

Abstract
Delayed rectifier K+-channels (Kv1.3) are predominantly expressed in T lymphocytes. Based on patch-clamp studies, the channels play crucial roles in facilitating the calcium influx necessary to trigger lymphocyte activation and proliferation. Using selective channel inhibitors in experimental animal models, in vivo studies then revealed the clinically relevant relationship between the channel expression and the pathogenesis of autoimmune diseases. In renal diseases, in which “chronic inflammation” or “the overstimulation of cellular immunity” is responsible for the pathogenesis, the overexpression of Kv1.3-channels in lymphocytes promotes their cellular proliferation and thus contributes to the progression of tubulointerstitial fibrosis. We recently demonstrated that benidipine, a potent dihydropyridine calcium channel blocker, which also strongly and persistently inhibits the lymphocyte Kv1.3-channel currents, suppressed the proliferation of kidney lymphocytes and actually ameliorated the progression of renal fibrosis. Based on the recent in vitro evidence that revealed the pharmacological properties of the channels, the most recent studies have revealed novel therapeutic implications of targeting the lymphocyte Kv1.3-channels for the treatment of renal diseases.
1. Introduction
T lymphocytes predominantly express delayed rectifier K+-channels (Kv1.3) in their plasma membranes [1–3]. Using selective channel inhibitors, patch-clamp studies revealed that the channels generate the K+-diffusion potential across the plasma membranes and play crucial roles in facilitating calcium influx necessary to trigger the lymphocyte activation and proliferation [3–6]. Previous studies demonstrated the involvement of inflammatory leukocytes, such as T lymphocytes, macrophages, and mast cells, in the pathogenesis of renal diseases, such as glomerulonephritis, chronic kidney disease (CKD), or tubulointerstitial fibrosis [7–11]. Since lymphocytes are actually activated [12] and serum cytokine levels are known to be elevated in patients with advanced-stage renal diseases [13, 14], Kv1.3-channels expressed in lymphocytes would contribute to the progression of the diseases. Regarding the molecular mechanisms by which lymphocytes are activated, the rise in the intracellular calcium concentration stimulates the phosphatase calcineurin activity, which then dephosphorylates nuclear factor of activated T cells (NFAT), enabling it to accumulate in the nucleus and bind to the promoter of the gene encoding interleukin 2 (IL-2) [6, 15] (Figure 1). Therefore, pharmacological targeting of calcineurin has been the main mechanism by which drugs, such as cyclosporine and tacrolimus, exert their immunosuppressive effects [16]. However, recent studies have also revealed that selective inhibition of lymphocyte Kv1.3-channels also represses lymphocyte activity and thus suppresses cellular immunity [17]. Recent patch-clamp studies, including ours, have shown that commonly used drugs, such as calcium channel blockers (CCBs) [18, 19], macrolide antibiotics, and HMG-CoA reductase inhibitors, effectively suppress the Kv1.3-channel currents in lymphocytes [20, 21]. Such studies suggested the therapeutic efficacy of these drugs for the treatment of renal diseases, in which “chronic inflammation” or “the overstimulation of cellular immunity” is responsible for the pathogenesis [22]. By summarizing the previous and recent findings obtained from studies in the relevant fields, this review provides an overview of the pathological roles of lymphocyte Kv1.3-channels in renal diseases. Based on the recent in vitro and in vivo evidence that revealed the pharmacological properties of the channels, this review also focuses on the novel therapeutic implications of targeting the channels for the treatment of renal diseases.
Figure 1.

Kv1.3-channel-induced activation pathway of T lymphocytes. Kv1.3-channels expressed in T lymphocytes facilitate the calcium influx necessary to trigger the lymphocyte activation and proliferation. The rise in the intracellular calcium concentration stimulates the phosphatase calcineurin activity, which then dephosphorylates nuclear factor of activated T cells (NFAT), enabling it to accumulate in the nucleus and bind to the promoter of the gene encoding interleukin 2 (IL-2).
2. Increased Numbers of Leukocytes in Rat Kidneys with Renal Diseases
Previous studies have described several laboratory models of renal diseases, including ligation of the renal artery branches or unilateral ureter [23, 24], ablation of renal mass by surgery [25, 26], toxic nephritis [27, 28], and immunologically induced nephritis [29, 30]. In the development of glomerulonephritis, inflammatory leukocytes are initially recruited from the bone marrow and infiltrate into the renal interstitium to produce proinflammatory cytokines [9]. Therefore, the kidneys from rat models with toxic or immunologically induced nephritis were characterized by the massive infiltration of T-lymphocytes or macrophages [9, 27–30]. On the other hand, in rat models with 5/6 nephrectomy (subtotal nephrectomy), the injured kidneys were mainly characterized by severe glomerulosclerosis, which was primarily caused by the renal hemodynamic changes, such as the increased glomerular pressure and the protein overload [31, 32]. However, with the increase in the serum creatinine, the kidneys from these subtotally nephrectomized rats were additionally characterized by diffuse interstitial fibrosis with the involvement of leukocyte infiltration [7, 8, 33]. In rats with subtotal nephrectomy followed by longer recovery periods, serum creatinine and blood urea nitrogen levels were markedly elevated, indicating advanced chronic renal failure (CRF) [11, 34]. In CRF rat kidneys with 8-week recovery period, the cortical interstitium was expanded with fibroedema (Figure 2(a)(B) versus Figure 2(a)(A)) and there was some infiltration of small round cells among spindle-shaped fibroblasts (Figure 2(a)(E) versus Figure 2(a)(D)). At 14 weeks, in addition to diffuse fibrosis in the cortical and medullary interstitium (Figure 2(a)(C)), the numbers of small round cells were dramatically increased in the cortical interstitium (Figure 2(a)(F)). Since the cortical expression of CD3 and ED-1, surface markers for T lymphocytes and macrophages, was markedly elevated [11], they were regarded as inflammatory leukocytes, such as T lymphocytes and macrophages. By immunohistochemistry, the CD3- or ED-1-positive small round cells were actually costained with Ki-67, a marker of cellular proliferation (Figures 2(b)(A) and 2(b)(B)). The findings indicated that T lymphocytes and macrophages were proliferating prominently within the cortical interstitium of advanced CRF rat kidneys [34].
Figure 2.

Histological features of sham-operated (sham), CRF, and advanced CRF rat kidneys. (a) Hematoxylin and eosin (H&E) and Masson's trichrome staining, in sham-operated (sham), CRF (8 weeks after nephrectomy), and advanced CRF (14 weeks after nephrectomy) rat kidneys. ((A), (B), and (C)) Low-power views of cortex, Masson's trichrome. Magnification, ×20. ((D), (E), and (F)) High-power views of cortical interstitium, H&E. Magnification, ×60. (b) Immunofluorescence staining using antibodies for CD3 ((A) green) and ED-1 ((B) green), costained with Ki-67 (red) in advanced CRF rat kidneys. High-power views of the cortical interstitium. Magnification, ×60. Modified from [34].
