

Abstract
A general protocol for the highly exo-selective macrocyclization of ynals using a nickel/N-heterocyclic carbene catalyst system has been developed. A series of 10- to 21-membered macrocycles bearing an exomethylene substituent was synthesized in good yields with excellent regioselectivity (exo:endo >95:5). Very high levels of long-range diastereocontrol can also be achieved for some classes of macrocycles. Complementary to previously reported endo-selective macrocyclizations, this method provides accesses to exo-alkylidene macrocycles from simple ynals in high selectivity.
Macrocycles are common structural motifs in many classes of natural products and bioactive molecules.1 While methods such as macrolactonization and ring-closing metathesis are commonly employed in their preparation,2–3 alternative strategies that allow fundamentally different disconnections and precursor functional groups are highly desirable. Nickel-catalyzed reductive coupling methods have proven to be useful in the preparation of macrocycles, since these methods allow the combination of functional groups not commonly employed in macrocycle-forming processes, and both a stereogenic center and a stereodefined alkene are established during assembly of the macrocyclic ring. Reports from our laboratory and the Jamison group have established highly selective reductive macrocyclization methods that were utilized in the synthesis of several natural products.4 These applications have typically involved control of regiochemistry by the electronic influence of the alkyne. For example, terminal alkynes display a bias toward endocyclization to provide internal 1,2-transdisubstituted alkenes (Scheme 1, structure 1), whereas aromatic alkynes display a bias toward exocyclization to provide exobenzylidene structural elements (structure 2).
Scheme 1.

Regiocontrol in nickel-catalyzed reductive macrocyclizations
In contrast to the ease of assembly of structure types 1 and 2, simple exomethylene substituents are commonly observed in numerous classes of macrolactones,1h-i but would require an unfavorable exocyclization from a simple terminal alkyne substrate to afford product 3. A single example of participation of a terminal alkyne in an exo-selective reductive macrocyclization was described by our laboratory while pursuing the total synthesis of 10-deoxymethynolide.4a However, the procedure required an N-heterocyclic carbene (NHC) ligand that was prepared in a multistep operation and that displays a somewhat limited scope when conducted with minimally functionalized ynal structural classes. Given the potential utility of directly accessing macrocycles of structure type 3 by the reductive exocyclization of simple ynals bearing terminal alkynes, we have conducted studies to provide a more broadly applicable method. As reported herein, a protocol that utilizes a readily available ligand has now been developed that is effective across a range of substrates and that additionally possesses unusual capabilities in long-range diastereocontrol. Mechanistic insights provide a basis for the highly exoselective outcome of this procedure.
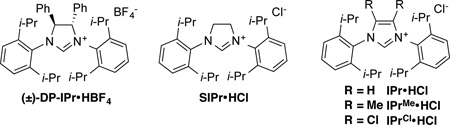
We began the study of exo-macrocyclization with ynal 4, by applying reaction conditions that were developed in our recent work with precursors leading to 10-deoxymethynolide.4a With ligand DP-IPr and triethylsilane (Et3SiH) as reducing agent (Table 1, entry 1), the desired macrocycle 5 was successfully synthesized with excellent regioselectivity (exo:endo 93:7) albeit in low yield (21%). In the course of conducting recent mechanistic studies of intermolecular aldehyde-alkyne reductive couplings, we observed that high temperatures paired with bulky silanes led to especially high levels of regiocontrol, where the more hindered alkyne terminus undergoes addition to the aldehyde.5a In exploring the options with DP-IPr, increasing the reaction temperature did not lead to effective macrocyclizations with substrate 4, and the Ni/DP-IPr catalyzed reaction with either Et3SiH or (i-Pr)3SiH as reducing agent (entries 2 and 3) failed to produce the desired macrocyclization product. Slight improvement of the reaction yield, however, was observed with SIPr ligand (entry 4). Encouraged by this result, other NHC ligands with 2,6-diisopropylphenyl substituents on nitrogen were examined, and the highest yield of macrocycle 5 was obtained with IPrCl as ligand (entries 5–7). IPrCl is easily accessible, highly stable, and is known to provide highly active catalysts in other catalytic processes.6,7 With IPrCl as a ligand, the reaction temperature, concentration and catalyst loading were then investigated. It was found that conducting the reaction at relatively high temperatures (90 °C) was indeed necessary, and lowering the temperature to 50 °C decreased the yield of compound 5 to 30% (entry 8). The concentration of the reaction could be increased to 5 mM (entry 9) but a decrease in yield was observed for the reaction performed at higher concentration (entry 10). The use of bulky (i-Pr)3SiH was essential for the regioselectivity (exo:endo), and large erosion of the regioselectivity was observed with Et3SiH as reducing agent (entry 11). The optimal catalyst loading for this reaction was 30 mol % of nickel/IPrCl. Lowering the catalyst loading decreased the yield, while increasing the catalyst loading to 40 mol % provided no benefits (entries 12 and 13).
