achieved target deconvolution in the haploid malaria parasite (see above) but this is more challenging in diploid parasites such as T. brucei. For T. brucei, RNAi libraries could be screened using new drugs to identify resistance mechanisms (118) and this can identify genes that contribute to drug action (see above) but this loss-of-function approach does not typically identify targets directly; knockdown of a target should render cells more susceptible to the drug. The alternative then would be a screen for ‘synthetic lethality’, combining RNAi and drug exposure to reveal an increased loss-of-fitness. This would be analogous to drug-induced haploinsufficient profiling (HIP) which has proven successful for the identification of drug targets in yeast; drug sensitivity is increased in cells with a decreased dosage of the gene encoding the drug target (139).
What about over-expression profiling? Multicopy suppression profiling (MSP) is essentially the reverse of HIP (81) and can confer resistance by increasing the dosage of a drug target (140). Reduced dosage is more likely to identify a target that is part of a multi-subunit complex, but increased dosage is less likely to lead to loss of the population due to growth insufficiency. Notwithstanding the caveats, genome-wide knockdown and over-expression screens combined (see Figure 3) could form part of a successful target deconvolution strategy. The over-expression approach should also identify genes involved in drug detoxification or efflux.
New genetic methods for target validation
Target validation can be carried out by genetic methods such as RNAi or conditional gene or protein expression. Although RNAi does not exist for P. falciparum, conditional knockdown methods are now being developed (70, 141). In addition a conditional knockout approach has now been described in P. falciparum which could be very useful for determining the function of essential genes (142). Most recently, zinc finger-based technology has been described for the rapid and specific integration of plasmid DNA, allowing for exquisite P. falciparum genome editing (143).
Use of heterologous systems
A screen of temperature sensitive mutants couple to genome sequencing in the genetically tractable T. gondii system led to the identification of parasites deficient in both egress and invasion into host cells resulting from a defect in exocytosis of the microneme organelles (144). Whole genome sequencing identified the etiological mutation in the T. gondii Doc2.1 protein. The homologous protein in P. falciparum was also shown to have a role in regulated exocytosis and invasion of RBCs. These studies indicate that screening in T. gondii promises to be a powerful means to identify the conserved essential roles of proteins in Apicomplexan parasites. In addition, suppressor mutations can be selected in genetic mutants to identify novel genes and pathways by whole genome sequencing.
The power of yeast genetics has similarly been harnessed to identify putative determinants of antimalarial susceptibility such as quinine (145). However, due to the evolutionary distance between these organisms it is not clear to what extent this approach will translate to P. falciparum. Among the trypanosomatids, including T. cruzi and leishmania spp., T. brucei is currently the most genetically tractable. Thus, T. brucei can provide a model to probe trypanosomatid drug targets. However, it will clearly be important to improve the tractability of these other important pathogens. One particularly notable advance here is the discovery of a functional RNAi pathway in Leishmania braziliensis (146).
Predicting behaviour in vivo
A major challenge is to establish assays that accurately predict behaviour in vivo once a target is perturbed. A focus on parasite specific biology, such as innate immune mechanisms, adaptive immune evasion, quorum sensing and developmental transitions, could be fruitful. The machineries underlying these processes can be identified through appropriate genetic screens and will likely yield druggable targets. Genetic screens in in vivo models could be used to identify those processes that are specifically required for survival in this hostile environment and to reveal differences between growth medium and the natural host environment. In some cases, drugs that target in vivo virulence mechanisms could co-opt the immune mechanisms of the host in clearing parasites. The development of assays that focus on distinct stages of the malaria life-cycle, improvements in growth assays (147) and the development of in vivo models that allow accurate quantification or parasite burden, and that reflect the second CNS-stage of HAT, will all be important here.
Combining target-based and whole-cell based screening
Whole-cell based assays can survey all potential cellular targets in their native physiological context, revealing enzyme inhibitors as well as inhibitors of macromolecular interactions, and also reflecting polypharmacology. In contrast, target-based screening would not be expected to probe mechanisms of drug uptake, mechanisms of activation in the case of pro-drugs, or nonspecific mechanisms of action by DNA intercalating agents, alkylating agents or detergents for example. Hits from target-based screens can also suffer from high attrition rates in terms of progression to chemical validation in whole cells. Thus, whole-cell based screens would be the favoured starting point if target deconvolution strategies could be improved.
Target-based and whole-cell screening can be achieved in parallel, using a whole-cell target-based screen (148) or, genetic screens and chemical screens for specific, ‘target-based’ phenotypes can be run in parallel (149). This approach could be used to probe virulence determinants, could greatly facilitate target deconvolution and has the potential to immediately yield lead compounds with whole cell chemical validation. Coverage of a broad chemical space may be necessary given the dual requirements for accessing and disrupting the function of a narrow set of targets. On the other hand, compounds disrupting the function of a single component of a protein complex, interactions among them or metabolites required for their function should all score as ‘hits’ in these screens.
An example of a potential target is the immune evasion strategies of African trypanosomes and malaria parasites. Targeting these mechanisms with small molecules should allow the host immune system to clear parasites. Drugs targeting these pathways may not be identified by conventional phenotypic screening because they may not have major impact on cell viability in culture in the absence of host immune effectors. Specifically, a read-out for loss of monoallelic VSG or var expression control should reflect disruption of an essential immune evasion process. The key here is that a high-throughput chemical screen and a high-throughput genetic screen could be conducted in parallel, scoring for this same phenotype. The allelic exclusion machinery would be identified in the genetic screen, while small molecules that target this same machinery should elicit the same phenotype. Other parasite functions and processes could potentially be targeted using this ‘chemical-genetic phenocopy’ approach, which could rapidly yield small molecules with known targets.
