

Abstract
Sirtuin 3 (SIRT3), an important regulator of energy metabolism and lipid oxidation, is induced in fasted liver mitochondria and implicated in metabolic syndrome. In fasted liver, SIRT3-mediated increases in substrate flux depend on oxidative phosphorylation (OXPHOS), but precisely how OXPHOS meets the challenge of increased substrate oxidation in fasted liver remains unclear. Here, we show that liver mitochondria in fasting mice adapt to the demand of increased substrate oxidation by increasing their OXPHOS efficiency. In response to cAMP signaling, SIRT3 deacetylated and activated leucine-rich protein 130 (LRP130; official symbol, LRPPRC), promoting a mitochondrial transcriptional program that enhanced hepatic OXPHOS. Using mass spectrometry, we identified SIRT3-regulated lysine residues in LRP130 that generated a lysine-to-arginine (KR) mutant of LRP130 that mimics deacetylated protein. Compared with wild-type LRP130 protein, expression of the KR mutant increased mitochondrial transcription and OXPHOS in vitro. Indeed, even when SIRT3 activity was abolished, activation of mitochondrial transcription and OXPHOS by the KR mutant remained robust, further highlighting the contribution of LRP130 deacetylation to increased OXPHOS in fasted liver. These data establish a link between nutrient sensing and mitochondrial transcription that regulates OXPHOS in fasted liver and may explain how fasted liver adapts to increased substrate oxidation.
Introduction
In normal fasted liver, an increase in ATP-dependent processes (such as gluconeogenesis and ureagenesis) and ketogenesis requires substrate oxidation, specifically β-oxidation of fatty acids. Presumably, oxidative phosphorylation (OXPHOS) must match this increased need for ATP during fasting. Precisely how coordination of this kind occurs in vivo remains an open question. Addressing this question is important, as several studies imply that a mismatch between substrate oxidation and OXPHOS may sustain or exacerbate metabolic disease (1–3). In principle, an increase in mitochondrial content (biogenesis) and/or OXPHOS efficiency (OXPHOS activity per mitochondrion) could accommodate increased ATP requirements in fasted liver. In terms of regulatory control, sirtuin 3 (SIRT3), a mitochondrial sensor of nutrients, is an attractive candidate, since SIRT3 influences both substrate oxidation and OXPHOS.
SIRT3 is a mitochondrial NAD+-dependent deacetylase that senses nutrient deprivation (4). Upon fasting, SIRT3 protein is induced in liver, where it activates enzyme systems involved in fatty acid oxidation and ketogenesis (5, 6). Both long-chain acyl-CoA dehydrogenase (ACADL; also known as LCAD) and 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2) are deacetylated by SIRT3, culminating in enhanced β-oxidation of fatty acids and ketogenesis, respectively (5, 6). In cell culture, SIRT3 influences energy metabolism; however, different mechanisms have been proposed for various cell types (7–10). Nevertheless, whether and how SIRT3 influences energy metabolism in vivo, and under what biological conditions, remains an open question. Given that SIRT3 is implicated in metabolic syndrome (10, 11), resolving this question might explain how defective mitochondria emerge in metabolic disease.
Mitochondrial transcription potently influences energy metabolism (12). The basal transcription machinery of mitochondria consists of mitochondrial transcription factor B2 (TFB2M) and mitochondrial RNA polymerase (POLRMT) (13–15). The basal machinery is activated by mitochondrial transcription factor A (TFAM), which also participates in mitochondrial DNA (mtDNA) packaging and replication (16–21). Recently, leucine-rich protein 130 (LRP130; official symbol, LRPPRC [leucine-rich PPR motif–containing]), a protein implicated in Leigh syndrome (22, 23), was found to stimulate transcription of the core mitochondrial machinery and induce mitochondrially encoded transcripts (12), including 13 polypeptides that encode core subunits of the electron transport chain (24, 25). Independent of mitochondrial biogenesis, LRP130 induced mitochondrially encoded gene expression, culminating in increased OXPHOS efficiency (i.e., greater OXPHOS per mitochondrion). Additionally, cells and liver replete with LRP130 had greater β-oxidation of fatty acid (12), which implies that increased OXPHOS facilitates greater substrate oxidation. If the transcription machinery of mitochondria is activated by nutrient deprivation to enhance energy metabolism, this might explain how fasted liver increases its substrate oxidation, a process critical for ATP-dependent pathways, such as gluconeogenesis and ureagenesis, as well as for ketogenesis.
Here, we investigated coordination between nutrient deprivation and OXPHOS activity, considering mitochondrial biogenesis and/or OXPHOS efficiency as potential mechanisms. We found that normal fasted liver used mitochondria with greater OXPHOS efficiency, rather than increasing mitochondrial mass. Greater OXPHOS efficiency was achieved by increasing mitochondrial transcription, regulatory control that was dependent on SIRT3 and LRP130. In fasted liver, SIRT3 deacetylated and activated LRP130, which stimulated mitochondrial transcription to promote OXPHOS. This process was triggered by glucagon/cAMP signaling. We propose that the transcription machinery of mitochondria senses nutrients via SIRT3, permitting augmentation of OXPHOS.
Results
In liver, fasting coordinately induces mitochondrially encoded transcripts and OXPHOS capacity.
Fasted liver is notable for ATP-consuming processes, and is thus dependent on OXPHOS. Given that induction of mitochondrially encoded transcripts stimulates OXPHOS, we wondered whether mitochondrially encoded transcripts and OXPHOS are coordinately induced in fasted liver of C57BL/6 mice. After fasting for 24 hours, mouse liver showed modestly increased mitochondrially encoded transcripts and OXPHOS capacity (Figure 1, A and C). Indicative of a normal fasted response, mRNA of Pepck and G6p, enzymes involved in gluconeogenesis, was induced, whereas that of Scd1 and Fasn, enzymes involved in lipogenesis, was attenuated (Figure 1B). Interestingly, the increase in complex IV activity that we observed (Figure 1C) paralleled that reported by another group; however, alterations in mitochondrial content were not explored in the previous report (26).
Figure 1. Fasting coordinately induces mitochondrially encoded transcripts and OXPHOS in liver.

(A) Hepatic gene expression of mitochondrially encoded transcripts in 24-hour fasted or fed C57BL/6 mice (n = 3). (B) Expression of genes that regulate mitochondrial biogenesis, mitochondrial transcription, lipogenesis, and gluconeogenesis (n = 3). (C) Complex activity of mitochondria isolated from liver of 24-hour fasted or fed C57BL/6 mice (n = 3). CI–CV, complexes I–V; CS, citrate synthase. (D) Biochemical assessment of mitochondrial content using citrate synthase activity in whole liver homogenate (n = 3). (E) Genetic assessment of mitochondrial content using mtDNA content (n = 3). (F) Assessment of mitochondrial content by immunoblotting citrate synthase protein in whole liver homogenate (n = 3–4). Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, 2-way ANOVA, with (B and C) or without (A) Bonferroni post-test, or 2-tailed unpaired Student’s t test (D and E).
Conceivably, changes in mitochondrially encoded transcripts and OXPHOS might be explained by increased mitochondrial content. We therefore quantified mitochondrial biogenesis using complementary genetic, biochemical, and protein assessments. While mRNA for Pgc1a, a potent regulator of mitochondrial biogenesis, was induced, there was no induction of Tfam, Polrmt, and Tfb2m, which comprise the basal transcription machinery of mitochondria, or of Lrp130, which stimulates the core machinery (Figure 1B). Because genes comprising the basal transcription machinery are induced during mitochondrial biogenesis, their lack of induction were indicative of stable mitochondrial content. mtDNA content was unchanged (Figure 1D), in support of unaltered mitochondrial mass. Because the activity of citrate synthase proved indistinguishable in isolated mitochondria (Figure 1E), its activity in whole-liver homogenate permitted biochemical assessment of mitochondrial content. In agreement with genetic data, the activity and protein level for citrate synthase were unchanged (Figure 1, E and F), indicative of stable mitochondrial content regardless of feeding status. Hence, induction of Pgc1a was not associated with altered mitochondrial content in fasted liver. As previously reported in other systems (27, 28), these data imply that a certain threshold effect of PGC-1α coactivator activity is necessary to induce mitochondrial biogenesis.
Next, we sought to understand which nutrient signaling pathway influences mitochondrially encoded gene expression. Broadly, the fasting response is notable for a decline in insulin and an increase in glucagon, the latter of which activates intracellular cAMP signaling. Using cultured primary hepatocytes, we evaluated these possibilities. Withdrawal of insulin had no effect on the fasting-responsive genes Pepck and G6p or on mitochondrially encoded genes (Figure 2, A and C). As reported by others, glucagon induced both Pepck and G6p (Figure 2C). Interestingly, mitochondrially encoded genes were also induced, while many nuclear encoded ETC genes were unaffected (Figure 2, A and B). Forskolin, which increases intracellular cAMP levels, induced mitochondrially encoded genes as well (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI69413DS1), which indicates that the cAMP pathway governed by glucagon is likely responsible. Even in Pgc1a null primary hepatocytes, glucagon was still sufficient to induce mitochondrially encoded genes, although the response was modestly blunted (Supplemental Figure 1, C and D). These data further support the observation that de novo biogenesis is not required. Induction of mitochondrially encoded genes was accompanied by an increase in mitochondrially encoded proteins (Figure 2, D–F). Even so, in both primary mouse hepatocytes and mouse liver, gene expression was largely unchanged for several nuclear encoded ETC subunits (Figure 2B and Figure 3A). Several nuclear encoded ETC proteins, however, were increased in fasting liver mitochondria (Figure 3, B and C), which indicates that they were modestly stabilized (or more efficiently translated) in the fasted state. Overall, these data are suggestive of a coordinated response that facilitates enhanced OXPHOS. Importantly, mitochondrially encoded proteins, which are central to the process, are regulated at the level of gene expression.