Recently, Liu et al. demonstrated that CD4+ T-lymphocytes, especially Th2 cells, contributed to the progression of renal fibrosis in a rat model of unilateral ureteral obstruction (UUO) [35]. In the development of the tubulointerstitial fibrosis in CRF rat kidneys, previous studies demonstrated that the inflammatory leukocytes were initially recruited from the bone marrow and infiltrated into the renal interstitium to trigger the proliferation of fibroblasts [36]. In this context, using the rat model of advanced CRF, the recent studies further demonstrated that the infiltrated leukocytes proliferated in situ in the cortical interstitium, and thus dramatically increased their numbers [11, 34].
3. Physiological Roles of Kv1.3-Channels Expressed in T-Lymphocytes
A variety of ion channels that are expressed in T lymphocytes include voltage-dependent K+-channels (Kv), Ca2+-activated K+-channels (KCa), Ca2+ release-activated Ca2+-channels (CRAC), Mg2+-inhibited Ca2+-permeable current (MIC) channels, and swelling-activated Cl− channels (Clswell) [37]. Among them, human T lymphocytes predominantly express Kv1.3-channels in their plasma membranes [1, 2]. In patch-clamp studies using thymus-derived murine lymphocytes (thymocytes), stepwise changes in the membrane potential evoked membrane currents characteristic to Kv-channels [18, 20, 21, 38, 39]. Since margatoxin, a selective inhibitor of Kv1.3-channels, almost totally abolished the currents in lymphocytes (Figure 3(a)) [38], these membrane currents were identified as Kv1.3-channel currents. The physiological roles of Kv1.3-channels in lymphocytes have been identified by several in vitro studies. Patch-clamp studies revealed that these channels generate the K+-diffusion potential across the plasma membranes and thus play a role in regulating the resting membrane potential and controlling the cell volume [40, 41]. The opening of Kv1.3-channels also brings about the membrane hyperpolarization and generates a driving force for the Ca2+ influx [4, 5, 37]. Consequently, it stimulates the following Ca2+ signaling pathway necessary to trigger the lymphocyte activation and proliferation.
Figure 3.

Effects of margatoxin and benidipine on Kv1.3 channel currents in murine thymocytes. Typical whole-cell current traces at different voltage steps recorded before and after the application of 100 nM margatoxin (a) or 10 μM benidipine ((b)(A)). The currents were elicited by voltage-steps from the holding potential of −80 mV to −40, 0, 40, and 80 mV, as depicted in the voltage protocol. Each pulse was applied for a 200-ms duration between 10-second intervals. (b)(B) Peak current densities (peak currents normalized by the membrane capacitance) obtained from the records in (A) at the voltage-step of 80 mV. (b)(C) Pulse-end current densities (pulse-end currents normalized by the membrane capacitance) obtained from the records in (a) at the voltage-step of 80 mV. # P < 0.05 versus before the drug application. Values are means ± SEM (n = 5). Differences were analyzed by ANOVA followed by Dunnett's or Student's t-test. Modified from [18, 38].
4. Pathological Roles of Lymphocyte Kv1.3-Channels in Renal Diseases
Peripheral lymphocytes are activated and the serum cytokine levels are known to be elevated in patients with end-stage renal disease [12–14]. Based on a previous patch-clamp study [42], the conductance of voltage-dependent K+-channels in lymphocytes was increased in such patients and the activity of the channels was strongly associated with the severity of renal dysfunction. In experimental animal models of renal diseases, such as renal allograft rejection [10] and glomerulonephritis [9], immunosuppression by the blockade of lymphocyte Kv1.3-channels actually prevented or ameliorated the progression of the diseases. By using selective Kv1.3-channel inhibitors, such as ShK and Psora-4, therapeutically [9, 10], these studies demonstrated the contribution of the channels to the pathogenesis of renal diseases.
In previous studies, the overexpression of Kv1.3-channnels was demonstrated in cells under various pathologic conditions, such as cancer [43, 44], ischemic heart disease [45], and autoimmune disorders [46, 47]. In autoimmune disorders, including multiple sclerosis, rheumatoid arthritis, and systemic lupus erythematosus, the inhibition of the Kv1.3-channel modulated the calcium influx patterns in T lymphocytes and thus exerted immunosuppressive effects [48–50]. Recently, using a rat model with advanced CRF, we demonstrated for the first time that Kv1.3-channels were overexpressed in proliferating leukocytes within fibrotic kidneys [11]. In our study, margatoxin, one of the most potent inhibitors of Kv1.3-channels which almost totally inhibits the channel currents in lymphocytes (Figure 3(a)) [38], decreased the number of proliferating lymphocytes and ameliorated the progression of renal fibrosis. These findings indicated that the overexpression of Kv1.3-channels in kidney lymphocytes promoted their cellular proliferation in advanced CKD. As previously demonstrated in cancer cells [51], the membrane hyperpolarization induced by the overexpression of the channels is thought to trigger the cell cycle progression in lymphocytes [52, 53]. Since the cytokines produced by lymphocytes stimulate the activity of fibroblasts to produce collagen [36], the proliferating lymphocytes in the interstitium would promote the progression of renal fibrosis in advanced CRF, contributing to the rapid deterioration of renal function [11].
5. Suppressive Effects of Benidipine on Lymphocyte Kv1.3-Channels
In addition to their cardiovascular effects on hypertension and ischemic heart disease [54], CCBs are also known to exert immunosuppressive properties in humans [55, 56]. According to several in vitro studies, CCBs, including nifedipine, verapamil, and diltiazem, repress the migration of leukocytes and inhibit their proliferation [57, 58]. Recently, using human peripheral leukocytes, Matsumori et al. demonstrated that CCBs also suppress the production of proinflammatory cytokines, such as IL-1β, tumor necrosis factor α (TNF-α), and interferon γ (IFN-γ) [59, 60]. According to their studies, 1,4-dihydropyridine (DHP) CCBs, including nifedipine, amlodipine, and benidipine, which are highly lipophilic compared to the other types of CCBs [61, 62], exert relatively stronger immunomodulatory effects. Among them, benidipine is one of the most lipophilic and longest acting DHP CCBs [63, 64]. In our patch-clamp study using murine thymocytes [18], benidipine almost totally and irreversibly inhibited the Kv1.3-channel currents (Figure 3(b)), indicating its usefulness as a potent Kv1.3-channel inhibitor. Although the effects of benidipine on cytokine production have not yet been directly examined [59, 60], the marked inhibition of the channel currents by this drug strongly suggested its higher immunosuppressive potency than the other CCBs. Moreover, the persistent effect of benidipine in decreasing the channel currents may suggest its longer duration of action in thymocytes, as previously demonstrated in cardiomyocytes [64] and isolated coronary arteries [65].