Host-directed therapeutics
Drug-resistance is likely to continue to be a serious problem during control efforts against malaria. By targeting the RBC, it might be possible to reduce the likelihood of drug-resistance. Host targets that have been identified in P. falciparum infections include RBC calpain (150) and the GPCR (151). The potential for targeting the latter with small molecule inhibitors has been explored (152). In contrast to P. falciparum, the RBC is also amenable to RNAi knockdown analysis (153) and hence functional screens are possible. In a similar fashion host determinants of infection of liver cells can also be identified (154).
CONCLUDING REMARKS
Improvements on existing antiparasitic chemotherapies, the development of new therapies and the ability to tackle resistance have been limited by a lack of knowledge in terms of understanding how drugs interact with, and ultimately kill, the parasite in question. The advent of genomics has presented a range of new opportunities to rapidly accelerate the process of filling this knowledge-gap. In particular, recent work on malaria parasites and trypanosomatids demonstrates what can now be achieved. Continuing work in this area will further illuminate mechanisms of drug transport, metabolism and the drug-targets themselves.
There is an emerging view that we need to move beyond conventional target-based screening for parasitic diseases. The genetic and genomic approaches we have outlined above, and resulting improvements in understanding drug-parasite interactions, should facilitate the rational design of improved drugs and drug combinations that are efficiently delivered to the parasite (targets), that disrupt novel targets and that minimise or delay the development of resistance. We envision specific targeting of parasite receptors and virulence mechanisms and co-opting parasite metabolic pathways and host immune effectors to increase potency and reduce toxicity. In addition to the genetic approaches discussed above, proteomic and metabolomic approaches also promise major advances in our understanding in this area.
The availability of multiple trypanosomatid and malarial genome sequences could facilitate the development of broad-spectrum drugs. Some would argue that this is unlikely due to the differences in the biology of the trypanosomatids and the diseases they cause. However, nifurtimox is already used to treat HAT and Chagas’ disease and this pro-drug is known to be activated in both parasites by a nitroreductase. Another nitro pro-drug, fexinidazole, shows promise for use against HAT and leishmaniasis (155). Thus, there may be a real possibility of developing broad spectrum antiparasitic agents. Even if the same compound cannot be used against multiple parasites, knowledge of the biology of any single parasite and how this biology relates to drug action will likely present opportunities for the development of drugs against related parasites. Actually, drugs developed for specific inhibition of a particular enzyme will have a narrow spectrum, while the nitro-drugs, for example, may cause more generalised damage to cellular components. This concept of network (or poly) pharmacology (5), action on multiple rather than single targets, could contribute to the development of more efficacious therapies with broader application. Some of the older antitrypanosomal drugs are thought to work in this way and this could lend support to approaches that perturb networks rather than individual targets.
The goal is to develop affordable, safe, easily administered, and effective antiparasitic therapies. Drug safety and efficacy are determined by interactions with intact cells rather than with individual proteins. It is now clear that drugs may display an affinity for transporters, metabolites, enzymes or other proteins and other macromolecules. Binding can block the transit of natural substrates and can be important for drug-uptake in the case of transporters, it can interfere with metabolic processes, inhibit enzymes or block protein-protein and other interactions, and any one of these affinities could kill parasites. Whole cell screening approaches show the greatest promise of effectively accessing this ‘druggable-space’ and, combined with genome-scale approaches, could help to deliver more effective drugs. Ultimately, the ability to translate genomic outputs into new and improved chemotherapies may determine whether diseases such as malaria and HAT can be eliminated as public health problems, goals recently stated by the WHO.
Acknowledgments
Work in the authors’ laboratories is funded by grants from The Wellcome Trust (093010/Z/10/Z and a Senior Investigator Award, 100320/Z/12/Z) to DH and from the National Institutes of Health (5R01AI091787 and 5R21AI088314) to MTD.
Contributor Information
David Horn, Email: d.horn@dundee.ac.uk.
Manoj T. Duraisingh, Email: mduraisi@hsph.harvard.edu.
References
- 1.Maser P, Wittlin S, Rottmann M, Wenzler T, Kaiser M, Brun R. Antiparasitic agents: new drugs on the horizon. Curr Opin Pharmacol. 2012;12:562–6. doi: 10.1016/j.coph.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–9. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 3.Stockwell BR. Chemical genetics: ligand-based discovery of gene function. Nat Rev Genet. 2000;1:116–25. doi: 10.1038/35038557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–20. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–90. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 6.Okell LC, Drakeley CJ, Bousema T, Whitty CJ, Ghani AC. Modelling the impact of artemisinin combination therapy and long-acting treatments on malaria transmission intensity. PLoS Med. 2008;5:e226. doi: 10.1371/journal.pmed.0050226. discussion e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature. 2013;496:528–32. doi: 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- 8.Taylor TE, Wills BA, Kazembe P, Chisale M, Wirima JJ, et al. Rapid coma resolution with artemether in Malawian children with cerebral malaria. Lancet. 1993;341:661–2. doi: 10.1016/0140-6736(93)90423-e. [DOI] [PubMed] [Google Scholar]