Table 1.
Optimization study
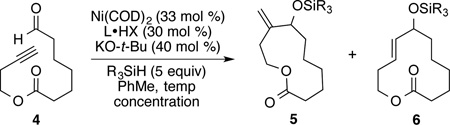 | |||||||
|---|---|---|---|---|---|---|---|
| entry | L•HX | temp (°C) |
R | C mM |
rra (5:6) |
NMRb yield % |
isolated yield % |
| 1 | DP-IPr | rt | Et | 3 | 93:7 | - | 21 |
| 2 | DP-IPr | 90 | Et | 3 | - | trace | - |
| 3 | DP-IPr | 90 | i-Pr | 3 | - | trace | - |
| 4 | SIPr | 90 | i-Pr | 3 | >95:5 | 27 | - |
| 5 | IPr | 90 | i-Pr | 3 | >95:5 | 18 | - |
| 6 | IPrMe | 90 | i-Pr | 3 | >95:5 | 35 | - |
| 7 | IPrCl | 90 | i-Pr | 3 | >95:5 | 67 | 66 |
| 8 | IPrCl | 50 | i-Pr | 3 | >95:5 | 30 | - |
| 9 | IPrCl | 90 | i-Pr | 5 | >95:5 | - | 69 |
| 10 | IPrCl | 90 | i-Pr | 10 | >95:5 | 32 | - |
| 11 | IPrCl | 90 | Et | 5 | 65:35 | - | 51c |
| 12d | IPrCl | 90 | i-Pr | 5 | >95:5 | 17 | - |
| 13e | IPrCl | 90 | i-Pr | 5 | >95:5 | - | 63 |
 | |||||||
Determined by 1H-NMR analysis of the crude mixture.
1,3,5-trimethoxybenzene was used as internal standard. Preparative experiments were not conducted with an internal standard due to complexity of removing the internal standard.
Combined yield.
Ni(COD)2 (23 mol %), IPrCl•HCl (20 mol %), KO-t-Bu (24 mol %).
Ni(COD)2 (43 mol %), IPrCl•HCl (40 mol %), KO-t-Bu (44 mol %).
After identifying the optimal reaction conditions (entry 9), the scope of ring size and functional groups tolerated in the exo-selective macrocyclization was investigated (Scheme 2). In all cases studied, high selectivity (>95:5) for the formation of exo-alkylidene macrocycles was observed. In general, the method worked well in generating molecules having ring size greater than 11, and 12-, 13-, 18- and 21-membered macrocycles 8b–8e were produced in good yields. Under the standard conditions, 10-membered macrocycle 8a was also successfully synthesized in moderate yield (45%). In addition to esters within the linear precursor, ethers are also tolerated as shown by the synthesis of macrocycle 8f. Unfortunately, further efforts to include an amide functional group or purely aliphatic chains only resulted in low yields. In these latter cases, dimers and higher oligomers were observed as major products.