6. Therapeutic Efficacy of Benidipine against the Progression of Tubulointerstitial Fibrosis in Advanced CRF
In our recent study, the overexpression of Kv1.3-channels in kidney lymphocytes was strongly associated with their in situ proliferation in advanced CRF rat kidneys [11], and the channel inhibition by margatoxin actually decreased the number of infiltrating leukocytes and slowed the progression of renal fibrosis. Based on one of our patch-clamp studies [18], since benidipine was also highly potent as a Kv1.3-channel inhibitor, the therapeutic effects of this drug on CKD were examined in our most recent study [34]. Previous studies have shown the therapeutic efficacy of antihypertensive drugs, such as CCBs [66] and angiotensin converting enzyme inhibitors (ACEIs) [67], for the prevention of glomerulosclerosis, since these drugs hemodynamically ameliorate glomerular hypertension. However, it is not well known how these pharmacological approaches slow the progression of tubulointerstitial fibrosis independently of their effects on glomerulosclerosis.
In advanced CRF rat kidneys with benidipine treatment, the size of the cortical interstitium, which included the areas of fibrosis, edema, and the inflammatory leukocyte infiltration, was smaller (Figure 4(a)(B) versus Figure 4(a)(A)), and the number of proliferating leukocytes was much reduced (Figure 4(b)(B) versus Figure 4(b)(A)) together with a significant decrease in the proinflammatory cytokine expression (Figure 4(c)). In these kidneys, Masson's trichrome staining and the immunohistochemistry for fibrosis markers, such as collagen III, demonstrated less staining in the cortical interstitium (Figures 5(a)(B) and 5(a)(D) versus Figures 5(a)(A) and 5(a)(C)). However, in the glomeruli, the amount of periodic acid Schiff positive material was a comparable with that in vehicle-treated CRF rat kidneys (Figure 5(b)(B) versus Figure 5(b)(A)). These results indicated that benidipine ameliorated the progression of renal fibrosis without affecting the deterioration of glomerulosclerosis. Benidipine, which blocks L- and T-types of calcium channels in the renal vasculature, is known to dilate both afferent and efferent arterioles of the glomeruli and to reduce glomerular hypertension [68]. In our recent study, however, benidipine did not decrease the severe proteinuria nor did it improve the systemic hypertension in advanced CRF rats [34]. Therefore, factors other than reducing the glomerular capillary pressure may also be involved in its pharmacological effects of ameliorating the renal injury [66]. Recently, benidipine was shown to reduce the circulating levels of inflammatory cytokines or proteins, such as IL-6 and high mobility group box-1 (HMGB-1), in patients with chronic kidney disease [69]. Since IL-6 and HMGB-1 accelerate the inflammation of systemic organs by promoting lymphocyte activation and proliferation [14, 70]; such effects of benidipine may have therapeutic potential for slowing the progression of renal fibrosis in advanced CRF.
Figure 4.

Histological features of vehicle- and benidipine-treated CRF rat kidneys and the expression of proinflammatory cytokines. (a) Hematoxylin and eosin staining (H&E) in vehicle-treated and benidipine-treated CRF rat kidneys. Low-power views of the cortex. Magnification, ×20. (b) Immunohistochemistry using antibody for Ki-67 (brown) in vehicle- and benidipine-treated CRF rat kidneys. High-power views of the cortical interstitium. Magnification, ×60. (c) The abundance of IL-2 mRNA (left) and interferon-γ (IFN-γ) (right) in the renal cortex of vehicle-treated and benidipine-treated CRF rat kidneys. * P < 0.05 versus vehicle-treated CRF rats. Values are means ± SEM (n = 5). Differences were analyzed by ANOVA followed by Dunnett's or Student's t-test. Modified from [34].
Figure 5.

Fibrotic marker expression and histological features of glomeruli in vehicle- and benidipine-treated CRF rat kidneys. (a) Masson's trichrome staining ((A) and (B)) and immunohistochemistry using antibody for collagen III ((C) and (D) brown) in vehicle-treated and benidipine-treated CRF rat kidneys. Low-power views of the cortex. Magnification, ×20. (b) Periodic acid Schiff (PAS) staining in vehicle-treated and benidipine-treated CRF rat kidneys. Low-power views of the cortex. Magnification, ×20. Modified from [34].
7. Novel Therapeutic Implications of Targeting Lymphocyte Kv1.3-Channels by Commonly Used Drugs
Grgic et al. demonstrated the therapeutic efficacy of blocking the intermediate-conductance Ca2+-activated K+-channels (KCa3.1) for renal fibrosis, since fibroblasts overexpressed the channels under a pathologic condition [24]. In a separate animal study, they also demonstrated the prophylactic efficacy of blocking lymphocyte Kv1.3-channels to prevent renal allograft rejection [10]. As an extension of these studies, our studies further suggested that targeting the Kv1.3-channels overexpressed in leukocytes would also be useful for the treatment of renal fibrosis in advanced CRF [11, 34]. In our series of patch-clamp studies, in addition to CCBs, such as benidipine and nifedipine [18], macrolide antibiotics and HMG-CoA reductase inhibitors (statins) also effectively suppressed lymphocyte Kv1.3-channel currents [20, 21] (Table 1). According to separate in vitro studies, these drugs exerted immunomodulatory properties besides their anti-inflammatory, antimicrobial, and anticholesterol effects [56, 71–75]. Since lymphocyte Kv1.3-channels trigger calcium influx, which is necessary for IL-2 synthesis [15] and since channel blockade by highly selective inhibitors, including margatoxin, ShK-Dap22, and PAP-1, actually repressed the immune response in lymphocytes [38, 76–78] (Table 2), the suppressive effects of NSAIDs, macrolide antibiotics, and statins on the channel currents were considered to contribute to their immunomodulatory properties. Additionally, as previously detected by the whole-cell patch-clamp technique [38], the amplitude of the peak channel currents was deeply associated with the “activation” of the channel currents [21]. Therefore, the stronger suppression of the peak currents by macrolide antibiotics or statins may indicate their higher immunosuppressive potency. In this regard, besides the use of the previously developed selective blockers for the channels [78–80], the use of CCBs, macrolide antibiotics, or statins could also potentially be useful as antifibrotic agents in patients with advanced CKD (Figure 6). The most likely side effects of the Kv1.3-channel inhibition include epileptic seizures or enteric twitches, since the channels are also expressed in the synapses of both central and enteric nervous systems [81, 82]. In normal rat kidneys, Kv1.3-channels are expressed in some proximal tubules [9] and predominantly in the basolateral membranes of the inner medullary collecting duct cells [83]. In these cells, the channels regulated the cellular or total body fluid volume by maintaining the driving force for Na+ reabsorption. Therefore, the channel inhibition may also affect such tubular functions and the urinary K+ secretion, which was actually demonstrated in previous studies using isolated collecting ducts [84]. However, compared to the highly selective inhibitors, which were originally derived from venom, scorpion, or sea anemone peptide toxins [49, 79, 85, 86], the drugs, such as CCBs, macrolide antibiotics, or statins, could be used more safely, since they have been employed in a common clinical practice for a longer period of time.