- 9.Baird JK. Chloroquine resistance in Plasmodium vivax. Antimicrob Agents Chemother. 2004;48:4075–83. doi: 10.1128/AAC.48.11.4075-4083.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hawkins VN, Auliff A, Prajapati SK, Rungsihirunrat K, Hapuarachchi HC, et al. Multiple origins of resistance-conferring mutations in Plasmodium vivax dihydrofolate reductase. Malar J. 2008;7:72. doi: 10.1186/1475-2875-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spillman NJ, Allen RJ, McNamara CW, Yeung BK, Winzeler EA, et al. Na(+) regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe. 2013;13:227–37. doi: 10.1016/j.chom.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steverding D. The development of drugs for treatment of sleeping sickness: a historical review. Parasit Vectors. 2010;3:15. doi: 10.1186/1756-3305-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkinson SR, Kelly JM. Trypanocidal drugs: mechanisms, resistance and new targets. Expert Rev Mol Med. 2009;11:e31. doi: 10.1017/S1462399409001252. [DOI] [PubMed] [Google Scholar]
- 14.Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet. 2009;374:56–64. doi: 10.1016/S0140-6736(09)61117-X. [DOI] [PubMed] [Google Scholar]
- 15.Simarro PP, Franco J, Diarra A, Postigo JA, Jannin J. Update on field use of the available drugs for the chemotherapy of human African trypanosomiasis. Parasitology. 2012;139:842–6. doi: 10.1017/S0031182012000169. [DOI] [PubMed] [Google Scholar]
- 16.Kotthaus J, Schade D, Schwering U, Hungeling H, Muller-Fielitz H, et al. New prodrugs of the antiprotozoal drug pentamidine. ChemMedChem. 2011;6:2233–42. doi: 10.1002/cmdc.201100422. [DOI] [PubMed] [Google Scholar]
- 17.Wenzler T, Boykin DW, Ismail MA, Hall JE, Tidwell RR, Brun R. New treatment option for second-stage African sleeping sickness: in vitro and in vivo efficacy of aza analogs of DB289. Antimicrob Agents Chemother. 2009;53:4185–92. doi: 10.1128/AAC.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thuita JK, Wang MZ, Kagira JM, Denton CL, Paine MF, et al. Pharmacology of DB844, an orally active aza analogue of pafuramidine, in a monkey model of second stage human African trypanosomiasis. PLoS Negl Trop Dis. 2012;6:e1734. doi: 10.1371/journal.pntd.0001734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaiser M, Bray MA, Cal M, Bourdin Trunz B, Torreele E, Brun R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob Agents Chemother. 2011;55:5602–8. doi: 10.1128/AAC.00246-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl Trop Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fairhurst RM, Nayyar GM, Breman JG, Hallett R, Vennerstrom JL, et al. Artemisinin-resistant malaria: research challenges, opportunities, and public health implications. Am J Trop Med Hyg. 2012;87:231–41. doi: 10.4269/ajtmh.2012.12-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.AlKadi HO. Antimalarial drug toxicity: a review. Chemotherapy. 2007;53:385–91. doi: 10.1159/000109767. [DOI] [PubMed] [Google Scholar]
- 23.malERA. A research agenda for malaria eradication: drugs. PLoS Med. 2011;8:e1000402. doi: 10.1371/journal.pmed.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salvidio E, Pannacciulli I, Tizianello A, Ajmar F. Nature of hemolytic crises and the fate of G6PD deficient, drug-damaged erythrocytes in Sardinians. N Engl J Med. 1967;276:1339–44. doi: 10.1056/NEJM196706152762402. [DOI] [PubMed] [Google Scholar]
- 25.Bowman ZS, Oatis JE, Jr, Whelan JL, Jollow DJ, McMillan DC. Primaquine-induced hemolytic anemia: susceptibility of normal versus glutathione-depleted rat erythrocytes to 5-hydroxyprimaquine. J Pharmacol Exp Ther. 2004;309:79–85. doi: 10.1124/jpet.103.062984. [DOI] [PubMed] [Google Scholar]
- 26.Iten M, Mett H, Evans A, Enyaru JC, Brun R, Kaminsky R. Alterations in ornithine decarboxylase characteristics account for tolerance of Trypanosoma brucei rhodesiense to D,L-α-difluoromethylornithine. Antimicrob Agents Chemother. 1997;41:1922–5. doi: 10.1128/aac.41.9.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuepfer I, Schmid C, Allan M, Edielu A, Haary EP, et al. Safety and efficacy of the 10-day melarsoprol schedule for the treatment of second stage Rhodesiense sleeping sickness. PLoS Negl Trop Dis. 2012;6:e1695. doi: 10.1371/journal.pntd.0001695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damper D, Patton CL. Pentamidine transport and sensitivity in brucei-group trypanosomes. J Protozool. 1976;23:349–56. doi: 10.1111/j.1550-7408.1976.tb03787.x. [DOI] [PubMed] [Google Scholar]
- 29.Frommel TO, Balber AE. Flow cytofluorimetric analysis of drug accumulation by multidrug-resistant Trypanosoma brucei brucei and T. b. rhodesiense. Mol Biochem Parasitol. 1987;26:183–91. doi: 10.1016/0166-6851(87)90142-3. [DOI] [PubMed] [Google Scholar]
- 30.Maser P, Luscher A, Kaminsky R. Drug transport and drug resistance in African trypanosomes. Drug Resist Updat. 2003;6:281–90. doi: 10.1016/j.drup.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Rollo IM, Williamson J. Acquired resistance to ‘Melarsen’, tryparsamide and amidines in pathogenic trypanosomes after treatment with ‘Melarsen’ alone. Nature. 1951;167:147–8. doi: 10.1038/167147a0. [DOI] [PubMed] [Google Scholar]
