

Abstract
The mechanisms underlying preferential atrophy of the striatum in Huntington's disease (HD) are unknown. One hypothesis is that a set of gene products preferentially expressed in the striatum could determine the particular vulnerability of this brain region to mutant huntingtin (mHtt). Here, we studied the striatal protein µ-crystallin (Crym). Crym is the NADPH-dependent p38 cytosolic T3-binding protein (p38CTBP), a key regulator of thyroid hormone (TH) T3 (3,5,3′-triiodo-l-thyronine) transportation. It has been also recently identified as the enzyme that reduces the sulfur-containing cyclic ketimines, which are potential neurotransmitters. Here, we confirm the preferential expression of the Crym protein in the rodent and macaque striatum. Crym expression was found to be higher in the macaque caudate than in the putamen. Expression of Crym was reduced in the BACHD and Knock-in 140CAG mouse models of HD before onset of striatal atrophy. We show that overexpression of Crym in striatal medium-size spiny neurons using a lentiviral-based strategy in mice is neuroprotective against the neurotoxicity of an N-terminal fragment of mHtt in vivo. Thus, reduction of Crym expression in HD could render striatal neurons more susceptible to mHtt suggesting that Crym may be a key determinant of the vulnerability of the striatum. In addition our work points to Crym as a potential molecular link between striatal degeneration and the THs deregulation reported in HD patients.
Introduction
The protein μ-crystallin (Crym) has been discovered as a highly enriched lens component in Marsupials (1). Crym was identified as the NADPH-dependent p38 cytosolic T3-binding protein [p38CTBP, also known as THBP for thyroid hormone (TH)-binding protein], which is likely a key regulator of the TH T3 (3,5,3′-triiodo-l-thyronine) transportation in cytoplasm and nucleus, a mechanism through which it is involved in regulating TH-related gene expression (2). Indeed, in the nucleus T3 binds to thyroid hormone receptor-containing dimers, which bind to genomic thyroid responsive elements to regulate gene transcription (3). Although Crym expression in the brain is high (4), its neurobiological function is unknown. Several observations indirectly suggest that Crym could be a crucial determinant of neuronal cell death/survival. Crym has been reported to be an actor of macular degeneration in non-human primates (5). Mutations in the Crym gene lead to non-syndromic deafness, possibly by the incapacity of the resulting mutants to be involved in the potassium ion recycling system with Na, K-ATPase (6,7). Recently, mutations in CRYM have been tentatively related to amyotrophic lateral sclerosis (ALS) (8). Given that many species-specific crystallins with NADPH-binding properties are protective against oxidation in the lens (2), upregulated Crym in microglia cells in late stages of ALS may have a protective effect against neurodegeneration as do other NADPH requiring enzymes such as thioredoxin (9). Further supporting the view that Crym plays a central role in the central nervous system, Hallen and collaborators recently demonstrated using purification and MS/MS identification that the enzyme ketimine reductase (E.C. 1.5.1.25) is actually Crym (10). Thus, Crym catalyzes the reduction of sulfur-containing substrates known as cyclic ketimines that derive from sulfur-containing amino acids and may play a potential role as neurotransmitters (10). Interestingly, the enzymatic activity of ketimine reductase is regulated by T3 levels. Thus, Crym may play a key role at the interface between metabolism, neurotransmission and cell survival.
We recently focused our interest on Crym because of its preferential expression in the striatum and its possible involvement in the pathogenesis of Huntington's disease (HD) (11,12). HD is an autosomal dominant hereditary neurodegenerative disorder characterized by a preferential atrophy of the striatum associated with cognitive, psychiatric and motor symptoms (13). HD is caused by an abnormal CAG repeat expansion in the huntingtin (htt) gene (14). Mutant huntingtin (mHtt) is neurotoxic to neurons, especially toward neurons of the striatum, through both a gain of function and a loss of function (15). mHtt produces anomalies in energy production and oxidative stress (16), Ca2+ deregulation and excitotoxicity (17), abnormal trophic factor expression, trafficking and release (15), alterations of cytoskeleton-mediated transport machinery (18) and defects in signaling pathways involved in cell survival (19). However, none of these mechanisms can explain per se the preferential vulnerability of the striatum in HD. One hypothesis is that a set of factors, selectively expressed in the striatum may confer to striatal GABAergic projection neurons their particular susceptibility in HD (for a review 20). For example, the presence of the dopamine subtype 2 receptors (21–23), the small GTPase Rhes through its E3-ubiquitin ligase activity (24) and the RGS2 protein (25) in striatal neurons have been shown to increase their susceptibility to mHtt in HD. On the contrary, the reduced expression of several proteins with preferential expression in the striatum, and shown to possess neuroprotective properties may also increase the vulnerability of striatal neurons, such as adenosine type 2 receptors (A2A-R) (26), cannabinoid type 1 receptors (CB1-R) (27,28) and mitogen- and stress-activated kinase-1 (MSK-1) (29,30).
We previously found that the level of mRNA expression of Crym, which we identified for its enriched expression in the striatum and accumbens, is significantly reduced by 50% in the striatum of R6/2 mice (11), a widely used transgenic model of HD. Retrospective analysis of gene array datasets of the striatum of HD patients and transgenic mouse models of HD confirms that Crym mRNA levels are reduced in HD (31,32). Thus, it is possible that Crym may be involved in striatal vulnerability through two possible ways: reduction of Crym expression could be a causal event leading to death/dysfunction, pointing to Crym as a neuroprotective/pro-survival protein. On the opposite, decreased expression of Crym in HD could be a regulatory/compensatory mechanism, if the presence Crym (like Rhes for instance) contributes to the vulnerability of striatal neurons.
The objective of the present study was to investigate whether the levels of Crym expression in the mouse striatum could change the toxicity of mHtt. We first characterized the enrichment of the protein Crym in the striatum in mouse, rat and cynomolgus monkey. Secondly, we examined the expression of Crym mRNA and protein in genetic mouse models of HD in absence of major striatal degeneration (BACHD transgenic mice and Knock-in 140CAG mice). Finally, we used a lentiviral vector (LV) approach (33) to overexpress recombinant Crym in striatal neurons and directly determine whether the protein could modulate mHtt toxicity in vivo in mice.
Results
Crym is expressed preferentially in the striatum and localized in the cytosol
While the expression of Crym mRNA was shown to be highly enriched in the striatum and accumbens, the expression of the protein had been rarely assessed. For this reason, a chicken IgY antibody was raised against Crym (anti-Crym). Western blot analysis of striatal and cortical extracts (total protein extracts) prepared from adult rats, mice and a male adult non-human primate (macaca fascicularis) showed that endogenous Crym, appearing at an apparent molecular weight of ∼37 kDa was preferentially expressed in the striatum (caudate/putamen) when compared with cerebral cortex (Fig. 1A and B). In a non-human primate, Crym expression was markedly higher in the caudate than in the putamen (Fig. 1B and C).
Figure 1.