Figure 2. Glucagon induces mitochondrially encoded genes and proteins.

(A) Neither insulin/dexamethasone (Insulin+Dex) nor their withdrawal (Vehicle) had an effect on mitochondrially encoded gene expression; however, glucagon induced mitochondrially encoded gene expression. (B) Effect of glucagon on several nuclear encoded ETC subunits. (C) Effect of glucagon on fasting-responsive genes and regulators of mitochondrial content and function. (D) [35S]-methionine labeling of cytoplasmic proteins (left 2 lanes) and mitochondrially encoded translation products, which were evident after inhibition of cytoplasmic translation with cycloheximide (CHX) (middle 6 lanes). Consistent with mitochondrial translation products, chloramphenicol (CAP) blocked their translation (right 2 lanes). GCG, glucagon. (E) Coomassie brilliant blue staining of the [35S]-labeled gel, showing equal protein loading. (F) Quantification of mitochondrially encoded [35S]-labeled proteins by scintillation courting (n = 3). Data are mean ± SEM. **P < 0.01, ***P < 0.001, 2-way ANOVA, with (B and C) or without (A) Bonferroni post-test, or 2-tailed unpaired Student’s t test (F).
Figure 3. Glucagon-mediated induction of mitochondrially encoded genes requires SIRT3.

(A) Fasted mouse liver showed no change in gene expression for several nuclear encoded ETC genes (n = 3–4). (B) Representative immunoblot of several nuclear encoded ETC subunits. Freshly isolated mitochondria were alkaline extracted with carbonate buffer, permitting assessment of the membrane fraction (Pellet) or soluble fraction. (C) Quantification of the several nuclear encoded ETC subunits (n = 3–4). (D–H) In Sirt3 knockout primary hepatocytes, glucagon-mediated induction of fasting-responsive genes was unaltered (D); however, induction of mitochondrially encoded gene expression was completely abrogated (E; n = 3) and no longer accompanied by increased mitochondrially encoded translation products (F and H). (G) Coomassie brilliant blue staining of [35S]-labeled gel, showing equal protein loading. Similar results were obtained using nicotinamide (see Supplemental Figure 1, E and F). Data are mean ± SEM, except in C (mean ± SD). *P < 0.05, **P < 0.01, ***P < 0.001, 2-way ANOVA with Bonferroni post-test (A, D, and E) or 2-tailed unpaired Student’s t test (C and H).
Finally, we used a different mouse strain to test the generality of the response. Similar to C57BL/6 mice, mitochondrially encoded genes were increased in 129S mice (Supplemental Figure 2), although the induction of Pepck, G6p and other genes was less robust. Similar to C57BL/6 mice, induction of mitochondrially encoded genes in 129S mice was not attributable to alterations in mitochondrial content (Supplemental Figure 2).
These observations indicated that the fasting response is accompanied by increases in mitochondrially encoded transcripts and OXPHOS, independent of de novo mitochondrial biogenesis. Given that increased mitochondrially encoded gene expression enhances OXPHOS (12), these data might imply that induction of mitochondrially encoded genes in cooperation with nuclear encoded subunits increases the capacity for OXPHOS in fasted liver.
SIRT3 is necessary and sufficient for fasting-mediated induction of mitochondrially encoded transcripts and OXPHOS.
By sensing the level of cellular NAD+, sirtuins are important sensors of nutrient deprivation. To globally assess this, we treated primary hepatocytes with nicotinamide, which inhibits NAD+-dependent sirtuin activity, and evaluated the response to glucagon. Nicotinamide had no effect on the fasting-responsive genes Pepck and G6p (Supplemental Figure 1E). In contrast, nicotinamide abrogated induction of mitochondrially encoded genes (Supplemental Figure 1F), which suggests that a NAD+-dependent pathway is necessary for their induction. These data indicate that a NAD+-dependent pathway, possibly via a sirtuin, mediates induction of mitochondrially encoded genes in the fasted response. SIRT3 protein is a sensor of nutrient deprivation that is induced in fasted liver mitochondria (5, 29). Moreover, prior studies in cell lines indicate that it influences cellular respiration (7–9); thus, SIRT3 might coordinate changes in energy metabolism with the fasted response. For these reasons, SIRT3 protein was an attractive candidate in the control of mitochondrially encoded gene expression and concomitant OXPHOS in fasted liver. We therefore challenged Sirt3 KO primary hepatocytes with glucagon. Similar to nicotinamide-treated primary hepatocytes, glucagon stimulated several fasting-responsive genes in the Sirt3 KO primary hepatocytes, but failed to induce mitochondrially encoded gene expression or mitochondrially encoded translation products (Figure 3, D–H).
Given that SIRT3 is implicated in energy metabolism in cell lines and is induced in fasted liver, we evaluated the fasting response in Sirt3 KO mouse liver. Neither mitochondrially encoded transcripts nor OXPHOS were induced in fasted Sirt3 KO liver, despite a slight increase in mitochondrial content (Figure 4, A–F). Again, there was no change in nuclear encoded ETC subunits (Supplemental Figure 3). Additionally, we depleted SIRT3 in C57BL/6 mouse liver using shRNA targeting Sirt3 (Supplemental Figure 4A). Again, both mitochondrially encoded gene expression and induction of OXPHOS were impaired, independent of mitochondrial content (Supplemental Figure 4). Moreover, SIRT3 knockdown did not influence the general fasting response, as these mice proved indistinguishable from wild-type C57BL/6 mice (compare Figure 1B and Supplemental Figure 4D). These findings in 2 mouse models of SIRT3 deficiency argue that SIRT3 is required for induction of mitochondrially encoded transcripts and OXPHOS in fasted liver.
Figure 4. In liver, SIRT3 is necessary and sufficient for fasting-mediated induction of mitochondrial encoded transcripts and OXPHOS.

(A) Hepatic gene expression of mitochondrially encoded transcripts in 24-hour fasted Sirt3 KO mice (129S background) (n = 3). (B) Expression of genes that regulate mitochondrial biogenesis, mitochondrial transcription, lipogenesis, and gluconeogenesis in Sirt3 KO liver (n = 3). (C) Complex activity from liver of 24-hour fasted or fed Sirt3 KO mice (n = 3). (D) Genetic assessment of mitochondrial content using mtDNA content (n = 3). (E) Biochemical assessment of mitochondrial content using citrate synthase activity in whole liver homogenate (n = 3). (F) Assessment of mitochondrial content by immunoblotting citrate synthase protein in whole liver homogenate (n = 3). (G and H) Expression of (G) mitochondrially encoded transcripts and (H) genes that regulate mitochondrial biogenesis and mitochondrial transcription in H2.35 hepatoma cells stably expressing LacZ or SIRT3 (n = 3). (I) Immunoblot showing expression of ectopically expressed myc-tagged SIRT3 protein. (J) Using a Clark-type oxygen electrode, cellular respiration — basal, proton leak, and maximal respiration (FCCP) — was assessed in H2.35 hepatoma cells stably expressing LacZ or SIRT3. (n = 4). See Supplemental Figure 6 for representative oxygen consumption curves. (K) Fluorescent assessment of mitochondrial content using MitoTracker Green FM (n = 3). (L) Genetic assessment of mitochondrial content using mtDNA content (n = 3). Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, 2-way ANOVA, with (A and G) or without (B and H) Bonferroni post-test, or 2-tailed unpaired Student’s t test (C–E and J–L).
Next, we evaluated whether SIRT3 is sufficient to induce mitochondrially encoded gene expression. SIRT3 was ectopically expressed in mouse liver using adenovirus encoding Sirt3 (Supplemental Figure 5, A–E). To minimize hormonal influences and genetic variations due to feeding status, food was briefly removed for 3 hours to clear the gastrointestinal tract of residual food. Brief food deprivation did not alter mitochondrially encoded transcripts or SIRT3 protein (data not shown), the latter of which is induced only after a longer fast (5). Even in the absence of potent fasting cues, ectopic expression of SIRT3 modestly induced mitochondrially encoded gene expression. Induction of mitochondrially encoded genes was not associated with alterations in mitochondrial content (Supplemental Figure 5, D and E). This action proved cell autonomous, as mitochondrially encoded gene expression was also induced in H2.35 murine hepatoma cells stably expressing SIRT3 as well as in primary hepatocytes transduced for 36 hours with Sirt3 adenovirus (Figure 4, G–L, and Supplemental Figure 5, F and G). Unlike primary hepatocytes, H2.35 hepatoma cells stably transduced with SIRT3 required supplementation with 500 μM NAD+, a cofactor for SIRT3. Even though mechanisms involving NAD+ uptake by mitochondria remain controversial, extracellular NAD+ does increase the concentration of NAD+ within mitochondria (30, 31). To make fair comparisons, LacZ H2.35 control cells were treated likewise.
Next, we evaluated OXPHOS in H2.35 cells stably expressing SIRT3. In cells replete with SIRT3, maximal cellular respiration was modestly induced about 12%, while basal respiration and proton leak were unchanged (Figure 4J). Increased respiratory capacity was independent of de novo mitochondrial biogenesis (Figure 4, K and L). Using galactose, a fuel in which very little ATP is derived from glycolysis, maximal cellular respiration was increased about 37% (Supplemental Figure 6). These data imply that, after a bout of fasting, induction of SIRT3 is both necessary and sufficient for the induction of mitochondrially encoded transcripts and OXPHOS.