- 32.Duraisingh MT, Cowman AF. Contribution of the pfmdr1 gene to antimalarial drug-resistance. Acta Trop. 2005;94:181–90. doi: 10.1016/j.actatropica.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Ecker A, Lehane AM, Clain J, Fidock DA. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012;28:504–14. doi: 10.1016/j.pt.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maser P, Sutterlin C, Kralli A, Kaminsky R. A nucleoside transporter from Trypanosoma brucei involved in drug resistance. Science. 1999;285:242–4. doi: 10.1126/science.285.5425.242. [DOI] [PubMed] [Google Scholar]
- 35.Stewart ML, Krishna S, Burchmore RJ, Brun R, de Koning HP, et al. Detection of arsenical drug resistance in Trypanosoma brucei with a simple fluorescence test. Lancet. 2005;366:486–7. doi: 10.1016/S0140-6736(05)66793-1. [DOI] [PubMed] [Google Scholar]
- 36.Kapetanaki S, Varotsis C. Fourier transform infrared investigation of non-heme Fe(III) and Fe(II) decomposition of artemisinin and of a simplified trioxane alcohol. J Med Chem. 2001;44:3150–6. doi: 10.1021/jm010848d. [DOI] [PubMed] [Google Scholar]
- 37.Meshnick SR. Artemisinin: mechanisms of action, resistance and toxicity. Int J Parasitol. 2002;32:1655–60. doi: 10.1016/s0020-7519(02)00194-7. [DOI] [PubMed] [Google Scholar]
- 38.Wilkinson SR, Taylor MC, Horn D, Kelly JM, Cheeseman I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc Natl Acad Sci U S A. 2008;105:5022–7. doi: 10.1073/pnas.0711014105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wyllie S, Patterson S, Fairlamb AH. Assessing the essentiality of Leishmania donovani nitroreductase and its role in nitro drug activation. Antimicrob Agents Chemother. 2013;57:901–6. doi: 10.1128/AAC.01788-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, et al. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:431–6. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sokolova AY, Wyllie S, Patterson S, Oza SL, Read KD, Fairlamb AH. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob Agents Chemother. 2010;54:2893–900. doi: 10.1128/AAC.00332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reeder JC, Rieckmann KH, Genton B, Lorry K, Wines B, Cowman AF. Point mutations in the dihydrofolate reductase and dihydropteroate synthetase genes and in vitro susceptibility to pyrimethamine and cycloguanil of Plasmodium falciparum isolates from Papua New Guinea. Am J Trop Med Hyg. 1996;55:209–13. doi: 10.4269/ajtmh.1996.55.209. [DOI] [PubMed] [Google Scholar]
- 43.Triglia T, Wang P, Sims PF, Hyde JE, Cowman AF. Allelic exchange at the endogenous genomic locus in Plasmodium falciparum proves the role of dihydropteroate synthase in sulfadoxine-resistant malaria. EMBO J. 1998;17:3807–15. doi: 10.1093/emboj/17.14.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Triglia T, Menting JG, Wilson C, Cowman AF. Mutations in dihydropteroate synthase are responsible for sulfone and sulfonamide resistance in Plasmodium falciparum. Proc Natl Acad Sci U S A. 1997;94:13944–9. doi: 10.1073/pnas.94.25.13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Srivastava IK, Vaidya AB. A mechanism for the synergistic antimalarial action of atovaquone and proguanil. Antimicrob Agents Chemother. 1999;43:1334–9. doi: 10.1128/aac.43.6.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edwards KJ, Jenkins TC, Neidle S. Crystal structure of a pentamidine-oligonucleotide complex: implications for DNA-binding properties. Biochemistry. 1992;31:7104–9. doi: 10.1021/bi00146a011. [DOI] [PubMed] [Google Scholar]
- 47.Shapiro TA, Englund PT. Selective cleavage of kinetoplast DNA minicircles promoted by antitrypanosomal drugs. Proc Natl Acad Sci U S A. 1990;87:950–4. doi: 10.1073/pnas.87.3.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schnaufer A. Evolution of dyskinetoplastic trypanosomes: how, and how often? Trends Parasitol. 2010;26:557–8. doi: 10.1016/j.pt.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanteri CA, Tidwell RR, Meshnick SR. The mitochondrion is a site of trypanocidal action of the aromatic diamidine DB75 in bloodstream forms of Trypanosoma brucei. Antimicrob Agents Chemother. 2008;52:875–82. doi: 10.1128/AAC.00642-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moreno SN. Pentamidine is an uncoupler of oxidative phosphorylation in rat liver mitochondria. Arch Biochem Biophys. 1996;326:15–20. doi: 10.1006/abbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- 51.Mathis AM, Holman JL, Sturk LM, Ismail MA, Boykin DW, et al. Accumulation and intracellular distribution of antitrypanosomal diamidine compounds DB75 and DB820 in African trypanosomes. Antimicrob Agents Chemother. 2006;50:2185–91. doi: 10.1128/AAC.00192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson WD, Tanious FA, Mathis A, Tevis D, Hall JE, Boykin DW. Antiparasitic compounds that target DNA. Biochimie. 2008;90:999–1014. doi: 10.1016/j.biochi.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fairlamb AH, Henderson GB, Cerami A. Trypanothione is the primary target for arsenical drugs against African trypanosomes. Proc Natl Acad Sci U S A. 1989;86:2607–11. doi: 10.1073/pnas.86.8.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jacobs RT, Nare B, Phillips MA. State of the art in African trypanosome drug discovery. Curr Top Med Chem. 2011;11:1255–74. doi: 10.2174/156802611795429167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frearson JA, Brand S, McElroy SP, Cleghorn LA, Smid O, et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature. 2010;464:728–32. doi: 10.1038/nature08893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gardner MJ, Hall N, Fung E, White O, Berriman M, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–22. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 58.Mackey ZB, Koupparis K, Nishino M, McKerrow JH. High-throughput analysis of an RNAi library identifies novel kinase targets in Trypanosoma brucei. Chem Biol Drug Des. 2011;78:454–63. doi: 10.1111/j.1747-0285.2011.01156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Naula C, Parsons M, Mottram JC. Protein kinases as drug targets in trypanosomes and Leishmania. Biochim Biophys Acta. 2005;1754:151–9. doi: 10.1016/j.bbapap.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Koning HP, Gould MK, Sterk GJ, Tenor H, Kunz S, et al. Pharmacological validation of Trypanosoma brucei phosphodiesterases as novel drug targets. J Infect Dis. 2012;206:229–37. doi: 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horn D. Histone deacetylases. Adv Exp Med Biol. 2008;625:81–6. doi: 10.1007/978-0-387-77570-8_7. [DOI] [PubMed] [Google Scholar]