Table 1.
Summary of changes in peak and pulse-end currents after application of CCBs, macrolide, and HMG-CoA reductase inhibitors (statins).
| Drugs | N | Peak current density (pA/pF) | Pulse-end current/peak current (I/I peak) (%) | ||
|---|---|---|---|---|---|
| Before | After | Before | After | ||
| Nifedipine (100 μM) | 5 | 297 ± 3.3 | 196 ± 2.6# | 61.3 ± 1.8 | 26.2 ± 5.4# |
| Benidipine (10 μM) | 5 | 384 ± 47 | 120 ± 15# | 53.4 ± 4.6 | 1.14 ± 0.21# |
| Clarithromycin (100 μM) | 5 | 277 ± 4.4 | 89.6 ± 10# | 48.5 ± 1.4 | 15.8 ± 1.0# |
| Pravastatin (1 mM) | 5 | 309 ± 16 | 278 ± 17 | 52.1 ± 2.4 | 33.4 ± 4.2# |
| Lovastatin (10 μM) | 5 | 301 ± 12 | 237 ± 5.0# | 55.1 ± 0.4 | 26.8 ± 0.7# |
| Simvastatin (10 μM) | 5 | 307 ± 3.7 | 225 ± 28# | 47.3 ± 3.9 | 12.1 ± 2.2# |
Table 2.
Summary of Kv1.3-channel inhibition and its functional outcomes.
| Authors | Cells | Kv1.3-channel inhibitors used | Functional outcomes of Kv1.3-channel inhibition |
|---|---|---|---|
| Villalonga et al. [76] | Raw 264.7 macrophages Jurkit T-lymphocytes |
Margatoxin Charybdotoxin |
Immunomodulatory effect |
| Kalman et al. [77] | L929 cell line stably expressing Kv1.3 |
ShK-Dap22 | Immunomodulatory effect |
| Schmitz et al. [78] | L929 cell line stably expressing Kv1.3 |
PAP-1 | Immunomodulatory effect |
| Kazama et al. [38] | Murine thymocytes | Margatoxin | Membrane stabilization Immunomodulatory effect |
| Leanza et al. [80] | Cancer cell lines (MCF-7, DLD-1 etc.) |
PAP-1 Psora-4 |
Cellular proliferation Apoptosis regulation |
| Hamilton et al. [79] | Skeletal muscle cell lines (C2C12, L6) |
Margatoxin PAP-1 Psora-4 |
Increased glucose uptake Increased AMPK activity |
ShK: Stichodactyla helianthus toxin; PAP-1: 5-(4-phenylbutoxy) psoralen.
AMPK: 5′ adenosine monophosphate-activated protein kinase.
Figure 6.
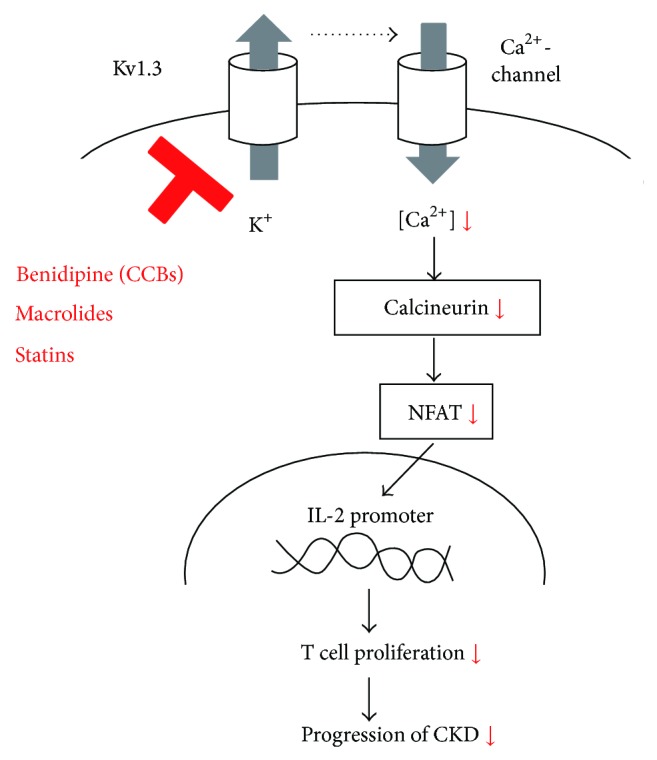
Therapeutic implications of targeting lymphocyte Kv1.3-channels in the treatment of CKD. In addition to benidipine, one of the calcium channel blockers (CCBs), macrolide antibiotics, and statins, which effectively inhibit lymphocyte Kv1.3-channels, could also potentially be useful in the treatment or the prevention of chronic kidney disease (CKD).
8. Conclusions and Perspectives
In a physiological condition, Kv1.3-channels expressed in T lymphocytes play crucial roles in the initiation of the immune response. In renal diseases, such as CKD, acute glomerulonephritis, and renal allograft rejection, the channels contribute to the pathogenesis of the diseases. In rat kidneys with advanced CRF, the overexpression of the channels in lymphocytes facilitated the progression of tubulointerstitial fibrosis by promoting lymphocyte proliferation, suggesting that the channel could be a potent therapeutic target for advanced-stage CKD. Benidipine, one of the most commonly used CCBs, which also strongly and persistently inhibits the lymphocyte Kv1.3-channel currents, suppressed the proliferation of kidney lymphocytes and actually ameliorated the progression of renal fibrosis. Since other drugs, such as NSAIDs, macrolide antibiotics, and statins, also effectively suppress the channel currents in lymphocytes, they may be useful for treating or preventing renal diseases.