SIRT3 activates mitochondrial transcription and promotes OXPHOS via LRP130.
Induction of mitochondrially encoded transcripts stimulates OXPHOS (12). Mitochondrially encoded gene expression is the summation of transcription and degradation. Presumably, SIRT3 influenced one or both of these pathways. Transcription and RNA degradation can be assessed by metabolic labeling, using pulse and pulse-chase experiments, respectively (Figure 5A). To assess mitochondrial transcription, we pulse-labeled cells with 4-thiouridine (4sU), a naturally occurring analog of UTP. After isolating RNA, 4sU-labeled transcripts were biotinylated, recovered on streptavidin beads, and quantified by quantitative PCR (qPCR). Because labeling is brief and much shorter than the half-life of most mitochondrially encoded transcripts, this method allows for quantitative assessment of transcription (32–34). Driven by a powerful CMV promoter, Sirt3 mRNA served as a positive control for transcription (Figure 5B). Several housekeeping genes were unchanged (Figure 5B), indicative of specific transcription. Ectopic expression of SIRT3 induced mitochondrial transcription, but failed to influence transcript degradation (Figure 5, C and D). Akin to transcription in the nucleus, these data support the general observation that changes in gene expression are largely driven by transcription (33). Overall, these data indicate that SIRT3 induces mitochondrially encoded gene expression by stimulating mitochondrial transcription. Given that mitochondrial mass and mtDNA content were stable (Figure 4 and Supplemental Figure 5), induction by SIRT3 signified increased transcription per mitochondrial genome.
Figure 5. SIRT3 induces mitochondrially encoded genes by stimulating mitochondrial transcription.

(A) Metabolic labeling with 4sU. This method quantifies de novo transcripts, permitting assessment of mitochondrial transcription in whole cells. It can also be used to monitor transcript degradation. (B) Transcription of Sirt3, driven by the viral CMV promoter, was induced in cells replete with SIRT3, whereas transcription of several housekeeping genes was unchanged (n = 3). (C) Cells stably expressing SIRT3 exhibited increased mitochondrial transcription (n = 3). (D) Uridine was used to chase the 4sU label and assess transcript half-life. Half-lives of mitochondrial transcripts in H2.35 hepatoma cells stably expressing SIRT3 were unchanged. Data are mean ± SEM. ****P < 0.0001, 2-way ANOVA, with (B) or without (C) Bonferroni post-test.
Next, we explored the mechanism by which SIRT3 influences mitochondrial transcription and OXPHOS. Independent of de novo mitochondrial biogenesis, it was previously reported that LRP130 interacts with POLRMT, culminating in enhanced mitochondrial transcription and attendant OXPHOS (12). We therefore wondered whether SIRT3 activates mitochondrial transcription and OXPHOS via LRP130. To test this, we ectopically expressed SIRT3 in cells deficient for LRP130. Using a previously validated RNAi (12, 35, 36), LRP130 knockdown in H2.35 cells was greater than 85% at the mRNA and protein levels (Figure 6, A and B). As previously reported (12, 35, 36), mitochondrially encoded transcripts were globally attenuated in LRP130-deficient cells, whereas genes involved in mitochondrial biogenesis and mitochondrial transcription were unchanged (Figure 6, C and D). In cells deficient for LRP130, ectopically expressed SIRT3 failed to induce either mitochondrially encoded transcripts or OXPHOS (Figure 6, C and E). In contrast, OXPHOS was increased in control shRNA LacZ cells replete with SIRT3 protein (Supplemental Figure 6), an induction similar to that shown in Figure 4J. Effects on mitochondrial transcription and energy metabolism were not caused by altered mitochondrial content, as genetic and fluorescent markers of mitochondrial content were unperturbed (Figure 6, F and G). Finally, effects on mitochondrial transcription and energy metabolism were not caused by perturbation of TFB2M or POLRMT, which collectively comprise the basal transcription machinery. Specifically, cells deficient or replete with LRP130 protein did not alter POLRMT or TFB2M protein levels (Supplemental Figure 7, A and B).
Figure 6. SIRT3-mediated induction of mitochondrial transcription requires LRP130.

(A–D) Gene expression of Lrp130 and Sirt3 (A), representative immunoblot of LRP130 and SIRT3 (B), gene expression of mitochondrially encoded transcripts (C), and expression of genes that regulate mitochondrial biogenesis and mitochondrial transcription (D) in H2.35 hepatoma cells replete with LacZ or SIRT3, superimposed with control (shCtrl) or Lrp130 knockdown (shLRP130) (n = 3). (E–G) H2.35 hepatoma cells replete with SIRT3 or LacZ, but deficient for LRP130, were used. (E) SIRT3 was no longer sufficient to influence maximal respiration, using a Clark-type oxygen electrode (n = 6). However, control shRNA SIRT3 cells retained increased respiratory capacity (see Supplemental Figure 6). Assessment of mitochondrial content was unchanged, as assessed by (F) MitoTracker Green FM or (G) mtDNA content (n = 3). (H–K) LRP130 liver-specific knockout (LRP130 LKO) mice were used. Hepatic gene (H) and protein (I) expression (n = 4). (J) Effect of fasting on mitochondrially encoded genes in liver. (K) Effect of fasting on fasting-responsive genes in liver. Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, 2-way ANOVA, with (A, D, H, and K) or without (C and J) Bonferroni post-test, or 2-tailed unpaired Student’s t test (E–G).
Next, we fasted mice in which Lrp130 was ablated in liver. To achieve liver-specific Lrp130 in adult mice, animals harboring floxed Lrp130 alleles were injected via tail vein with adeno-associated virus containing a vector coding for liver-specific cre recombinase (AAV-Cre), yielding liver-specific LRP130 KO mice (referred to herein as LRP130 LKO mice; Figure 6, H and I). After a 24-hour fast, mitochondrially encoded genes were induced in wild-type mice, but not LRP130 LKO mice (Figure 6J), which indicates that LRP130 is required for induction of mitochondrially encoded genes in fasted liver. As previously reported, mitochondrially encoded gene expression was globally decreased in LRP130 LKO liver (Figure 6J). Details of the Lrp130flox/flox allele are shown in Supplemental Figure 7C. We additionally ectopically expressed SIRT3 in Lrp130 KO primary hepatocytes. The ability of SIRT3 to induce mitochondrially encoded genes was abrogated in Lrp130 KO hepatocytes (Supplemental Figure 8). Finally, we evaluated fatty acid in H2.35 cells stably expressing SIRT3 but deficient for LRP130, since fatty acid oxidation depends on OXPHOS. Antimycin A (which inhibits electron transfer and, hence, OXPHOS) served as a positive control. In the absence of LRP130, fatty acid oxidation was significantly impaired (Supplemental Figure 8), which suggests that LRP130 is required for fatty acid oxidation. In summary, SIRT3 activated mitochondrial transcription and increased OXPHOS. This action was dependent on LRP130, a regulator of mitochondrial transcription and OXPHOS.
SIRT3 deacetylates LRP130 in fasted liver and increases its transcriptional activity.
Given that nutrient deprivation induces mitochondrial SIRT3, we evaluated whether SIRT3 influences acetylation of LRP130 protein in fasted liver mitochondria. Compared with fed liver, LRP130 was deacetylated in fasted liver mitochondria, a state in which SIRT3 activity (and protein) was increased (Figure 7A). Supporting regulation by SIRT3, LRP130 was hyperacetylated in SIRT3-deficient mouse liver (Figure 7B). In accordance with cell-autonomous control, NAD+ promoted deacetylation of LRP130 in H2.35 cells replete with SIRT3 (Figure 7C). Buttressing these data, LRP130 coimmunoprecipitated with SIRT3 (Figure 7D), but failed to immunoprecipitate with SIRT5 (data not shown), another — albeit less potent — deacetylase localized to mitochondria. To assess a functional interaction, we evaluated whether SIRT3 deacetylates purified LRP130 protein. Since mitochondrial acetylases have not been characterized, we used CREB-binding protein (CBP) to acetylate purified LRP130 protein, an approach that is standard in the field (37, 38). In the presence of purified SIRT3 protein, acetylated LRP130 C-terminal protein was robustly deacetylated by SIRT3 (Figure 7E). Similar, but less robust, results were obtained using N-terminal fragments of LRP130 (data not shown). Collectively, these data argue that SIRT3 deacetylates LRP130 in fasted liver. Finally, the conclusion that LRP130 is a bona fide target of SIRT3 was buttressed by a review of proteomic analyses (39–41), in which SIRT3 was found to be deficient in either cells or mouse liver (Supplemental Figure 9).
Figure 7. During the fasted response, SIRT3 deacetylates LRP130 in liver mitochondria.

(A) Representative immunoblot and protein quantification showing reduced acetylation of LRP130 (Ac-LRP130) in mitochondria isolated from fasted mouse liver (n = 3 experiments). (B) Immunoblot and protein quantification showing hyperacetylation of LRP130 in mitochondria isolated from liver deficient for SIRT3 (n = 4). (C) Immunoblot showing deacetylation of LRP130 in H2.35 cells stably expressing SIRT3 upon treatment with 500 μM NAD+. (D) SIRT3 and LRP130 coimmunoprecipitated, using ectopically expressed SIRT3-FLAG protein and endogenous LRP130. (E) Using purified proteins, SIRT3 robustly deacetylated the C terminus of LRP130. Deacetylation of LRP130 was inhibited by 12.5 mM nicotinamide (NAM). Shown is 1 representative of 4 independent experiments. Similar, but less robust, deacetylation was obtained for the N terminus of LRP130 (not shown). (F) Acetylated LRP130 fragments were incubated with control buffer or purified SIRT3 protein, then subjected to mass spectrometry. Lysines showing greater than 50% deacetylation by SIRT3 are graphed (gray and black bars). Percent deacetylation was calculated as 1 – (SIRT3 signal/control signal). 7 lysines were mutated to arginines (gray bars), generating LRP130-7KR, which mimics deacetylated protein. Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, 2-tailed unpaired Student’s t test (A and B).