- 62.McKerrow JH, Engel JC, Caffrey CR. Cysteine protease inhibitors as chemotherapy for parasitic infections. Bioorg Med Chem. 1999;7:639–44. doi: 10.1016/s0968-0896(99)00008-5. [DOI] [PubMed] [Google Scholar]
- 63.Nkemngu NJ, Rosenkranz V, Wink M, Steverding D. Antitrypanosomal activities of proteasome inhibitors. Antimicrob Agents Chemother. 2002;46:2038–40. doi: 10.1128/AAC.46.6.2038-2040.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aguero F, Al-Lazikani B, Aslett M, Berriman M, Buckner FS, et al. Genomic-scale prioritization of drug targets: the TDR Targets database. Nat Rev Drug Discov. 2008;7:900–7. doi: 10.1038/nrd2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, et al. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011;21:915–24. doi: 10.1101/gr.115089.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wirtz E, Leal S, Ochatt C, Cross GA. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 67.Magarinos MP, Carmona SJ, Crowther GJ, Ralph SA, Roos DS, et al. TDR Targets: a chemogenomics resource for neglected diseases. Nucleic Acids Res. 2012;40:D1118–27. doi: 10.1093/nar/gkr1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 69.Militello KT, Refour P, Comeaux CA, Duraisingh MT. Antisense RNA and RNAi in protozoan parasites: working hard or hardly working? Mol Biochem Parasitol. 2008;157:117–26. doi: 10.1016/j.molbiopara.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 70.Pino P, Sebastian S, Kim EA, Bush E, Brochet M, et al. A tetracycline-repressible transactivator system to study essential genes in malaria parasites. Cell Host Microbe. 2012;12:824–34. doi: 10.1016/j.chom.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dvorin JD, Martyn DC, Patel SD, Grimley JS, Collins CR, et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science. 2010;328:910–2. doi: 10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chong CR, Chen X, Shi L, Liu JO, Sullivan DJ., Jr A clinical drug library screen identifies astemizole as an antimalarial agent. Nat Chem Biol. 2006;2:415–6. doi: 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]
- 73.Weisman JL, Liou AP, Shelat AA, Cohen FE, Guy RK, DeRisi JL. Searching for new antimalarial therapeutics amongst known drugs. Chem Biol Drug Des. 2006;67:409–16. doi: 10.1111/j.1747-0285.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–5. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–10. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 76.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci U S A. 2008;105:9059–64. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baniecki ML, Wirth DF, Clardy J. High-throughput Plasmodium falciparum growth assay for malaria drug discovery. Antimicrob Agents Chemother. 2007;51:716–23. doi: 10.1128/AAC.01144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jensen K, Plichta D, Panagiotou G, Kouskoumvekaki I. Mapping the genome of Plasmodium falciparum on the drug-like chemical space reveals novel anti-malarial targets and potential drug leads. Mol Biosyst. 2012;8:1678–85. doi: 10.1039/c2mb00008c. [DOI] [PubMed] [Google Scholar]
- 79.Brown LE, Chih-Chien Cheng K, Wei WG, Yuan P, Dai P, et al. Discovery of new antimalarial chemotypes through chemical methodology and library development. Proc Natl Acad Sci U S A. 2011;108:6775–80. doi: 10.1073/pnas.1017666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mackey ZB, Baca AM, Mallari JP, Apsel B, Shelat A, et al. Discovery of trypanocidal compounds by whole cell HTS of Trypanosoma brucei. Chem Biol Drug Des. 2006;67:355–63. doi: 10.1111/j.1747-0285.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- 81.Cong F, Cheung AK, Huang SM. Chemical genetics-based target identification in drug discovery. Annu Rev Pharmacol Toxicol. 2012;52:57–78. doi: 10.1146/annurev-pharmtox-010611-134639. [DOI] [PubMed] [Google Scholar]
- 82.Terstappen GC, Schlupen C, Raggiaschi R, Gaviraghi G. Target deconvolution strategies in drug discovery. Nat Rev Drug Discov. 2007;6:891–903. doi: 10.1038/nrd2410. [DOI] [PubMed] [Google Scholar]
- 83.Walliker D, Quakyi IA, Wellems TE, McCutchan TF, Szarfman A, et al. Genetic analysis of the human malaria parasite Plasmodium falciparum. Science. 1987;236:1661–6. doi: 10.1126/science.3299700. [DOI] [PubMed] [Google Scholar]
- 84.Su X, Kirkman LA, Fujioka H, Wellems TE. Complex polymorphisms in an approximately 330 kDa protein are linked to chloroquine-resistant P. falciparum in Southeast Asia and Africa. Cell. 1997;91:593–603. doi: 10.1016/s0092-8674(00)80447-x. [DOI] [PubMed] [Google Scholar]
- 85.Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–3. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferdig MT, Cooper RA, Mu J, Deng B, Joy DA, et al. Dissecting the loci of low-level quinine resistance in malaria parasites. Mol Microbiol. 2004;52:985–97. doi: 10.1111/j.1365-2958.2004.04035.x. [DOI] [PubMed] [Google Scholar]