“Chronic inflammatory diseases” are a category of diseases, in which “chronic inflammation” or “the overstimulation of cellular immunity” is responsible for the pathogenesis [22]. Besides infectious diseases and autoimmune disorders, a number of diseases, such as cancer, neuroinflammatory diseases, and metabolic disorders, nowadays fall into this category [87]. Recently, in addition to renal diseases, the involvement of lymphocyte Kv1.3-channels has also been demonstrated in the development of these “chronic inflammatory diseases” [43–45, 48–50, 52, 88–91]. Therefore, our future tasks would include revealing the as yet unknown significance of the channels in the pathogenesis of these diseases and revealing novel therapeutic applications based on targeting these channels.
Acknowledgments
This work was supported by MEXT KAKENHI Grant no. 25860155 and Miyagi Kidney Foundation grant.
Conflict of Interests
The author declares that he has no conflict of interests.
References
- 1.Cahalan M. D., Chandy K. G., DeCoursey T. E., Gupta S. A voltage-gated potassium channel in human T lymphocytes. Journal of Physiology. 1985;358:197–237. doi: 10.1113/jphysiol.1985.sp015548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeCoursey T. E., Chandy K. G., Gupta S., Cahalan M. D. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307(5950):465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- 3.Lewis R. S., Cahalan M. D. Potassium and calcium channels in lymphocytes. Annual Review of Immunology. 1995;13:623–653. doi: 10.1146/annurev.iy.13.040195.003203. [DOI] [PubMed] [Google Scholar]
- 4.Cahalan M. D., Wulff H., Chandy K. G. Molecular properties and physiological roles of ion channels in the immune system. Journal of Clinical Immunology. 2001;21(4):235–252. doi: 10.1023/A:1010958907271. [DOI] [PubMed] [Google Scholar]
- 5.Hu L., Pennington M., Jiang Q., Whartenby K. A., Calabresi P. A. Characterization of the functional properties of the voltage-gated potassium channel Kv1.3 in human CD4+ T lymphocytes. Journal of Immunology. 2007;179(7):4563–4570. doi: 10.4049/jimmunol.179.7.4563. [DOI] [PubMed] [Google Scholar]
- 6.Kazama I. Physiological significance of delayed rectifier K+ channels (Kv1.3) expressed in T lymphocytes and their pathological significance in chronic kidney disease. The Journal of Physiological Sciences. 2015;65(1):25–35. doi: 10.1007/s12576-014-0331-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumano K., Kogure K., Tanaka T., Sakai T. A new method of inducing experimental chronic renal failure by cryosurgery. Kidney International. 1986;30(3):433–436. doi: 10.1038/ki.1986.203. [DOI] [PubMed] [Google Scholar]
- 8.Jones S. E., Kelly D. J., Cox A. J., Zhang Y., Gow R. M., Gilbert R. E. Mast cell infiltration and chemokine expression in progressive renal disease. Kidney International. 2003;64(3):906–913. doi: 10.1046/j.1523-1755.2003.00183.x. [DOI] [PubMed] [Google Scholar]
- 9.Hyodo T., Oda T., Kikuchi Y., et al. Voltage-gated potassium channel Kv1.3 blocker as a potential treatment for rat anti-glomerular basement membrane glomerulonephritis. The American Journal of Physiology—Renal Physiology. 2010;299(6):F1258–F1269. doi: 10.1152/ajprenal.00374.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grgic I., Wulff H., Eichler I., Flothmann C., Köhler R., Hoyer J. Blockade of T-lymphocyte KCa3.1 and Kv1.3 channels as novel immunosuppression strategy to prevent kidney allograft rejection. Transplantation Proceedings. 2009;41(6):2601–2606. doi: 10.1016/j.transproceed.2009.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kazama I., Maruyama Y., Endo Y., et al. Overexpression of delayed rectifier K+ channels promotes in situ proliferation of leukocytes in rat kidneys with advanced chronic renal failure. International Journal of Nephrology. 2012;2012:8. doi: 10.1155/2012/581581.581581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chatenoud L., Dugas B., Beaurain G., et al. Presence of preactivated T cells in hemodialyzed patients: their possible role in altered immunity. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(19):7457–7461. doi: 10.1073/pnas.83.19.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaysen G. A., Kumar V. Inflammation in ESRD: causes and potential consequences. Journal of Renal Nutrition. 2003;13(2):158–160. doi: 10.1053/jren.2003.50021. [DOI] [PubMed] [Google Scholar]
- 14.Barreto D. V., Barreto F. C., Liabeuf S., et al. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney International. 2010;77(6):550–556. doi: 10.1038/ki.2009.503. [DOI] [PubMed] [Google Scholar]
- 15.Chandy K. G., Wulff H., Beeton C., Pennington M., Gutman G. A., Cahalan M. D. K+ channels as targets for specific immunomodulation. Trends in Pharmacological Sciences. 2004;25(5):280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugiyama K., Isogai K., Toyama A., et al. Correlation between the pharmacological efficacy of cyclosporine and tacrolimus as evaluated by the lymphocyte immunosuppressant sensitivity test (LIST) and the MTT assay procedure in patients before and after renal transplantation. International Journal of Clinical Pharmacology and Therapeutics. 2011;49(2):145–152. doi: 10.5414/CPP49145. [DOI] [PubMed] [Google Scholar]
- 17.Rangaraju S., Chi V., Pennington M. W., Chandy K. G. Kv1.3 potassium channels as a therapeutic target in multiple sclerosis. Expert Opinion on Therapeutic Targets. 2009;13(8):909–924. doi: 10.1517/14728220903018957. [DOI] [PubMed] [Google Scholar]
- 18.Kazama I., Maruyama Y., Matsubara M. Benidipine persistently inhibits delayed rectifier K+-channel currents in murine thymocytes. Immunopharmacology and Immunotoxicology. 2013;35(1):28–33. doi: 10.3109/08923973.2012.723011. [DOI] [PubMed] [Google Scholar]
- 19.Baba A., Tachi M., Maruyama Y., Kazama I. Suppressive effects of diltiazem and verapamil on delayed rectifier K+-channel currents in murine thymocytes. Pharmacological Reports. 2015 doi: 10.1016/j.pharep.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Kazama I., Maruyama Y. Differential effects of clarithromycin and azithromycin on delayed rectifier K+-channel currents in murine thymocytes. Pharmaceutical Biology. 2013;51(6):760–765. doi: 10.3109/13880209.2013.764539. [DOI] [PubMed] [Google Scholar]