Next, we used mass spectrometry to map 17 lysine residues of LRP130 sensitive to deacetylation by SIRT3 (Figure 7F and Supplemental Table 1). Sites with greater than 2-fold change were deemed positive. The mass spectrometry signal from the control reaction containing acetylated LRP130 protein was divided into the signal from the SIRT3 reaction. This ratio was subtracted from 1, yielding the arbitrary metric of percent deacetylation.
To evaluate the genetic and functional implications of LRP130 protein acetylation, we mutated 7 of 17 acetylated lysines to arginine (referred to herein as LRP130-7KR; Figure 7F and Supplemental Table 1). By mutating a lysine to an arginine, a mutated site retains its positive charge and functionally mimics deacetylated protein. Similarly, we generated a 7KQ mutant of LRP130 (LRP130-7KQ). Mutating a lysine to glutamine retains a neutral charge at the site and often mimics acetylated protein.
2 groups previously reported that LRP130 stimulates mitochondrial transcription (12, 42). In cell lysates, LRP130 interacted with both TFB2M and POLRMT (12), both of which comprise the basal transcription machinery of mitochondria (15). We therefore reasoned that the acetylation status of LRP130 might influence its interaction with TFB2M and/or POLRMT. Because POLRMT and LRP130 migrate at similar molecular weights, we refined the level of interaction to aas 340–710 of POLRMT (Supplemental Figure 10). This enabled us to use a 1–710 aa fragment of POLRMT, which is easily distinguished from LRP130. We evaluated whether LRP130-7KR (which mimics deacetylated protein) differentially interacts with either TFB2M or the 1–710 aa fragment of POLRMT. Compared with murine LRP130-WT, LRP130-7KR preferentially interacted with POLRMT, whereas differential binding of TFB2M was less apparent (Figure 8, A and B).
Figure 8. LRP130-7KR stimulates mitochondrial transcription and promotes OXPHOS.

(A) LRP130-7KR, which mimics deacetylated protein, showed increased affinity for aas 1–710 of POLRMT, a fragment that contains the catalytic domain. See Supplemental Figure 10 for more detailed mapping. (B) In contrast, compared with LRP130-WT, LRP130-7KR showed no differential affinity for TFB2M. (C, D, and F) Using 293T cells, endogenous human LRP130 was knocked down >95%, then reconstituted with murine LRP130-WT or LRP130-7KR (see Supplemental Figure 11). The shRNA targeting human LRP130 does not target mouse Lrp130 (not shown). Endogenous NAD+-dependent sirtuin activity was inhibited with 10 mM nicotinamide for 16 hours. LRP130-7KR had greater (C) mitochondrially encoded gene expression (n = 3), (D) mitochondrial transcription (n = 3), and (F) maximal respiration (n = 5) versus LRP130-WT. (E) Immunoblot showing similar levels of ectopically expressed LRP130-WT and LRP130-7KR. See Supplemental Figure 11 for total LRP130 protein. Data are mean ± SEM. *P < 0.05, ***P < 0.001, 2-way ANOVA (C and D) or 2-tailed unpaired Student’s t test (F).
Next, we evaluated whether LRP130-7KR and LRP130-7KQ exhibited differential activity. We transiently expressed LRP130-WT or the murine LRP130 mutants in 293T cells deficient for endogenous human LRP130. We used this strategy to mitigate competition with endogenous LRP130 and to evaluate activity using physiologically relevant levels of LRP130 protein. Although human and mouse LRP130 share more than 75% homology at the protein level, the shRNA targeting human LRP130 mRNA does not target mouse Lrp130 mRNA. Knockdown of human LRP130 in 293T cells and reconstitution with murine LRP130 is shown in Supplemental Figures 11 and 12. Endogenous SIRT3 activity was inhibited using 10 mM nicotinamide, a dose previously shown to impair NAD+-dependent sirtuin activity in 293T cells. Compared with LRP130-WT, LRP130-7KR exhibited greater activity, as assessed by mitochondrial gene expression, mitochondrial transcription, and oxygen consumption (Figure 8, C, D, and F). The level of ectopically expressed LRP130 protein was comparable (Figure 8E). Interestingly, the effect on transcription was much greater than that on respiration, indicative of either a saturation effect or limitations of our experimental system. In contrast, LRP130-7KQ did not increase mitochondrially encoded gene expression (Supplemental Figure 12), which suggests that deacetylation of LRP130 (as reflected by LRP130-7KR) increases its activity. Since SIRT3-dependent pathways were inhibited by nicotinamide, in terms of mitochondrial transcription and OXPHOS, these data indicate that LRP130 is a crucial mediator of SIRT3 action. Taken together, our results imply that deacetylation of LRP130 by SIRT3 enhances the activity of LRP130, which in turn stimulates mitochondrial transcription and OXPHOS.
In summary, we have identified a mechanism whereby nutrient sensing by SIRT3 induces mitochondrial transcription and enhances OXPHOS via LRP130 (Figure 9). Moreover, our present findings indicate that SIRT3 coordinates activation of enzyme systems involved in fatty acid oxidation as well as OXPHOS. In fasted liver, our data support a model whereby activation of mitochondrial transcription increases OXPHOS capacity, and this enhanced bioenergetic efficiency would be predicted to support increased substrate oxidation.
Figure 9. In fasted liver, the transcription machinery of mitochondria sense nutrient deprivation via SIRT3, culminating in enhanced energy metabolism.

In liver, SIRT3 is induced by nutrient deprivation. Specifically, glucagon, which activates cAMP signaling, increases SIRT3 activity, perhaps by indirectly increasing mitochondrial NAD+ levels. LRP130 is then deacetylated and activated by SIRT3, which strengthens the LRP130-POLRMT interaction. This culminates in increased mitochondrial transcription and attendant OXPHOS. Presumably, processes dependent on OXPHOS — fatty acid oxidation, gluconeogenesis, ketogenesis, and ureagenesis — are augmented by increased respiratory capacity. To simplify the model, deacetylation of enzymes involved in fatty acid oxidation and ketogenesis, as well as SIRT3 action on individual OXPHOS subunits, are not illustrated.
Discussion
Here, we identified an unanticipated link between nutrient sensing and mitochondrial transcription that was critical for fasting-mediated induction of OXPHOS. In fasted liver, ATP synthesis is accompanied by a 20% increase in oxygen consumption (43, 44), enabling ATP-dependent processes such as gluconeogenesis and ureagenesis. While increases in nutrient flux and oxidation are essential, OXPHOS capacity is critical factor as well. We showed here that SIRT3 deacetylated LRP130 during the fasted response. Once deacetylated, LRP130 stimulated mitochondrial transcription to increase mitochondrially encoded gene expression and OPXHOS, independent of de novo mitochondrial biogenesis. In fasted liver, SIRT3 protein has previously been shown to plateau at 18–24 hours and activate enzyme systems involved in fatty acid oxidation, culminating in greater β-oxidation of fatty acids (5). At 24 hours, we observed SIRT3-mediated deacetylation of LRP130. We therefore propose that SIRT3 couples enhanced β-oxidation of fatty acids to increase OXPHOS capacity via LRP130. Presumably, this strategy permits metabolic matching between 2 enzyme systems that are linked by coenzyme cycling: shuttling of NAD+ and NADH between fatty oxidative pathways and OXPHOS.
Governed by SIRT3 and LRP130, nutrient control of OXPHOS via mitochondrial transcription is a simple, yet elegant, model. Regarding control of OXPHOS, prior studies on SIRT3 implicated modifications of various subunits of the electron transport chain or alterations in mitochondrial content. In SIRT3-deficient cell lines, respiration is impaired and there is hyperacetylation of several respiratory complexes, including complex I (7, 10), succinate dehydrogenase (9), and complexes III and V (10). The stabilization of several nuclear encoded subunits may be related to their acetylation status, which might explain the differential regulation of complex activity previously reported by others (7). One limitation to interpreting these studies is that mutant subunits that mimic acetylated or deacetylated protein were not evaluated. It is therefore difficult to discern the functional relevance of these modifications.