- 87.Desai SA. Ion and nutrient uptake by malaria parasite-infected erythrocytes. Cell Microbiol. 2012;14:1003–9. doi: 10.1111/j.1462-5822.2012.01790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tait A, Morrison LJ, Duffy CW, Cooper A, Turner CM, Macleod A. Trypanosome genetics: populations, phenotypes and diversity. Vet Parasitol. 2011;181:61–8. doi: 10.1016/j.vetpar.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 89.Mu J, Duan J, Makova KD, Joy DA, Huynh CQ, et al. Chromosome-wide SNPs reveal an ancient origin for Plasmodium falciparum. Nature. 2002;418:323–6. doi: 10.1038/nature00836. [DOI] [PubMed] [Google Scholar]
- 90.Park DJ, Lukens AK, Neafsey DE, Schaffner SF, Chang HH, et al. Sequence-based association and selection scans identify drug resistance loci in the Plasmodium falciparum malaria parasite. Proc Natl Acad Sci U S A. 2012;109:13052–7. doi: 10.1073/pnas.1210585109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Van Tyne D, Park DJ, Schaffner SF, Neafsey DE, Angelino E, et al. Identification and functional validation of the novel antimalarial resistance locus PF10_0355 in Plasmodium falciparum. PLoS Genet. 2011;7:e1001383. doi: 10.1371/journal.pgen.1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Takala-Harrison S, Clark TG, Jacob CG, Cummings MP, Miotto O, et al. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc Natl Acad Sci U S A. 2013;110:240–5. doi: 10.1073/pnas.1211205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yuan J, Cheng KC, Johnson RL, Huang R, Pattaradilokrat S, et al. Chemical genomic profiling for antimalarial therapies, response signatures, and molecular targets. Science. 2011;333:724–9. doi: 10.1126/science.1205216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mehlotra RK, Henry-Halldin CN, Zimmerman PA. Application of pharmacogenomics to malaria: a holistic approach for successful chemotherapy. Pharmacogenomics. 2009;10:435–49. doi: 10.2217/14622416.10.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dharia NV, Sidhu AB, Cassera MB, Westenberger SJ, Bopp SE, et al. Use of high-density tiling microarrays to identify mutations globally and elucidate mechanisms of drug resistance in Plasmodium falciparum. Genome Biol. 2009;10:R21. doi: 10.1186/gb-2009-10-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eastman RT, Dharia NV, Winzeler EA, Fidock DA. Piperaquine resistance is associated with a copy number variation on chromosome 5 in drug-pressured Plasmodium falciparum parasites. Antimicrob Agents Chemother. 2011;55:3908–16. doi: 10.1128/AAC.01793-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nam TG, McNamara CW, Bopp S, Dharia NV, Meister S, et al. A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem Biol. 2011;6:1214–22. doi: 10.1021/cb200105d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–80. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harbut MB, Patel BA, Yeung BK, McNamara CW, Bright AT, et al. Targeting the ERAD pathway via inhibition of signal peptide peptidase for antiparasitic therapeutic design. Proc Natl Acad Sci U S A. 2012;109:21486–91. doi: 10.1073/pnas.1216016110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Baker N, Alsford S, Horn D. Genome-wide RNAi screens in African trypanosomes identify the nifurtimox activator NTR and the eflornithine transporter AAT6. Mol Biochem Parasitol. 2011;176:55–7. doi: 10.1016/j.molbiopara.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schumann Burkard G, Jutzi P, Roditi I. Genome-wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol Biochem Parasitol. 2011;175:91–4. doi: 10.1016/j.molbiopara.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 102.Vincent IM, Creek D, Watson DG, Kamleh MA, Woods DJ, et al. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. 2010;6:e1001204. doi: 10.1371/journal.ppat.1001204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baker N, Glover L, Munday JC, Aguinaga Andres D, Barrett MP, et al. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc Natl Acad Sci U S A. 2012;109:10996–1001. doi: 10.1073/pnas.1202885109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hoepfner D, McNamara CW, Lim CS, Studer C, Riedl R, et al. Selective and specific inhibition of the Plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe. 2012;11:654–63. doi: 10.1016/j.chom.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maier AG, Rug M, O’Neill MT, Brown M, Chakravorty S, et al. Exported proteins required for virulence and rigidity of Plasmodium falciparum-infected human erythrocytes. Cell. 2008;134:48–61. doi: 10.1016/j.cell.2008.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tewari R, Straschil U, Bateman A, Bohme U, Cherevach I, et al. The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe. 2010;8:377–87. doi: 10.1016/j.chom.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dorin-Semblat D, Schmitt S, Semblat JP, Sicard A, Reininger L, et al. Plasmodium falciparum NIMA-related kinase Pfnek-1: sex specificity and assessment of essentiality for the erythrocytic asexual cycle. Microbiology. 2011;157:2785–94. doi: 10.1099/mic.0.049023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Langridge GC, Phan MD, Turner DJ, Perkins TT, Parts L, et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 2009;19:2308–16. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Murry JP, Rubin EJ. New genetic approaches shed light on TB virulence. Trends Microbiol. 2005;13:366–72. doi: 10.1016/j.tim.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 110.Balu B, Shoue DA, Fraser MJ, Jr, Adams JH. High-efficiency transformation of Plasmodium falciparum by the lepidopteran transposable element piggyBac. Proc Natl Acad Sci U S A. 2005;102:16391–6. doi: 10.1073/pnas.0504679102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ikadai H, Shaw Saliba K, Kanzok SM, McLean KJ, Tanaka TQ, et al. Transposon mutagenesis identifies genes essential for Plasmodium falciparum gametocytogenesis. Proc Natl Acad Sci U S A. 2013;110:1676–84. doi: 10.1073/pnas.1217712110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Graves PR, Kwiek JJ, Fadden P, Ray R, Hardeman K, et al. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol Pharmacol. 2002;62:1364–72. doi: 10.1124/mol.62.6.1364. [DOI] [PubMed] [Google Scholar]