Scheme 2.

Scope of macrocyclizationsa
arr is ratio of regioisomers.
In addition to terminal alkynes, the reaction of internal alkynes was also explored. In our previous study on nickel-catalyzed reductive macrocyclizations, a larger ring (21-membered) exo-alkylidene macrocycle was prepared using IPr as ligand and Et3SiH as reducing reagent (exo:endo = 5:1).4b Based on the high regioselectivity observed in this study, we envisioned that higher selectivity toward the synthesis of exoalkylidene macrocycles could be achieved with IPrCl ligand. Indeed, only exocyclization products 8g and 8h were obtained from the Ni/IPrCl catalyzed reaction of ynal 7g (Scheme 2). In contrast to the need for bulky silanes to achieve high levels of regiocontrol with terminal alkynes, excellent regioselectivity was obtained with both Et3SiH and (i-Pr)3SiH using IPrCl as ligand.
Prior advances from Jamison and our laboratory have described high levels of diastereoselectivity in macrocyclizations utilizing chirality adjacent to the aldehyde or alkyne.4 The Jamison approach to amphidinolides T1 and T4 in particular described a detailed conformational analysis of the diastereoselectivity of macrocyclizations of aromatic alkynes.4e To more fully explore the potential of diastereoselective macrocyclizations, including those potentially involving long-range diastereocontrol, we examined the reactivity of ynals 9a–9f (Scheme 3). Interestingly, the 11-membered macrocycles 10a–10c were all formed as single diastereomers. Remarkably, despite the remote positioning of substituents, macrocycle 10d was generated with a high level of 1,5-diastereocontrol (90:10).
Scheme 3.

Diastereoselectivity studya
arr is the ratio of regioisomers. bCombined yield.
The stereochemistry of 10a–10c was deduced by COSY and NOE experiments. The diol derived from 10d, by removing the two triisopropylsilyl groups, was crystalline, and its structure was unambiguously assigned by X-ray crystallography.8 Further study showed that the diastereoselectivity of the macrocyclization strongly depends on the ring size formed. With a methyl substituent at the homopropargylic position, 13- and 16-membered macrocycles 10e and 10f were synthesized in good yields employing the standard conditions. Compared to the 11-membered macrocycle 10b (dr >95:5), the diastereoselectivity of 13-membered macrocycle 10e dropped to 76:24, and a completely non-selective reaction was observed for the synthesis of 16-membered macrocycle 10f (dr 50:50).
Coupled with the previously reported advances allowing endo-selective macrocyclizations of terminal alkynes,4b the exocyclization protocol described herein now allows either regioisomer to be effectively obtained from a common substrate. To demonstrate the versatility of this feature, substrate 9d was subjected to the macrocyclization conditions, allowing either endocyclization or exocyclization to proceed (Scheme 4). Whereas the exocyclization procedure requires a bulky ligand IPrCl and bulky silane (i-Pr)3SiH to produce 10d, the endocyclization proceeds best with IMes as ligand and Et3SiH as reductant to produce 12-membered macrocycle 11.
Scheme 4.