In liver, the salutary actions of SIRT3 on OXPHOS do not require an increase in mitochondrial content. This is in contrast to immortalized, brown fat–like HIB1B cells, in which ectopic expression of SIRT3 was associated with increased Pgc1a, mitochondrial biogenesis, and concomitant OXPHOS (45). Using mouse models and cell culture, we did not observe changes in mitochondrial mass due to SIRT3, which suggests that the effect of SIRT3 on mitochondrial mass is restricted to certain cellular contexts. Notably, our results indicate a prominent role for SIRT3 in mitochondrial transcription, which promotes OXPHOS. Ectopic expression of SIRT3 in cells deficient for LRP130 protein failed to induce OXPHOS or mitochondrially encoded genes. Similarly, in liver deficient for LRP130, fasting failed to induce mitochondrially encoded gene expression. These data suggest that mechanisms independent of LRP130 are either trivial or operate downstream of mitochondrial transcription. For instance, in LRP130-deficient cells, reduction of mitochondrially encoded genes is associated with reduced OXPHOS subunits (12, 35, 36, 46), attenuation that might offset deacetylation of individual OXPHOS subunits. If normal mitochondrially encoded gene expression is a prerequisite for SIRT3-mediated activation of individual OXPHOS subunits, then ablation of LRP130 would abrogate any downstream effect. Additionally, LRP130 might influence factors involved in mitochondrial transcription. In our studies, neither POLRMT nor TFB2M, which comprise the initiation complex, were unchanged. Even so, we cannot exclude the possibility that other factors involved in transcription are not secondarily perturbed. Nevertheless, LRP130-deficient cells still retained some OXPHOS complexes, albeit less than those of wild-type cells. If activation of individual OXPHOS subunits were a predominant action of SIRT3, one would anticipate some enhancement of OXPHOS even in the absence of LRP130, a finding we failed to observe. Additionally, SIRT3 regulates several hundred mitochondrial proteins. Indeed, in other cellular systems, variegated actions of SIRT3 are not readily intuitive (47); hence, without further investigation, our findings predominantly relate to the fasted response. Nonetheless, even when NAD+-dependent sirtuins were pharmacologically inhibited, LRP130-7KR had greater activity in terms of transcription and OXPHOS capacity. This indicates that isolated deacetylation of LRP130 is sufficient to stimulate mitochondrial transcription and OXPHOS. Although we cannot exclude direct effects of acetylation on OXPHOS complexes per se, our data highlight a significant pathway involving mitochondrial transcription and attendant OXPHOS mediated by SIRT3 and LRP130.
Our results indicate that SIRT3 influences mitochondrial transcription via LRP130. In concert with prior reports by us and others (12, 42), these data further support a role for LRP130 in the control of mitochondrial transcription. Some studies have reported that LRP130 stabilizes mitochondrial transcripts; however, such a role for LRP130 in SIRT3-mediated control of mitochondrial gene expression was not supported here. As stated earlier, the LRP130-7KR mutant increased mitochondrially encoded transcripts, even when endogenous SIRT3 was inhibited. This suggests that deacetylation of basal mitochondrial transcription is not strictly required. Thus far, no differential acetylation has been reported for POLRMT or TFB2M, which collectively constitute the initiation complex. While our results suggest that deacetylation of LRP130 is sufficient to stimulate mitochondrial transcription and attendant OXPHOS, we cannot definitely exclude parallel deacetylation and activation of the basal transcription machinery. TFAM, a regulator of both mitochondrial transcription and replication, is acetylated in cells and mouse liver (40, 48), which is dependent on SIRT3 (40). After caloric restriction, there is a slight decrease in acetylation of TFAM; however, the functional and biological significance of this slight change is unknown, as an acetylation mutant was not evaluated (40). Although interesting and important, evaluating the basal transcription machinery in the context of biology will prove challenging, since ablation of its components completely interdicts OXPHOS, while in our study, ectopic expression of either TFB2M or POLRMT paradoxically attenuated mitochondrially encoded gene expression. This implies a finely tuned balance, limiting the tools and approaches available to decipher mechanisms. In terms of mitochondrially encoded gene expression, ablation of LRP130 still permitted some basal transcription, while its ectopic expression induced transcription. We therefore believe LRP130 is uniquely poised to specifically evaluate mitochondrial transcription and OXPHOS.
What are the broader questions pertinent to integrative physiology and disease? An increasing number of reports suggests that a mismatch between nutrient flux and oxidative capacity promotes and/or exacerbates insulin resistance (1–3). In human and mouse studies, increased fatty acid flux in the setting of impaired tricarboxylic acid and/or OXPHOS activity is associated with insulin resistance (1, 3, 49, 50). SIRT3 activity is impaired in mice fed a high-fat diet for 16–24 weeks that develop insulin resistance (51). Additionally, ablation of SIRT3 leads to metabolic syndrome (10, 11), suggestive of a causal role. Although the notion is speculative, a decline in SIRT3 activity might lead to impaired OXPHOS in liver and exacerbate insulin resistance. One additional question to consider is whether pathways involved in OXPHOS and fatty acid oxidation exhibit differential sensitivity to SIRT3 activity, which might produce a mismatch between fatty acid oxidation and OXPHOS.
In conclusion, induction of SIRT3 by fasting induced OXPHOS by increasing mitochondrial gene expression. Upon deacetylation by SIRT3, LRP130 induced mitochondrial transcription, culminating in greater OXPHOS capacity. We hypothesize that greater OXPHOS capacity improves mitochondrial bioenergetic efficiency, thereby supporting parallel and downstream biochemical pathways.
Methods
Cell culture and animal experiments
Stable transduction of H2.35 cells with SIRT3 or LacZ was achieved using pQCXIP vectors (Clontech), as previously described (12). H2.35 cells stably expressing SIRT3 or LacZ were treated with 500 μM NAD+ 16 hours prior to harvest. Knockdown of murine Lrp130 was performed as previously described (35, 36). The shRNA sequence targeted against human LRP130 was 5′-ATGTGAGTCACTATAATGCT-3′. Primary hepatocytes were isolated from mice as previously described (52). Briefly, approximately 400,000 hepatocytes were seeded per well of a 6-well plate and infected with adenovirus encoding the gene of interest. Virus sufficient to produce a multiplicity of infection of 400 in AD-293 cells (Stratagene, catalog no. 240085) was used to infect 400,000 hepatocytes. After 16 hours, virus was removed and replaced with fresh medium. Hepatocytes were further incubated for 20 hours, after which cells were harvested. For transduction of liver, 8 × 109 live adenoviral particles were tail vein injected into C57BL/6 mice on day 0 in 100 μl PBS. On day 5, liver tissue was harvested. Mice were maintained in 12-hour light/12-hour dark cycle.
Lrp130flox/flox mice, which harbor loxP sites flanking exons 3 and 4 of Lrp130, were generated by the University of Massachusetts Medical School Transgenic Animal Modeling Core from KOMP ES cell clone CSD33081. Prior to experimentation, mice were backcrossed for at least 8 generations onto the C57BL/6 background. Lrp130flox/flox mice and wild-type controls aged 8–12 weeks were infected with AAV-Cre at 1.0 × 1011 GC/mouse in 100 μl PBS. The vector for liver-specific cre recombinase driven by the human thyroxine-binding globulin promoter was packaged in adeno-associated virus by the Viral Vector Core of the University of Massachusetts Medical School as previously described (53). 3 weeks after injection, mice were singly housed beginning at 10 am for exactly 24 hours in a fed or fasted state, after which they were euthanized for protein and genetic studies. Pgc1aflox/flox mice (The Jackson Laboratory, stock no. 009666) were injected with AAV-Cre as described above. 3 weeks after infection, primary hepatocytes were isolated for experimentation.
Assessment of mitochondrial mass using MitoTracker Green FM
Mitochondrial mass was evaluated by flow cytometric analysis of cells stained with MitoTracker Green FM (Invitrogen). Briefly, cells were stained with Mitotracker Green FM at 100 nM for 30 minutes at 37°C. Cells were rinsed with PBS, trypsinized, and centrifuged at 130 g for 4 minutes. Cell pellets were resuspended in PBS containing 1% FBS for FACS. To quantify mitochondrial mass, 20,000 live cell counts were gated. Since MitoTracker Green FM is excited at 490 nm and emits at 516 nm, the FITC channel (excitation at 494 nm, emission at 520 nm) was used to detect signal.
Quantification of transcription and RNA degradation in whole cells
4sU was used to quantify transcription and RNA degradation as previously described (32), with some modifications. For transcription assays, 1.77 × 106 H2.35 cells stably expressing LacZ or SIRT3 were plated in 10-cm dishes and allowed to attach overnight. Cells were treated with 500 μM NAD+ for 16 hours and labeled with 2 mM 4sU (Sigma-Aldrich, catalog no. T4509) for 15 minutes at 37°C. RNA was isolated from cells using TRIzol and Qiagen’s RNeasy mini kit. 50 μg RNA was labeled with EZ-link Biotin-HPDP for 1.5 hours at room temperature. Unbound Biotin-HPDP was removed by chloroform/isoamylalcohol (24:1) extraction at 16,000 g for 5 minutes. A 1:10 volume of 5 M NaCl and an equal volume of isopropanol were added. Precipitated RNA was pelleted at 16,100 g for 20 minutes at 4°C. The pellet was washed with 75% ethanol and again pelleted at 16,100 g for 5 minutes at 4°C. The pellet was resuspended in 100 μl TE buffer (10 mM Tris-Cl pH 8.0, 1 mM EDTA). RNA samples were denatured at 65°C for 10 minutes, then rapidly cooled on ice for 5 minutes. Subsequently, denatured biotinylated RNA was captured using Dynabeads M-270 streptavidin (Invitrogen) with rotation at room temperature for 15 minutes. Beads were washed 3 times with 55°C wash buffer (100 mM Tris pH 7.5, 10 mM EDTA, 1 M NaCl, 0.1% Tween-20), followed by 3 washes at room temperature. Biotin-labeled RNA was eluted with 100 mM DTT by rotating at room temperature for 10 minutes. De novo transcripts were cleaned up using Qiagen’s RNeasy mini kit. Recovered transcripts were quantified using Ribogreen (Invitrogen). Approximately 120 ng RNA was used for reverse transcription and subsequent qPCR (RT-qPCR). To assess specificity, a reaction containing no 4sU label was performed in parallel. In general, 4sU transcripts were enriched more than 60-fold.