- 113.Kim HS, Park SH, Gunzl A, Cross GA. MCM-BP is required for repression of life-cycle specific genes transcribed by RNA polymerase I in the mammalian infectious form of Trypanosoma brucei. PLoS One. 2013;8:e57001. doi: 10.1371/journal.pone.0057001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leal S, Acosta-Serrano A, Morris J, Cross GA. Transposon mutagenesis of Trypanosoma brucei identifies glycosylation mutants resistant to concanavalin A. J Biol Chem. 2004;279:28979–88. doi: 10.1074/jbc.M403479200. [DOI] [PubMed] [Google Scholar]
- 115.Judson N, Mekalanos JJ. Transposon-based approaches to identify essential bacterial genes. Trends Microbiol. 2000;8:521–6. doi: 10.1016/s0966-842x(00)01865-5. [DOI] [PubMed] [Google Scholar]
- 116.Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat Rev Drug Discov. 2010;9:690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pan X, Reissman S, Douglas NR, Huang Z, Yuan DS, et al. Trivalent arsenic inhibits the functions of chaperonin complex. Genetics. 2010;186:725–34. doi: 10.1534/genetics.110.117655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Alsford S, Eckert S, Baker N, Glover L, Sanchez-Flores A, et al. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature. 2012;482:232–6. doi: 10.1038/nature10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Baker N, de Koning HP, Maser P, Horn D. Drug resistance in African trypanosomiasis: the melarsoprol and pentamidine story. Trends Parasitol. 2013;29:110–8. doi: 10.1016/j.pt.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Alsford S, Field MC, Horn D. Receptor-mediated endocytosis for drug delivery in African trypanosomes: fulfilling Paul Ehrlich’s vision of chemotherapy. Trends Parasitol. 2013;29:207–12. doi: 10.1016/j.pt.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 121.Mok S, Imwong M, Mackinnon MJ, Sim J, Ramadoss R, et al. Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics. 2011;12:391. doi: 10.1186/1471-2164-12-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hu G, Cabrera A, Kono M, Mok S, Chaal BK, et al. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat Biotechnol. 2010;28:91–8. doi: 10.1038/nbt.1597. [DOI] [PubMed] [Google Scholar]
- 123.Siegel TN, Gunasekera K, Cross GA, Ochsenreiter T. Gene expression in Trypanosoma brucei: lessons from high-throughput RNA sequencing. Trends Parasitol. 2011;27:434–41. doi: 10.1016/j.pt.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ubeda JM, Legare D, Raymond F, Ouameur AA, Boisvert S, et al. Modulation of gene expression in drug resistant Leishmania is associated with gene amplification, gene deletion and chromosome aneuploidy. Genome Biol. 2008;9:R115. doi: 10.1186/gb-2008-9-7-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang K, Yang T, Wu Q, Zhao X, Nice EC, Huang C. Chemistry-based functional proteomics for drug target deconvolution. Expert Rev Proteomics. 2012;9:293–310. doi: 10.1586/epr.12.19. [DOI] [PubMed] [Google Scholar]
- 126.Barton V, Ward SA, Chadwick J, Hill A, O’Neill PM. Rationale design of biotinylated antimalarial endoperoxide carbon centered radical prodrugs for applications in proteomics. J Med Chem. 2010;53:4555–9. doi: 10.1021/jm100201j. [DOI] [PubMed] [Google Scholar]
- 127.Morita M, Sanai H, Hiramoto A, Sato A, Hiraoka O, et al. Plasmodium falciparum endoplasmic reticulum-resident calcium binding protein is a possible target of synthetic antimalarial endoperoxides, N-89 and N-251. J Proteome Res. 2012;11:5704–11. doi: 10.1021/pr3005315. [DOI] [PubMed] [Google Scholar]
- 128.Greenbaum DC, Baruch A, Grainger M, Bozdech Z, Medzihradszky KF, et al. A role for the protease falcipain 1 in host cell invasion by the human malaria parasite. Science. 2002;298:2002–6. doi: 10.1126/science.1077426. [DOI] [PubMed] [Google Scholar]
- 129.Lucumi E, Darling C, Jo H, Napper AD, Chandramohanadas R, et al. Discovery of potent small-molecule inhibitors of multidrug-resistant Plasmodium falciparum using a novel miniaturized high-throughput luciferase-based assay. Antimicrob Agents Chemother. 2010;54:3597–604. doi: 10.1128/AAC.00431-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Harbut MB, Velmourougane G, Dalal S, Reiss G, Whisstock JC, et al. Bestatin-based chemical biology strategy reveals distinct roles for malaria M1- and M17-family aminopeptidases. Proc Natl Acad Sci U S A. 2011;108:E526–34. doi: 10.1073/pnas.1105601108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kuettel S, Mosimann M, Maser P, Kaiser M, Brun R, et al. Adenosine Kinase of T. b. Rhodesiense identified as the putative target of 4-[5-(4-phenoxyphenyl)-2H-pyrazol-3-yl]morpholine using chemical proteomics. PLoS Negl Trop Dis. 2009;3:e506. doi: 10.1371/journal.pntd.0000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mercer L, Bowling T, Perales J, Freeman J, Nguyen T, et al. 2,4-Diaminopyrimidines as potent inhibitors of Trypanosoma brucei and identification of molecular targets by a chemical proteomics approach. PLoS Negl Trop Dis. 2011;5:e956. doi: 10.1371/journal.pntd.0000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pieretti S, Haanstra JR, Mazet M, Perozzo R, Bergamini C, et al. Naphthoquinone derivatives exert their antitrypanosomal activity via a multi-target mechanism. PLoS Negl Trop Dis. 2013;7:e2012. doi: 10.1371/journal.pntd.0002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Urbaniak MD, Mathieson T, Bantscheff M, Eberhard D, Grimaldi R, et al. Chemical proteomic analysis reveals the drugability of the kinome of Trypanosoma brucei. ACS Chem Biol. 2012;7:1858–65. doi: 10.1021/cb300326z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Creek DJ, Anderson J, McConville MJ, Barrett MP. Metabolomic analysis of trypanosomatid protozoa. Mol Biochem Parasitol. 