- 21.Kazama I., Baba A., Maruyama Y. HMG-CoA reductase inhibitors pravastatin, lovastatin and simvastatin suppress delayed rectifier K(+)-channel currents in murine thymocytes. Pharmacological Reports. 2014;66(4):712–717. doi: 10.1016/j.pharep.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Heap G. A., van Heel D. A. The genetics of chronic inflammatory diseases. Human Molecular Genetics. 2009;18(1):R101–R106. doi: 10.1093/hmg/ddp001. [DOI] [PubMed] [Google Scholar]
- 23.Avioli L. V., Scott S., Lee S. W., de Luca H. F. Intestinal calcium absorption: Nature of defect in chronic renal disease. Science. 1969;166(3909):1154–1156. doi: 10.1126/science.166.3909.1154. [DOI] [PubMed] [Google Scholar]
- 24.Grgic I., Kiss E., Kaistha B. P., et al. Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(34):14518–14523. doi: 10.1073/pnas.0903458106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Platt R., Roscoe M. H., Smith F. W. Experimental renal failure. Clinical Science. 1952;11(3):217–231. [PubMed] [Google Scholar]
- 26.Morrison A. B. Experimentally induced chronic renal insufficiency in the rat. Laboratory Investigation. 1962;11:321–332. [PubMed] [Google Scholar]
- 27.Bencosme S. A., Stone R. S., Latta H., Madden S. C. Acute tubular and glomerular lesions in rat kidneys after uranium. Archives of Pathology. 1960;69:470–476. [PubMed] [Google Scholar]
- 28.Fukuda S., Kopple J. D. Chronic uremia syndrome in dogs induced with uranyl nitrate. Nephron. 1980;25(3):139–143. doi: 10.1159/000181770. [DOI] [PubMed] [Google Scholar]
- 29.Allison M. E. M., Wilson C. B., Gottschalk C. W. Pathophysiology of experimental glomerulonephritis in rats. The Journal of Clinical Investigation. 1974;53(5):1402–1423. doi: 10.1172/jci107689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kondo Y., Shigematsu H., Kobayashi Y. Cellular aspects of rabbit Masugi nephritis. II. Progressive glomerular injuries with crescent formation. Laboratory Investigation. 1972;27(6):620–631. [PubMed] [Google Scholar]
- 31.Michimata M., Kazama I., Mizukami K., et al. Urinary concentration defect and limited expression of sodium cotransporter, rBSC1, in a rat model of chronic renal failure. Nephron. 2003;93(2):p34–p41. doi: 10.1159/000068526. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki K., Hatano R., Michimata M., et al. Residual urinary concentrating ability and AQP2 expression in a rat model for chronic renal failure. Nephron Physiology. 2005;99(1):p16–p22. doi: 10.1159/000081798. [DOI] [PubMed] [Google Scholar]
- 33.Sanada S., Toyama H., Ejima Y., Matsubara M. Potential for erythropoietin synthesis in kidney of uraemic rat alters depending on severity of renal failure. Nephrology. 2009;14(8):735–742. doi: 10.1111/j.1440-1797.2009.01110.x. [DOI] [PubMed] [Google Scholar]
- 34.Kazama I., Baba A., Matsubara M., Endo Y., Toyama H., Ejima Y. Benidipine suppresses in situ proliferation of leukocytes and slows the progression of renal fibrosis in rat kidneys with advanced chronic renal failure. Nephron Experimental Nephrology. 2014;128(1-2):67–79. doi: 10.1159/000368080. [DOI] [PubMed] [Google Scholar]
- 35.Liu L., Kou P., Zeng Q., et al. CD4+ T lymphocytes, especially Th2 cells, contribute to the progress of renal fibrosis. The American Journal of Nephrology. 2012;36(4):386–396. doi: 10.1159/000343283. [DOI] [PubMed] [Google Scholar]
- 36.El Nahas A. M., Anderson S., Harris K. P. G., editors. Mechanisms and Management of Progressive Renal Failure. London, UK: Oxford University Press; 2000. [Google Scholar]
- 37.Cahalan M. D., Chandy K. G. The functional network of ion channels in T lymphocytes. Immunological Reviews. 2009;231(1):59–87. doi: 10.1111/j.1600-065X.2009.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kazama I., Maruyama Y., Murata Y., Sano M. Voltage-dependent biphasic effects of chloroquine on delayed rectifier K+-channel currents in murine thymocytes. Journal of Physiological Sciences. 2012;62(3):267–274. doi: 10.1007/s12576-012-0195-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kazama I., Maruyama Y., Murata Y. Suppressive effects of nonsteroidal anti-inflammatory drugs diclofenac sodium, salicylate and indomethacin on delayed rectifier K+-channel currents in murine thymocytes. Immunopharmacology and Immunotoxicology. 2012;34(5):874–878. doi: 10.3109/08923973.2012.666249. [DOI] [PubMed] [Google Scholar]
- 40.Ishida Y., Chused T. M. Lack of voltage sensitive potassium channels and generation of membrane potential by sodium potassium ATPase in murine T lymphocytes. Journal of Immunology. 1993;151(2):610–620. [PubMed] [Google Scholar]
- 41.Deutsch C., Chen L. Q. Heterologous expression of specific K+ channels in T lymphocytes: functional consequences for volume regulation. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(21):10036–10040. doi: 10.1073/pnas.90.21.10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teisseyre A., Zmonarski S. C., Klinger M., Mozrzymas J. W., Miȩkisz S. Patch-clamp study on T-lymphocyte potassium conductance in patients with chronic renal failure. Nephron. 1996;72(4):587–594. doi: 10.1159/000188944. [DOI] [PubMed] [Google Scholar]
- 43.Abdul M., Hoosein N. Expression and activity of potassium ion channels in human prostate cancer. Cancer Letters. 2002;186(1):99–105. doi: 10.1016/S0304-3835(02)00348-8. [DOI] [PubMed] [Google Scholar]
- 44.Jang S. H., Kang K. S., Ryu P. D., Lee S. Y. Kv1.3 voltage-gated K+channel subunit as a potential diagnostic marker and therapeutic target for breast cancer. BMB Reports. 2009;42(8):535–539. doi: 10.5483/bmbrep.2009.42.8.535. [DOI] [PubMed] [Google Scholar]
- 45.Huang S., Zhang C.-T., Tang J.-R., Cai L., Zhang Z., Zhou M.-G. Upregulated voltage-gated potassium channel Kv1.3 on CD4+CD28null T lymphocytes from patients with acute coronary syndrome. Journal of Geriatric Cardiology. 2010;7(1):40–46. [Google Scholar]