For RNA degradation studies, on day 0 at 9 am, H2.35 cells stably expressing LacZ or SIRT3 were plated at a density of 2.8 × 105 cells/well of a 6-well plate. On day 1 at 9 am, cells were labeled with 0.1 mM 4sU, labeling that continued for 24 hours. On day 1 at 5 pm, labeling medium was supplemented with 500 μM NAD+. The 0-hour time point was defined as 9 am on day 2. At the 1- and 2-hour time points, cells were washed with PBS, after which medium containing 20 mM uridine (Sigma-Aldrich, catalog no. U3003) was used to chase the 4sU label. RNA harvesting and processing proceeded as described above.
Immunopurification of acetylated proteins and detection of acetylated LRP130 protein
Acetylated proteins were immunopurified as previously described (54). Briefly, crude mitochondria were extracted from approximately 400 mg of mouse liver, then frozen on liquid nitrogen. The frozen mitochondrial pellet, about 100 μl volume, was lysed in 500 μl FLAG lysis buffer (50 mM Tris-Cl pH 7.8, 137 mM NaCl, 10 mM NaF, 1 mM EDTA, 1% Triton X-100, 0.2% sarkosyl, 10% glycerol) supplemented with 10 μM TSA and 5 mM nicotinamide. The typical lysate yielded a protein concentration of about 5 mg/ml. 2–10 mg mitochondrial lysate was incubated with 30 μl anti–acetylated lysine antibody–conjugated beads (Immunechem, catalog no. ICP0388) at 4°C overnight. The beads were washed with FLAG lysis buffer 4 times, 1 minute per wash, at room temperature. Acetylated protein was eluted with 49 μl freshly prepared glycine (0.1 M, pH 2.5) for 5–10 minutes at room temperature. 1 elution usually proved sufficient, yielding the highest protein concentration. 1 μl saturated Tris, prepared by dissolving Tris base in ddH2O until saturated, was added to the eluate to achieve a pH of 7.0. To detect acetylated LRP130 protein, immunoenriched acetylated fractions were immunoblotted for LRP130 using anti-LRP130 rabbit polyclonal sera, as previously reported (12).
qPCR of mtDNA content and RT-qPCR
To isolate total DNA, approximately 20 mg frozen liver was lysed in 200 μl tissue lysis buffer (50 mM Tris-Cl pH 7.5, 50 mM EDTA pH 8.0, 100 mM NaCl, 1% Triton X-100, 5 mM DTT, 100 mg/ml proteinase K) at 56°C overnight. The lysate was extracted once with phenol/chloroform, after which the aqueous phase was precipitated with 0.1 volumes of 3 M sodium acetate (pH 5.2) and 2.5 volumes of 100% ethanol. Total DNA was quantified using Quant-iT Picogreen dsDNA assay kit (Invitrogen). In a qPCR reaction volume of 15 μl, 400 pg total DNA was used to quantify mtDNA content. Showing strong agreement, 4 mtDNA genes — ND1, ND6, ATP6, and COX1 — were used to quantify mtDNA content. 18S and 28S rRNA served as nuclear reference genes (unless otherwise indicated). In addition, Tbp served as a reference gene (unless otherwise indicated); however, 10 ng total DNA in a reaction volume of 15 μl was used. For RT-qPCR, total RNA was isolated and processed as previously described (12). See Supplemental Tables 2 and 3 for primers.
Citrate synthase activity in whole liver homogenate
Citrate synthase assay was performed as previously described (55), with slight modifications. 30 mg frozen liver was placed in 300 μl RIPA buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 5 mM EDTA pH 8.0, 0.1% SDS, 1% sodium deoxycholate, 1% NP40) supplemented with PMSF and protease inhibitor cocktail (Sigma-Aldrich), followed by homogenization using a bead-mill homogenizer (Qiagen GmbH) at 25 Hz for 120 seconds. The liver homogenate was incubated on ice for 30 minutes, then centrifuged at 16,100 g for 10 minutes at 4°C. The protein concentration of the cleared homogenate was determined using a BCA kit (Thermo Scientific). 10 μg protein was loaded in citrate synthase assay buffer (100 mM Tris-Cl pH 8.0, 0.1 mM DTNB, 0.1 mM acetyl-CoA). The reaction was started by adding 0.25 mM freshly prepared oxaloacetate. A blank sample contained all reagents except liver homogenate. The increase in absorbance due to the formation of 5-thio-2-nitrobenzene anion at 412 nm was measured every 30 seconds for 15 minutes at room temperature. The activity of citrate synthase was expressed as the blank-corrected slope of the absorbance versus time plot within the linear range. Activity was presented as absorbance units per second per milligram protein (AU/s•mg protein), obtained by multiplying AU/s•10 μg protein by 1,000 μg/mg protein.
Cloning and construction of LRP130-7KR
Lysine-to-arginine (KR) mutations were made using a Quick Change II XL site-directed mutagenesis kit (Stratagene, catalog no. 200521). First, a 5KR mutant (K1036R, K1059R, K1250R, K1348R, and K1355R) was generated by serially mutating a C-terminal fragment of LRP130 (1,011–1,392 aa) contained in the pET30a vector. After PCR amplification, the mutated fragment was digested with BlpI/PacI and ligated into the BlpI/PacI site of a pQCXIP vector encoding full-length murine LRP130. Second, a 2KR mutant (K225R and K452R) was generated by serially mutating an N-terminal fragment of LRP130 (60–1,010 aa) contained in the pET30a vector. After PCR amplification, the fragment was digested with BspE1 and ligated into the BspE1 site of LRP130-5KR/pQCXIP, yielding LRP130-7KR (K225R, K452R, K1036R, K1059R, K1250R, K1348R, and K1355R). Confirmation of mutations was assessed using Sanger method sequencing.
Cloning and construction of LRP130-7KQ
Lysine-to-glutamine (KQ) mutations were made using a Quick Change II XL site-directed mutagenesis kit (Stratagene, catalog no. 200521). First, a 2KQ mutant (K1036Q and K1250Q) was generated by serially mutating a C-terminal fragment of LRP130 (1,011–1,392 aa) contained in the pET30a vector. After PCR amplification, the mutated fragment was digested with BlpI/PacI and ligated into the BlpI/PacI site of a pQCXIP vector encoding full-length murine LRP130. Second, we removed a segment of the above LRP130-2KQ/pQCXIP plasmid by digestion with SnaBI, yielding the 7.6-kb truncation product LRP130-2KQ/ΔpQCXIP. Then, 3 additional C-terminal KQ mutations (K1059Q, K1348Q, and K1355Q) were generated by serially mutating the LRP130-2KQ/ΔpQCXIP vector. This mutated fragment was digested with XhoI/PacI and reinserted into a similarly digested, full-length LRP130/pQCXIP vector, producing a 5KQ mutant of LRP130. A third 2KQ mutant (K225Q and K452Q) was generated by serially mutating an N-terminal fragment of LRP130 (60–1,010 aa) contained in the pET30a vector. After PCR amplification, the fragment was digested with BspE1 and ligated into the BspE1 site of LRP130-5KQ/pQCXIP, yielding LRP130-7KQ (K225Q, K452Q, K1036Q, K1059Q, K1250Q, K1348Q, and K1355Q). Confirmation of mutations was assessed using Sanger sequencing.
Purification of recombinant proteins
For purification of LRP130 fragments contained in pET30a, a single colony was cultured in 6 ml kanamycin LB media at 37°C overnight. 5 ml of overnight culture was transferred into 100 ml of LB media and shaken at 37°C for 3 hours until OD600 was about 0.4. The culture was then induced with 1 mM IPTG for 5 hours at 37°C. Bacteria were harvested by centrifugation at 1,500 g for 15 minutes, and the pellet was frozen at –80°C until further processing. After thawing, the pellet was resuspended in 10 ml lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 0.5 mM PMSF, pH 8.0). 1 mg/ml lysozyme and 100 μl of 10% Triton X-100 was added into the lysis buffer and incubated at 37°C for 10 minutes. Bacterial DNA was sheared by sonicating (Sonic Dismemberator 60, Fisher, maximum output) twice for 10 seconds. Cell debris was pelleted at 9,300 g for 20 minutes at 4°C. 100 μl Ni-NTA beads were added to the supernatant and incubated at 4°C overnight. The beads were washed 3–4 times with 1 ml wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0), 1 minute each, at room temperature. His-tagged LRP130 protein was eluted 2 hours at 4°C with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole). The buffer was exchanged to BC100 (100 mM NaCl, 20% glycerol, 20 mM Tris-Cl pH 7.9, 0.1% NP-40) using a NucAway spin column (Invitrogen).
For purification of CBP-GST fusion protein, E. coli Rosetta (DE3) cells were transformed with plasmid encoding CBP-HAT-GST (pGEX-4T1-CBP-HAT). Bacteria were cultured in 6 ml LB medium containing carbencillin (100 μg/ml). After overnight culture at 37°C with vigorous shaking, 1,000 ml fresh LB medium was inoculated, and bacteria were allowed to grow for 3–5 hours, until OD600 reached 0.4–0.6. Expression of GST-CBP fusion protein was induced with overnight at room temperature using 0.5 mM IPTG. Bacteria were harvested by centrifugation at 1,500 g for 15 minutes. The pellet was resuspended in 10–15 ml ice-cold PBS and an equal volume of BC100 buffer. Following sonication, Triton X-100 was added until reaching a final concentration of 1%. Lysates were clarified at 13,300 g for 30 minutes. The supernatant was transferred to a new tube. 100 μl glutathione agarose beads (Pierce, catalog no. 16110) was added to the supernatant and incubated at 4°C overnight. The beads were washed 3 times with 20 ml BC500 (500 mM NaCl, 20% glycerol, 20 mM Tris-Cl pH 7.9, 0.1% NP-40) and 2 times with 20 ml BC100. GST-tagged CBP protein was eluted for 2 hours at 4°C with 20 mM reduced glutathione (Thermo Scientific, catalog no. 78259).