Regiochemical alterations
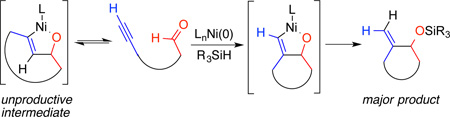
Recent mechanistic investigations of intermolecular nickel-catalyzed reductive couplings have elucidated the operative mechanistic pathways according to catalyst and silane structure. The typical pathway for silane-mediated reductive couplings involves rate-determining formation of a nickel metallacycle, which thus governs the regioselectivity of the process.9 Alternatively, the specific combination of a large NHC ligand, a bulky silane, and high temperature was observed to follow a different kinetic description, where formation of the metallacycle leading to the minor product is reversible.5a This outcome leads to very high regioselectivity for the product that possesses the more hindered C-C bond. The conditions (large ligand, bulky silane, high temperature, slow addition) favoring formation of the more hindered exocyclization-derived macrocyclization products (Schemes 2–3) closely pattern the conditions that favored reversible formation of the metallacycle in our recent mechanistic investigations. On this basis, it is likely that the procedure reported herein involves reversible formation of the endocyclic metallacycle 15, but due to the more hindered nature of the Ni-C bond, its capture by the bulky silane [i.e., (i-Pr)3SiH] is inefficient (Scheme 5). Reversion back to the Ni(0) ynal complex 14, reorientation of the alkyne to intermediate 12, and then formation of exocyclic metallacycle 13 that allows a productive σ-bond metathesis leading to preferred formation of the exocyclic product.
Scheme 5.

Mechanistic rationale
According to the above mechanistic hypothesis, regioselectivity for the exocyclic product should erode as the silane steric hindrance decreases. To address this question, the macrocyclization of ynal 9b was conducted with (i-Pr)3SiH and also with Et3SiH under otherwise identical conditions (Scheme 6). Compared to the highly regioselective synthesis of 10b using (i-Pr)3SiH, the reaction of 9b with Et3SiH generated the products as a mixture of exo- and endocyclization products 16 and 17 with a ratio of 32:68 favoring macrocycle 17 derived from endocyclization. These results support the above hypothesis for the origin of regiocontrol since the use of a smaller silane promotes a faster addition of silane to the hindered endocyclic metallacycle 15, thus rendering the production of endocyclic products more efficient.
Scheme 6.

Influence of silane sterics on regioselectivity
Whereas exocyclization products 10b or 16 were obtained with high levels of diastereoselectivity regardless of silane structure, modest levels of diastereocontrol were observed for the endocyclization product 17 (Scheme 6). The choice of silane reducing agents is thus important for the reaction to achieve high regioselectivity (exo vs endo) but has negligible influences on the diastereoselectivity. The diastereoselectivity of the exocyclization reaction is likely controlled by transannular interactions associated with the 12 to 13 transformation. As demonstrated across varying ring sizes, the impact of these interactions, and their resulting influence on diastereoselectivity, strongly depends on the size of the macrocyclic ring formed (Scheme 3, compounds 10b, 10e and 10f).