2012;181:73–84. doi: 10.1016/j.molbiopara.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 136.Vincent IM, Creek DJ, Burgess K, Woods DJ, Burchmore RJ, Barrett MP. Untargeted metabolomics reveals a lack of synergy between nifurtimox and eflornithine against Trypanosoma brucei. PLoS Negl Trop Dis. 2012;6:e1618. doi: 10.1371/journal.pntd.0001618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Creek DJ, Nijagal B, Kim DH, Rojas F, Matthews KR, Barrett MP. Metabolomics guides rational development of a simplified cell culture medium for drug screening against Trypanosoma brucei. Antimicrob Agents Chemother. 2013 doi: 10.1128/AAC.00044-13. [DOI] [PMC free article] [PubMed]
- 138.Olszewski KL, Morrisey JM, Wilinski D, Burns JM, Vaidya AB, et al. Host-parasite interactions revealed by Plasmodium falciparum metabolomics. Cell Host Microbe. 2009;5:191–9. doi: 10.1016/j.chom.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Giaever G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, et al. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1999;21:278–83. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- 140.Ranade RM, Gillespie JR, Shibata S, Verlinde CL, Fan E, et al. Induced Resistance to Methionyl-tRNA Synthetase Inhibitors in Trypanosoma brucei is Due to Overexpression of the Target. Antimicrob Agents Chemother. 2013 doi: 10.1128/AAC.02578-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Armstrong CM, Goldberg DE. An FKBP destabilization domain modulates protein levels in Plasmodium falciparum. Nat Methods. 2007;4:1007–9. doi: 10.1038/nmeth1132. [DOI] [PubMed] [Google Scholar]
- 142.Collins CR, Das S, Wong EH, Andenmatten N, Stallmach R, et al. Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol Microbiol. 2013 doi: 10.1111/mmi.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Straimer J, Lee MC, Lee AH, Zeitler B, Williams AE, et al. Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nat Methods. 2012;9:993–8. doi: 10.1038/nmeth.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Farrell A, Thirugnanam S, Lorestani A, Dvorin JD, Eidell KP, et al. A DOC2 protein identified by mutational profiling is essential for apicomplexan parasite exocytosis. Science. 2012;335:218–21. doi: 10.1126/science.1210829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dos Santos SC, Sa-Correia I. A genome-wide screen identifies yeast genes required for protection against or enhanced cytotoxicity of the antimalarial drug quinine. Mol Genet Genomics. 2011;286:333–46. doi: 10.1007/s00438-011-0649-5. [DOI] [PubMed] [Google Scholar]
- 146.Lye LF, Owens K, Shi H, Murta SM, Vieira AC, et al. Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog. 2010;6:e1001161. doi: 10.1371/journal.ppat.1001161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.De Rycker M, O’Neill S, Joshi D, Campbell L, Gray DW, Fairlamb AH. A static-cidal assay for Trypanosoma brucei to aid hit prioritisation for progression into drug discovery programmes. PLoS Negl Trop Dis. 2012;6:e1932. doi: 10.1371/journal.pntd.0001932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Raju RM, Goldberg AL, Rubin EJ. Bacterial proteolytic complexes as therapeutic targets. Nat Rev Drug Discov. 2012;11:777–89. doi: 10.1038/nrd3846. [DOI] [PubMed] [Google Scholar]
- 149.Roemer T, Boone C. Systems-level antimicrobial drug and drug synergy discovery. Nat Chem Biol. 2013;9:222–31. doi: 10.1038/nchembio.1205. [DOI] [PubMed] [Google Scholar]
- 150.Chandramohanadas R, Davis PH, Beiting DP, Harbut MB, Darling C, et al. Apicomplexan parasites co-opt host calpains to facilitate their escape from infected cells. Science. 2009;324:794–7. doi: 10.1126/science.1171085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Harrison T, Samuel BU, Akompong T, Hamm H, Mohandas N, et al. Erythrocyte G protein-coupled receptor signaling in malarial infection. Science. 2003;301:1734–6. doi: 10.1126/science.1089324. [DOI] [PubMed] [Google Scholar]
- 152.Murphy SC, Harrison T, Hamm HE, Lomasney JW, Mohandas N, Haldar K. Erythrocyte G protein as a novel target for malarial chemotherapy. PLoS Med. 2006;3:e528. doi: 10.1371/journal.pmed.0030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bei AK, Brugnara C, Duraisingh MT. In vitro genetic analysis of an erythrocyte determinant of malaria infection. J Infect Dis. 2010;202:1722–7. doi: 10.1086/657157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Prudencio M, Mota MM. Targeting host factors to circumvent anti-malarial drug resistance. Curr Pharm Des. 2013;19:290–9. doi: 10.2174/138161213804070276. [DOI] [PubMed] [Google Scholar]
- 155.Wyllie S, Patterson S, Stojanovski L, Simeons FR, Norval S, et al. The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci Transl Med. 2012;4:119re1. doi: 10.1126/scitranslmed.3003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huang Z, Chen K, Zhang J, Li Y, Wang H, et al. A functional variomics tool for discovering drug-resistance genes and drug targets. Cell Rep. 2013;3:577–85. doi: 10.1016/j.celrep.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]