In vitro acetylation and deacetylation assays
Acetylation reactions of 10 μl volume contained 50 mM HEPES (pH 8.0), 10% glycerol, 1 mM DTT, 1 mM PMSF, 10 mM sodium butyrate, 100 μM acetyl-CoA, 2 μg LRP130 purified protein, and 100 ng CBP-HAT domain protein (1,319–1,710 aa). The reaction mixture was incubated at 30°C for 1.5 hours. Subsequently, an equal volume of 2× LDS loading buffer was added, and the reactions were analyzed by immunoblotting overnight at 4°C with anti–acetylated lysine antibody (Cell Signaling, catalog no. 9441).
For in vitro deacetylation assay, acetylated LRP130 protein was recovered by binding to Ni-NTA beads (Qiagen, catalog no. 30410) and washed 3–4 times with 1 ml wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0), 1 minute per wash, at room temperature. Acetylated LRP130 protein was eluted with 50 μl of 250 mM imidazole at 4°C for 2 hours. The elution buffer was changed to BC100 using a NucAway spin column (Invitrogen). Deacetylation was performed in a 20-μl reaction containing approximately 1 μg acetylated LRP130 protein, 2 μl purified SIRT3 (Cyclex kit, catalog no. CY-1153), and 500 μM NAD+. The reactions were incubated at 30°C for 1.5 hours in a buffer containing 50 mM Tris-HCl (pH 9.0), 50 mM NaCl, 4 mM MgCl2, 0.5 mM DTT, 0.2 mM PMSF, 0.02% NP-40, and 5% glycerol. Subsequently, the reaction was stopped by adding 4× LDS loading buffer, and the reactions were analyzed by immunoblotting overnight at 4°C using anti–acetylated lysine antibody (Cell Signaling, catalog no. 9441).
Identification of peptides using tandem mass spectrometry
Mass spectrometry studies were performed at the Proteomics and Mass Spectrometry Facility of University of Massachusetts Medical School, as previously reported (56). In brief, silver stained gel bands were rinsed in water and briefly incubated with ammonium bicarbonate (100 mM). Gel bands were next destained with a 1:1 ratio of potassium ferricyanide (30 mM) and sodium thiosulfate (100 mM), and transferred to new tubes containing 1 ml water for 1 hour. After removal of the water, gel slices were subjected to addition of 50 μl of 250 mM ammonium bicarbonate, reduced with 5 μl of 45 mM DTT for 30 minutes at 50°C, and then alkylated with 5 μl of 100 mM iodoacetamide for 30 minutes at room temperature. The slices were then washed twice with 1 ml water, incubated in a 1:1 solution of 50 mM ammonium bicarbonate/acetonitrile at room temperature for 1 hour, soaked in 200 μl acetonitrile, removed, and dried in a SpeedVac. Samples were then digested with 50 μl of 2 ng/μl trypsin (Sigma-Aldrich) in 0.01% ProteaseMAX Surfactant (Promega) in 50 mM ammonium bicarbonate at 37°C for 21 hours. The supernatant of each sample was then removed and placed in a separate 0.5-ml tube, and each slice was further extracted with 100 μl acetonitrile/1% (v/v) formic acid (4:1), combined with respective supernatants of each sample, taken to dryness in a SpeedVac, and reconstituted in 20 μl of 0.1% TFA. LC-MS/MS analysis was done on a LTQ Orbitrap Velos mass spectrometer (Thermo Scientific) as previously reported (56), with the exception of using a 90-minute gradient for peptide elution. Raw data files were processed with either Mascot Distiller (Matrix Science) or Extract_MSN (Thermo Scientific), searched against the SwissProt database using the Mascot Search engine (Matrix Science). In brief, parent mass tolerances were set to 10 ppm, while fragment mass tolerances were set to 0.5 Da. Full tryptic specificity with 2 missed cleavages was used, and variable modifications of acetylation (protein N-term, lysine), pyro-glutamination (N-term glutamine), oxidation (methionine), and carbamidomethylation (cysteine) were considered. Further peptide annotation was achieved using Scaffold (Proteome Software), while relative quantification of acetylated peptides was performed using ProteoIQ (Nusep Inc.). Spectra of acetylated peptides were manually inspected to further validate assignments.
Metabolic labeling of mitochondrial proteins
Mouse primary hepatocytes were isolated as previously described (36). Cells were seeded in 6-well plates at 4.5 × 105 cells/well. After 16 hours, cells were treated with vehicle or 20 nM glucagon (Bachem) for 24 hours in starvation media (DMEM, 0.2% BSA, 2 mM sodium pyruvate). Cells were pretreated with 100 μg/ml cycloheximide (Sigma-Aldrich) and/or 100 μg/ml chloramphenicol (Sigma-Aldrich) as indicated for 10 minutes in methionine and cysteine-free starvation media containing vehicle or glucagon. Cells were then labeled with [35S]-methionine and [35S]-cysteine (50 μCi/ml, PerkinElmer) for 60 minutes and chased for 10 minutes in regular DMEM. Cells were washed with PBS twice and lysed in RIPA buffer supplemented with protease inhibitor cocktail (Sigma-Aldrich), followed by 3 freeze-thaw cycles. Lysates were cleared by centrifugation at 16,100 g for 10 minutes at 4°C. Protein concentration was determined by BCA assay (Thermo Scientific). 80 μg of protein (20 μg for total translation) were separated on a 15% polyacrylamide gel at 50 mA. The gel was fixed and stained using colloidal blue staining kit (Invitrogen) to confirm equal protein loading. The gel was then dried at 70°C for 3 hours and exposed to a Biomax MR film (Carestream Health). Radioactivity in 5 μl of lysates was quantified using a liquid scintillation counter (Beckman Coulter) and normalized to protein concentration.
Palmitate oxidation assay
Palmitate oxidation was performed as previously described (12). Briefly, H2.35 mouse hepatoma cells were incubated in starvation medium (1% BSA, 25 mM HEPES, 1 mM sodium pyruvate in DMEM) in the presence of 0.5 mM NAD+ for 16 hours. Cells were then incubated for 2 hours in preincubation medium (1% BSA, 25 mM HEPES, 0.25 mM sodium palmitate, 0.5 mM carnitine in DMEM), and subsequently, 0.5 μCi/well of 14C-palmitate was added for 90 minutes. The plate was frozen in liquid nitrogen, and 250 μl perchloric acid was added to each well. Filter paper moistened with 3N NaOH was immediately placed over each well and released CO2.
Comparison of acetylated LRP130 sites across studies
To estimate the probability that, by chance, 2 studies (Study A and Study B) would identify a given number of acetylated residues in common, we used the hypergeometric distribution. If, of a total N acetylated LRP130 peptides, Study A identified a peptides and Study B identified b peptides, the probability that the sets of peptides identified by the 2 studies would have exactly c peptides in common, 0 ≤ c ≤ min{a,b}, is as follows.
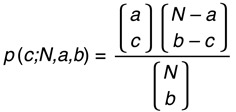 (Equation 1)
(Equation 1)
Derivation.
Conditioning on the peptides identified in Study A induces a separation of the peptides in Study B into 2 groups: the a peptides identified in Study A and the N – a peptides not identified in Study A. If the 2 studies identify exactly c peptides in common, Study B must identify c peptides among the a peptides and its remaining b – c peptides among the N – a peptides. The number of ways in which this can occur is the product of the binomial coefficients in the numerator. The total number of ways in which Study B can identify b peptides among the N is equal to the binomial coefficient in the denominator. This conditional probability applies to each of the possible sets of a peptides that Study A could identify. Because those sets are equally likely, the conditional probability is also the unconditional probability.
Assumptions.
The application of the probabilities assumes there are 102 potentially acetylatable lysine residues in LRP130 and that, if acetylated, each residue would have equal probability of detection by mass spectrometry. It also assumes that mass spectrometry of LRP130 protein approaches 100% for each study. Because these conditions may not hold for every study, the probabilities are approximations.
Domain mapping and in vitro binding assay
Truncated mouse POLRMT clones expressing aas 1–340, 1–500, 1–650, and 1–710 were each generated by PCR. The products of these reactions were digested with BamHI and XbaI, then ligated in frame into pcDNA3.1 myc His B plasmid. Clones containing full-length and truncated POLRMT were subjected to in vitro transcription/translation using a TnT kit (Promega, catalog no. L4610). Full-length mouse LRP130 fused with an S-tag was immobilized on S-protein agarose (Novagen, catalog no. 69704), then washed with 25 mM HEPES and 0.125% Tween20 containing protease inhibitors. The translated products were subsequently treated with DNaseI and RNase for 10 minutes at room temperature and centrifuged for 10 minutes at 15,700 g at 4°C. The supernatants were incubated with immobilized S-tag LRP130 agarose. After incubation for 2 hours at 4°C, complexes were extensively washed with 25 mM HEPES, 0.125% Tween-20, and 0.125 M KCl, and c-myc was detected by immunoblotting.
Cellular respiration, complex activity, and isolation of mitochondria
Cellular respiration (12, 35), isolation of mitochondria (12, 35, 36), complex activity (12), and fractionation of mitochondria by alkaline treatment (57) were performed as previously described.