In conclusion, a general protocol for the exo-selective macrocyclization of ynals has been developed. Employing a Ni/IPrCl catalyst system paired with (i-Pr)3SiH as reducing agent, high selectivity for the synthesis of exo-alkylidene macrocycles was achieved. Although IPrCl has been successfully applied to other chemical transformations,7 its use in reductive coupling reactions of this type has not been previously described. Under the optimized conditions, 10- to 21-membered macrocycles were prepared in synthetically useful yields with high regioselectivity. Complementary to previously published endo-selective macrocyclizations, this study provides access to exo-alkylidene macrocycles with high selectivity from similar ynal substrates. Excellent diastereoselectivity of the exoselective macrocyclization was also observed for macrocycles of certain ring sizes. High levels of diastereocontrol were obtained for macrocycles with substituents quite remote from the reacting alkyne and aldehyde functional groups. Future efforts will involve increasingly complex illustrations of this macrocyclization method and utilization of the novel structures obtained in enzymatic oxidation processes.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NIH grants GM-57014 (which supports catalytic ligand-controlled regioreversals) and GM-78553 (which supports the examination of novel macrolide analogues in enzymatic hydroxylations). SN acknowledges support from the NIH Chemistry-Biology Interface Training Grant (GM008597). We thank Dr. Jeff Kampf (University of Michigan) for the X-ray crystallographic structure determination. Evan P. Jackson (University of Michigan) is thanked for the initial synthesis of IPrCl ligand.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Synthetic details, spectral data and CIF file for the X-ray crystallographic data. These materials are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Giordanetto F, Kihlberg J. J. Med. Chem. 2014;57:278. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]; (b) Vendeville S, Cummings MD. Annu. Rep. Med. Chem. 2013;48:371. [Google Scholar]; (c) Yu X, Sun D. Molecules. 2013;18:6230. doi: 10.3390/molecules18066230. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Madsen CM, Clausen MH. Eur. J. Org. Chem. 2011:3107. [Google Scholar]; (e) Marsault E, Peterson ML. J. Med. Chem. 2011;54:1961. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]; (f) Li Y, Pattenden G. Nat. Prod. Rep. 2011;28:429. doi: 10.1039/c0np00029a. [DOI] [PubMed] [Google Scholar]; (g) Gaul C, Njardarson JT, Shan D, Dorn DC, Wu KD, Tong WP, Huang XY, Moore MAS, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:11326. doi: 10.1021/ja048779q. [DOI] [PubMed] [Google Scholar]; (h) Kobayashi J, Tsuda M. Nat. Prod. Rep. 2004;21:77. doi: 10.1039/b310427n. [DOI] [PubMed] [Google Scholar]; (i) Blunt JW, Copp BR, Munro MHG, Northcote PT, Prinsep MR. Nat. Prod. Rep. 2004;21:1. doi: 10.1039/b305250h. [DOI] [PubMed] [Google Scholar]
- 2.Selected reviews on macrocyclization: Xie J, Bogliotti N. Chem. Rev. 2014;114:7678. doi: 10.1021/cr400035j. Yudin AK. Chem. Sci. 2015;6:30. doi: 10.1039/c4sc03089c. White CJ, Yudin AK. Nat. Chem. 2011;3:509. doi: 10.1038/nchem.1062. Tanaka K, Tajima Y. Eur. J. Org. Chem. 2012:3715. Prunet J. Eur. J. Org. Chem. 2011:3634. Crane EA, Scheidt KA. Angew. Chem. Int. Ed. 2010;49:8316. doi: 10.1002/anie.201002809. Gradillas A, Pérez-Castells J. Angew. Chem. Int. Ed. 2006;45:6086. doi: 10.1002/anie.200600641. Blankenstein J, Zhu JP. Eur. J. Org. Chem. 2005:1949. Parenty A, Moreau X, Campagne JM. Chem. Rev. 2006;106:911. doi: 10.1021/cr0301402.
- 3.Selected examples of macrocyclizations: Zhang H, Yu EC, Torker S, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2014;136:16493. doi: 10.1021/ja510768c. Mangold SL, O’Leary DJ, Grubbs RH. J. Am. Chem. Soc. 2014;136:12469. doi: 10.1021/ja507166g. Santandrea J, Bédard A-C, Collins SK. Org. Lett. 2014;16:3892. doi: 10.1021/ol501898b. Bai Y, Davis DC, Dai M. Angew. Chem. Int. Ed. 2014;53:6519. doi: 10.1002/anie.201403006. Zhao W, Li Z, Sun J. J. Am. Chem. Soc. 2013;135:4680. doi: 10.1021/ja400883q. Lumbroso A, Abermil N, Breit B. Chem. Sci. 2012;3:789. Bédard A-C, Collins SK. J. Am. Chem. Soc. 2011;133:19976. doi: 10.1021/ja208902t. Hili R, Rai V, Yudin AK. J. Am. Chem. Soc. 2010;132:2889. doi: 10.1021/ja910544p. Oppolzer W, Radinov RN. J. Am. Chem. Soc. 1993;115:1593.