- 46.Toldi G., Bajnok A., Dobi D., et al. The effects of Kv1.3 and IKCa1 potassium channel inhibition on calcium influx of human peripheral T lymphocytes in rheumatoid arthritis. Immunobiology. 2013;218(3):311–316. doi: 10.1016/j.imbio.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 47.Rus H., Pardo C. A., Hu L., et al. The voltage-gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(31):11094–11099. doi: 10.1073/pnas.0501770102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orbán C., Biró E., Grozdics E., Bajnok A., Toldi G. Modulation of T lymphocyte calcium influx patterns via the inhibition of Kv1.3 and IKCa1 potassium channels in autoimmune disorders. Frontiers in Immunology. 2013;4, article 234 doi: 10.3389/fimmu.2013.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han S., Yi H., Yin S. J., et al. Structural basis of a potent peptide inhibitor designed for Kv1.3 channel, a therapeutic target of autoimmune disease. The Journal of Biological Chemistry. 2008;283(27):19058–19065. doi: 10.1074/jbc.m802054200. [DOI] [PubMed] [Google Scholar]
- 50.Beeton C., Wulff H., Barbaria J., et al. Selective blockade of T lymphocyte K+ channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(24):13942–13947. doi: 10.1073/pnas.241497298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wonderlin W. F., Strobl J. S. Potassium channels, proliferation and G1 progression. Journal of Membrane Biology. 1996;154(2):91–107. doi: 10.1007/s002329900135. [DOI] [PubMed] [Google Scholar]
- 52.Jang S. H., Choi S. Y., Ryu P. D., Lee S. Y. Anti-proliferative effect of Kv1.3 blockers in A549 human lung adenocarcinoma in vitro and in vivo. European Journal of Pharmacology. 2011;651(1–3):26–32. doi: 10.1016/j.ejphar.2010.10.066. [DOI] [PubMed] [Google Scholar]
- 53.Ouadid-Ahidouch H., Ahidouch A. K+ channel expression in human breast cancer cells: Involvement in cell cycle regulation and carcinogenesis. Journal of Membrane Biology. 2008;221(1):1–6. doi: 10.1007/s00232-007-9080-6. [DOI] [PubMed] [Google Scholar]
- 54.Braunwald E. Mechanism of action of calcium-channel-blocking agents. The New England Journal of Medicine. 1982;307(26):1618–1627. doi: 10.1056/nejm198212233072605. [DOI] [PubMed] [Google Scholar]
- 55.Palamaras I., Kyriakos K. Calcium antagonists in dermatology: a review of the evidence and research-based studies. Dermatology Online Journal. 2005;11(2) [PubMed] [Google Scholar]
- 56.Horvath M., Mezey Z., Josfay A., Nanay I., Varsanyi M., Gero S. The effect of some drugs on in vitro cellular immune reactions and on circulating immune complexes in patients with myocardial infarction. Journal of Investigational Allergology and Clinical Immunology. 1991;1:404–410. [PubMed] [Google Scholar]
- 57.Birx D. L., Berger M., Fleischer T. A. The interference of T cell activation by calcium channel blocking agents. Journal of Immunology. 1984;133(6):2904–2909. [PubMed] [Google Scholar]
- 58.Bacon K. B., Westwick J., Camp R. D. R. Potent and specific inhibition of IL-8-, IL-1 alpha- and IL-1 beta-induced in vitro human lymphocyte migration by calcium channel antagonists. Biochemical and Biophysical Research Communications. 1989;165(1):349–354. doi: 10.1016/0006-291x(89)91076-0. [DOI] [PubMed] [Google Scholar]
- 59.Matsumori A., Nishio R., Nose Y. Calcium channel blockers differentially modulate cytokine production by peripheral blood mononuclear cells. Circulation Journal. 2010;74(3):567–571. doi: 10.1253/circj.CJ-09-0467. [DOI] [PubMed] [Google Scholar]
- 60.Liu W., Matsumori A. Calcium channel blockers and modulation of innate immunity. Current Opinion in Infectious Diseases. 2011;24(3):254–258. doi: 10.1097/QCO.0b013e3283463e5b. [DOI] [PubMed] [Google Scholar]
- 61.Furukawa T., Yamakawa T., Midera T., Sagawa T., Mori Y., Nukada T. Selectivities of dihydropyridine derivatives in blocking Ca2+ channel subtypes expressed in Xenopus oocytes . Journal of Pharmacology and Experimental Therapeutics. 1999;291(2):464–473. [PubMed] [Google Scholar]
- 62.Herbette L. G., Chester D. W., Rhodes D. G. Structural analysis of drug molecules in biological membranes. Biophysical Journal. 1986;49(1):91–94. doi: 10.1016/S0006-3495(86)83605-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitakaze M., Node K., Minamino T., Asanuma H., Kuzuya T., Hori M. A Ca channel blocker, benidipine, increases coronary blood flow and attenuates the severity of myocardial ischemia via NO-dependent mechanisms in dogs. Journal of the American College of Cardiology. 1999;33(1):242–249. doi: 10.1016/S0735-1097(98)00556-7. [DOI] [PubMed] [Google Scholar]
- 64.Yamamoto M., Gotoh Y., Imaizumi Y., Watanabe M. Mechanisms of long-lasting effects of benidipine on Ca current in guinea-pig ventricular cells. British Journal of Pharmacology. 1990;100(4):669–676. doi: 10.1111/j.1476-5381.1990.tb14074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karasawa A., Kubo K. Calcium antagonistic effects and the in vitro duration of actions of KW-3049, a new 1,4-dihydropyridine derivative, in isolated canine coronary arteries. Japanese Journal of Pharmacology. 1988;47(1):35–44. doi: 10.1254/jjp.47.35. [DOI] [PubMed] [Google Scholar]
- 66.Hayashi K., Homma K., Wakino S., et al. T-type Ca channel blockade as a determinant of kidney protection. Keio Journal of Medicine. 2010;59(3):84–95. doi: 10.2302/kjm.59.84. [DOI] [PubMed] [Google Scholar]
- 67.Remuzzi G. Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia) The Lancet. 1996;349(9069):1857–1863. doi: 10.1016/s0140-6736(96)11445-8. [DOI] [PubMed] [Google Scholar]
- 68.Hayashi K., Wakino S., Homma K., Sugano N., Saruta T. Pathophysiological significance of T-type Ca2+ channels: Role of T-type Ca2+ channels in renal microcirculation. Journal of Pharmacological Sciences. 2005;99(3):221–227. doi: 10.1254/jphs.fmj05002x6. [DOI] [PubMed] [Google Scholar]
- 69.Nakamura T., Sato E., Fujiwara N., et al. Comparative effects of benidipine and amlodipine on proteinuria, urinary 8-OHdG, urinary L-FABP, and inflammatory and atherosclerosis markers in early-stage chronic kidney disease. The American Journal of the Medical Sciences. 2010;339(2):157–163. doi: 10.1097/maj.0b013e3181c643df. [DOI] [PubMed] [Google Scholar]