Statistics
2-tailed Student’s t tests were used to analyze complex activity, mitochondrial content (using measures of mtDNA or fluorescence), protein quantification, and cellular respiration data. P values less than 0.05 were considered significant. 2-way ANOVA was used to assess differences in gene expression. Because mitochondrially encoded genes are transcribed as long polycistronic transcripts and thus tightly coordinated as a “gene set,” we evaluated the entire set using a 2-way ANOVA, reporting a P value comparing differences among various groups (or treatments). P values less than 0.05 were considered significant. In contrast, nuclear-encoded genes were subjected to post-hoc analyses (as indicated in the figure legends), since they are not polycistronic genes that do not necessarily fit into a specific gene set. For these studies, Bonferroni-corrected post-tests were implemented using a multiple comparison–adjusted α. All analyses were conducted using GraphPad Prism version 6.00.
Unedited blots
See complete unedited blots in the supplemental material.
Study approval
All animal experiments were approved by the IACUC of University of Massachusetts Medical School.
Supplementary Material
Acknowledgments
We thank J. Leszyk and S. Shaffer for mass spectrometry studies performed at the Proteomics and Mass Spectrometry Facility of University of Massachusetts Medical School; Silvia Corvera (University of Massachusetts Medical School) for critical review and comments; and Sharina Person (Department of Quantitative Health Sciences, University of Massachusetts Medical School) for assistance with statistical analysis. This work was supported by NIDDK grant 5R01DK089185 (to M.P. Cooper); the DERC Pilot and Feasibility Program at University of Massachusetts Medical School (to M.P. Cooper); and NHLBI grants HL092122 (to J.F. Keaney Jr.), P01 HL059407 (to G. Gao), and R21EB015684-01 (to G. Gao). M.P. Cooper is a member of the University of Massachusetts DERC (DK32520), and core resources supported by Diabetes Research Center grant DK32520 were also used.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(2):768–784. doi:10.1172/JCI69413.
References
- 1.Finck BN, et al. A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell Metab. 2005;1(2):133–144. doi: 10.1016/j.cmet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Koves TR, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7(1):45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Satapati S, et al. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res. 2012;53(6):1080–1092. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13(4):225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirschey MD, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464(7285):121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimazu T, et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010;12(6):654–661. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn BH, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105(38):14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49(2):304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finley LW, et al. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One. 2011;6(8):e23295. doi: 10.1371/journal.pone.0023295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jing E, et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A. 2011;108(35):14608–14613. doi: 10.1073/pnas.1111308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirschey MD, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44(1):177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, et al. LRP130 protein remodels mitochondria and stimulates fatty acid oxidation. J Biol Chem. 2011;286(48):41253–41264. doi: 10.1074/jbc.M111.276121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiranti V, et al. Identification of the gene encoding the human mitochondrial RNA polymerase (h-mtRPOL) by cyberscreening of the Expressed Sequence Tags database. Hum Mol Genet. 1997;6(4):615–625. doi: 10.1093/hmg/6.4.615. [DOI] [PubMed] [Google Scholar]
- 14.Masters BS, Stohl LL, Clayton DA. Yeast mitochondrial RNA polymerase is homologous to those encoded by bacteriophages T3 and T7. Cell. 1987;51(1):89–99. doi: 10.1016/0092-8674(87)90013-4. [DOI] [PubMed] [Google Scholar]
- 15.Shutt TE, Lodeiro MF, Cotney J, Cameron CE, Shadel GS. Core human mitochondrial transcription apparatus is a regulated two-component system in vitro. Proc Natl Acad Sci U S A. 2010;107(27):12133–12138. doi: 10.1073/pnas.0910581107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisher RP, Clayton DA. A transcription factor required for promoter recognition by human mitochondrial RNA polymerase. Accurate initiation at the heavy- and light-strand promoters dissected and reconstituted in vitro. J Biol Chem. 1985;260(20):11330–11338. [PubMed] [Google Scholar]
- 17.Fisher RP, Clayton DA. Purification and characterization of human mitochondrial transcription factor 1. Mol Cell Biol. 1988;8(8):3496–3509. doi: 10.1128/mcb.8.8.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher RP, Lisowsky T, Parisi MA, Clayton DA. DNA wrapping and bending by a mitochondrial high mobility group-like transcriptional activator protein. J Biol Chem. 1992;267(5):3358–3367. [PubMed] [Google Scholar]
- 19.Ekstrand MI, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13(9):935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- 20.Kanki T, et al. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol Cell Biol. 2004;24(22):9823–9834. doi: 10.1128/MCB.24.22.9823-9834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaufman BA, et al. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Cell Biol. 2007;18(9):3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morin C, et al. Clinical, metabolic, and genetic aspects of cytochrome C oxidase deficiency in Saguenay-Lac-Saint-Jean. Am J Hum Genet. 1993;53(2):488–496. [PMC free article] [PubMed] [Google Scholar]
- 23.Mootha VK, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A. 2003;100(2):605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 25.Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26(2 pt 2):167–180. doi: 10.1016/0092-8674(81)90300-7. [DOI] [PubMed] [Google Scholar]
- 26.Casas F, et al. New molecular aspects of regulation of mitochondrial activity by fenofibrate and fasting. FEBS Lett. 2000;482(1–2):71–74. doi: 10.1016/S0014-5793(00)02023-8. [DOI] [PubMed] [Google Scholar]
- 27.St-Pierre J, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 28.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 29.Caton PW, Holness MJ, Bishop-Bailey D, Sugden MC. PPARalpha-LXR as a novel metabolostatic signalling axis in skeletal muscle that acts to optimize substrate selection in response to nutrient status. Biochem J. 2011;437(3):521–530. doi: 10.1042/BJ20110702. [DOI] [PubMed] [Google Scholar]
- 30.Pittelli M, et al. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol Pharmacol. 2011;80(6):1136–1146. doi: 10.1124/mol.111.073916. [DOI] [PubMed] [Google Scholar]
- 31.Nikiforov A, Dolle C, Niere M, Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem. 2011;286(24):21767–21778. doi: 10.1074/jbc.M110.213298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolken L, et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 2008;14(9):1959–1972. doi: 10.1261/rna.1136108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rabani M, et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 2011;29(5):436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwanhausser B, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 35.Cooper MP, Uldry M, Kajimura S, Arany Z, Spiegelman BM. Modulation of PGC-1 coactivator pathways in brown fat differentiation through LRP130. J Biol Chem. 2008;283(46):31960–31967. doi: 10.1074/jbc.M805431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooper MP, et al. Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC-1α/LRP130 complex. Genes Dev. 2006;20(21):2996–3009. doi: 10.1101/gad.1483906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103(27):10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008;28(20):6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sol EM, et al. Proteomic investigations of lysine acetylation identify diverse substrates of mitochondrial deacetylase sirt3. PLoS One. 2012;7(12):e50545. doi: 10.1371/journal.pone.0050545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hebert AS, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. 2013;49(1):186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rardin MJ, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A. 2013;110(16):6601–6606. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sondheimer N, Fang JK, Polyak E, Falk MJ, Avadhani NG. Leucine-rich pentatricopeptide-repeat containing protein regulates mitochondrial transcription. Biochemistry. 2010;49(35):7467–7473. doi: 10.1021/bi1008479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsumura T, Thurman RG. Measuring rates of O2 uptake in periportal and pericentral regions of liver lobule: stop-flow experiments with perfused liver. Am J Physiol. 1983;244(6):G656–G659. doi: 10.1152/ajpgi.1983.244.6.G656. [DOI] [PubMed] [Google Scholar]
- 44.Olson MJ, Thurman RG. Quantitation of ketogenesis in periportal and pericentral regions of the liver lobule. Arch Biochem Biophys. 1987;253(1):26–37. doi: 10.1016/0003-9861(87)90633-3. [DOI] [PubMed] [Google Scholar]
- 45.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280(14):13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 46.Gohil VM, Nilsson R, Belcher-Timme CA, Luo B, Root DE, Mootha VK. Mitochondrial and nuclear genomic responses to loss of lrpprc expression. J Biol Chem. 2010;285(18):13742–13747. doi: 10.1074/jbc.M109.098400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Y, et al. NAD+-dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J Biol Chem. 2010;285(10):7417–7429. doi: 10.1074/jbc.M109.053421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dinardo MM, et al. Acetylation and level of mitochondrial transcription factor A in several organs of young and old rats. Biochem Biophys Res Commun. 2003;301(1):187–191. doi: 10.1016/S0006-291X(02)03008-5. [DOI] [PubMed] [Google Scholar]
- 49.Adams SH, et al. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr. 2009;139(6):1073–1081. doi: 10.3945/jn.108.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mihalik SJ, et al. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring). 2010;18(9):1695–1700. doi: 10.1038/oby.2009.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kendrick AA, et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433(3):505–514. doi: 10.1042/BJ20100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin J, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119(1):121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 53.Gao GP, Sena-Esteves M. Molecular Cloning. Introducing Genes into Mammalian Cells: Viral Vectors. . Green MR, Sambrook J, eds. Cold Spring Harbor, New York, USA: Cold Spring Harbor Laboratory Press; 2012. [DOI] [PubMed] [Google Scholar]
- 54.Fan W, Luo J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell. 2010;39(2):247–258. doi: 10.1016/j.molcel.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 55.Baur JA, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444(7117):337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paquette N, et al. Serine/threonine acetylation of TGFbeta-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc Natl Acad Sci U S A. 2012;109(31):12710–12715. doi: 10.1073/pnas.1008203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158(4):647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.