- 4.Nickel-catalyzed macrocyclizations: Shareef A-R, Sherman DH, Montgomery J. Chem. Sci. 2012;3:892. doi: 10.1039/C2SC00866A. Knapp-Reed B, Mahandru GM, Montgomery J. J. Am. Chem. Soc. 2005;127:13156. doi: 10.1021/ja054590i. Chrovian CC, Knapp-Reed B, Montgomery J. Org. Lett. 2008;10:811. doi: 10.1021/ol702961v. Trenkle JD, Jamison TF. Angew. Chem. Int. Ed. 2009;48:5366. doi: 10.1002/anie.200902079. Colby EA, O’Brien KC, Jamison TF. J. Am. Chem. Soc. 2005;127:4297. doi: 10.1021/ja042733f. Chan J, Jamison TF. J. Am. Chem. Soc. 2004;126:10682. doi: 10.1021/ja0470968. Colby EA, O'Brien KC, Jamison TF. J. Am. Chem. Soc. 2004;126:998. doi: 10.1021/ja039716v.
- 5.For other regiochemistry reversal in nickel catalysis: Jackson EP, Montgomery J. J. Am. Chem. Soc. 2015;137:958. doi: 10.1021/ja511778a. Malik HA, Sormunen GJ, Montgomery J. J. Am. Chem. Soc. 2010;132:6304. doi: 10.1021/ja102262v. Miller ZD, Li W, Belderrain TR, Montgomery J. J. Am. Chem. Soc. 2013;135:15282. doi: 10.1021/ja407749w. Moragas T, Cornella J, Martin R. J. Am. Chem. Soc. 2014;136:17702. doi: 10.1021/ja509077a. Köpfer A, Sam B, Breit B, Krische MJ. Chem. Sci. 2013;4:1876. Bausch CC, Patman RL, Breit B, Krische MJ. Angew. Chem. Int. Ed. 2011;50:5686. doi: 10.1002/anie.201101496. Zhao Y, Weix DJ. J. Am. Chem. Soc. 2013;136:48. doi: 10.1021/ja410704d. Donets PA, Cramer N. Angew. Chem. Int. Ed. 2015;54:633. doi: 10.1002/anie.201409669. (i) For a copper-catalyzed process: Tani Y, Fujihara T, Terao J, Tsuji Y. J. Am. Chem. Soc. 2014;136:17706. doi: 10.1021/ja512040c. (j) For a review of regiochemistry reversals reactions: Mahatthananchai J, Dumas AM, Bode JW. Angew. Chem. Int. Ed. 2012;51:10954. doi: 10.1002/anie.201201787.
- 6.For the synthesis of IPrCl: Gaillard S, Slawin AMZ, Bonura AT, Stevens ED, Nolan SP. Organometallics. 2009;29:394. Arduengo AJ, III, Krafczyk R, Schmutzler R, Craig HA, Goerlich JR, Marshall WJ, Unverzagt M. Tetrahedron. 1999;55:14523.
- 7. Semba K, Fujihara T, Terao J, Tsuji Y. Chem. Eur. J. 2012;18:4179. doi: 10.1002/chem.201103612. Arduengo AJ, III, Davidson F, Dias HVR, Goerlich JR, Khasnis D, Marshall WJ, Prakasha TK. J. Am. Chem. Soc. 1997;119:12742. Semba K, Fujihara T, Xu T, Terao J, Tsuji Y. Adv. Synth. Catal. 2012;354:1542. Fujihara T, Xu T, Semba K, Terao J, Tsuji Y. Angew. Chem. Int. Ed. 2011;50:523. doi: 10.1002/anie.201006292. Akana JA, Bhattacharyya KX, Müller P, Sadighi JP. J. Am. Chem. Soc. 2007;129:7736. doi: 10.1021/ja0723784.
- 8.The crystal structure has been deposited at the Cambridge Crystallographic Data Centre, and the deposition number CCDC 1045615 has been allocated.
- 9.(a) Liu P, Montgomery J, Houk KN. J. Am. Chem. Soc. 2011;133:6956. doi: 10.1021/ja202007s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baxter RD, Montgomery J. J. Am. Chem. Soc. 2011;133:5728. doi: 10.1021/ja200867d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.