- 70.Andersson U., Wang H., Palmblad K., et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. The Journal of Experimental Medicine. 2000;192(4):565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sugiyama K., Shirai R., Mukae H., et al. Differing effects of clarithromycin and azithromycin on cytokine production by murine dendritic cells. Clinical and Experimental Immunology. 2007;147(3):540–546. doi: 10.1111/j.1365-2249.2007.03299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morikawa K., Zhang J., Nonaka M., Morikawa S. Modulatory effect of macrolide antibiotics on the Th1- and Th2-type cytokine production. International Journal of Antimicrobial Agents. 2002;19(1):53–59. doi: 10.1016/S0924-8579(01)00457-5. [DOI] [PubMed] [Google Scholar]
- 73.Khan A. A., Slifer T. R., Araujo F. G., Remington J. S. Effect of clarithromycin and azithromycin on production of cytokines by human monocytes. International Journal of Antimicrobial Agents. 1999;11(2):121–132. doi: 10.1016/S0924-8579(98)00091-0. [DOI] [PubMed] [Google Scholar]
- 74.Bessler H., Salman H., Bergman M., Straussberg R., Djaldetti M. In vitro effect of statins on cytokine production and mitogen response of human peripheral blood mononuclear cells. Clinical Immunology. 2005;117(1):73–77. doi: 10.1016/j.clim.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 75.Rosenson R. S., Tangney C. C., Casey L. C. Inhibition of proinflammatory cytokine production by pravastatin. The Lancet. 1999;353(9157):983–984. doi: 10.1016/S0140-6736(98)05917-0. [DOI] [PubMed] [Google Scholar]
- 76.Villalonga N., David M., Bielańska J., et al. Immunomodulatory effects of diclofenac in leukocytes through the targeting of Kv1.3 voltage-dependent potassium channels. Biochemical Pharmacology. 2010;80(6):858–866. doi: 10.1016/j.bcp.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 77.Kalman K., Pennington M. W., Lanigan M. D., et al. Shk-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. The Journal of Biological Chemistry. 1998;273(49):32697–32707. doi: 10.1074/jbc.273.49.32697. [DOI] [PubMed] [Google Scholar]
- 78.Schmitz A., Sankaranarayanan A., Azam P., et al. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory t cells in autoimmune diseases. Molecular Pharmacology. 2005;68(5):1254–1270. doi: 10.1124/mol.105.015669. [DOI] [PubMed] [Google Scholar]
- 79.Hamilton D. L., Beall C., Jeromson S., Chevtzoff C., Cuthbertson D. J., Ashford M. L. J. Kv1.3 inhibitors have differential effects on glucose uptake and AMPK activity in skeletal muscle cell lines and mouse ex vivo skeletal muscle. The Journal of Physiological Sciences. 2014;64(1):13–20. doi: 10.1007/s12576-013-0285-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leanza L., O'Reilly P., Doyle A., et al. Correlation between potassium channel expression and sensitivity to drug-induced cell death in tumor cell lines. Current Pharmaceutical Design. 2014;20(2):189–200. doi: 10.2174/13816128113199990032. [DOI] [PubMed] [Google Scholar]
- 81.Lewis R. J., Garcia M. L. Therapeutic potential of venom peptides. Nature Reviews Drug Discovery. 2003;2(10):790–802. doi: 10.1038/nrd1197. [DOI] [PubMed] [Google Scholar]
- 82.Judge S. I. V., Lee J. M., Bever C. T., Jr., Hoffman P. M. Voltage-gated potassium channels in multiple sclerosis: overview and new implications for treatment of central nervous system inflammation and degeneration. Journal of Rehabilitation Research and Development. 2006;43(1):111–122. doi: 10.1682/jrrd.2004.09.0116. [DOI] [PubMed] [Google Scholar]
- 83.Escobar L. I., Martínez-Téllez J. C., Salas M., et al. A voltage-gated K+ current in renal inner medullary collecting duct cells. The American Journal of Physiology—Cell Physiology. 2004;286(4):C965–C974. doi: 10.1152/ajpcell.00074.2003. [DOI] [PubMed] [Google Scholar]
- 84.Carrisoza-Gaytán R., Salvador C., Satlin L. M., et al. Potassium secretion by voltage-gated potassium channel Kv1.3 in the rat kidney. The American Journal of Physiology - Renal Physiology. 2010;299(1):F255–F264. doi: 10.1152/ajprenal.00697.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.George Chandy K., Cahalan M., Pennington M., Norton R. S., Wulff H., Gutman G. A. Potassium channels in T lymphocytes: toxins to therapeutic immunosuppressants. Toxicon. 2001;39(9):1269–1276. doi: 10.1016/s0041-0101(01)00120-9. [DOI] [PubMed] [Google Scholar]
- 86.Mouhat S., Teodorescu G., Homerick D., et al. Pharmacological profiling of Orthochirus scrobiculosus toxin 1 analogs with a trimmed N-terminal domain. Molecular Pharmacology. 2006;69(1):354–362. doi: 10.1124/mol.105.017210. [DOI] [PubMed] [Google Scholar]
- 87.Kazama I., Maruyama Y., Baba A. Amphipath-induced plasma membrane curvature controls microparticle formation from adipocytes: novel therapeutic implications for metabolic disorders. Medical Hypotheses. 2014;82(2):196–198. doi: 10.1016/j.mehy.2013.11.037. [DOI] [PubMed] [Google Scholar]
- 88.Kazama I., Sasagawa N., Nakajima T. Complete remission of human parvovirus B19 associated symptoms by loxoprofen in patients with atopic predispositions. Case Reports in Medicine. 2012;2012:4. doi: 10.1155/2012/703281.703281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kazama I., Tamada T., Nakajima T. Resolution of migratory pulmonary infiltrates by moxifloxacin in a patient with dual infection of Mycoplasma pneumoniae and Bordetella pertussis . Infezioni in Medicina. 2012;20(4):288–292. [PubMed] [Google Scholar]
- 90.Kazama I., Nakajima T. Dual infection of Mycoplasma pneumoniae and Chlamydophila pneumoniae in patients with atopic predispositions successfully treated by moxifloxacin. Infezioni in Medicina. 2014;22(1):41–46. [PubMed] [Google Scholar]
- 91.Kazama I., Nakajima T. A case of fitz-hugh-curtis syndrome complicated by appendicitis conservatively treated with antibiotics. Clinical Medicine Insights: Case Reports. 2013;6:35–40. doi: 10.4137/CCRep.S11522. [DOI] [PMC free article] [PubMed] [Google Scholar]