Western blot analysis of the expression of Crym protein in the mammalian brain. Representative western blots of total protein extracts prepared from adult mouse (Ms) and rat striatum (St) and cerebral cortex (Cx) (parietal) (A) and from macaque caudate (Cd), putamen (Put) and cerebral cortex (motor cortex) (B). (C) Semi-quantitative analysis of western blots of macaque brain extracts indicating preferential expression of Crym in caudate and putamen when compared with cerebral cortex. *P < 0.006; one-way ANOVA (P < 0.01) and post hoc Bonferroni test.
Experiments using differential centrifugation of rat brain samples followed by western blot analysis showed that Crym was mainly localized in the cytosol with low expression in the membrane fraction (Fig. 2). Similar results were obtained in mouse cerebral cortex and striatum (not shown). Thus, these results confirmed that Crym is a cytosolic protein preferentially expressed in the striatum when compared with cerebral cortex in the adult brain. In the primate striatum, levels of the protein are higher in caudate than in putamen, consistent with mRNA levels described for the macaque and human brain (see Allen Brain Atlas).
Figure 2.

Cytosolic localization of Crym in neurons. Western blot analysis of the cytosolic (cyt) and membrane (mb) fractions after differential centrifugation of homogenates prepared from the rat striatum and cerebral cortex (parietal). (A) Representative western blot. (B) Quantitative analysis of western blots. Results are expressed as mean ± S.E.M. #P < 0.004 ; *P < 0.003; one-way ANOVA (P < 0.001) and post hoc Bonferroni test.
Recombinant Crym protein is localized in the cytoplasm and nucleus
Little is known about the expression of Crym in cells, especially neurons. The cDNA for mouse Crym was cloned and a sequence encoding an hemagglutinin (HA) tag was added to its 3′ end. After transfection of HEK293T cell using this construct, expression of the recombinant protein was assessed by western blot using an anti-HA antibody. A single band migrating at ∼37 kDa was detected, which is consistent with the theoretical apparent molecular weight of full-length Crym and with the signal obtained using the anti-Crym antibody (Fig. 3A). Confocal microscopy analysis of HEK cells transfected with the Crym-HA construct using immunofluorescence detection of the HA tag indicated that Crym was located ubiquitously throughout the cell, including the nucleus and cell processes (Fig. 3B). Transfection of primary cultures of striatal neurons was also performed with a construct expressing Crym fused to the green fluorescent protein (GFP-Crym). Confocal microscopy analysis of the transfected neurons also indicated that GFP-Crym was located ubiquitously throughout the soma, the distal portions of neurites and within the nucleus (Fig. 3C).
Figure 3.

Recombinant Crym in cultured cells. HEK293T cells were transfected with a Crym-HA vector (A, B). Neurons were transfected with eGFP-Crym construct. (A) Western blot showing the detection of recombinant Crym-HA protein as a single band using either an anti-HA or an anti-Crym antibody. (B) Immunofluorescence detection of Crym-HA in HEK293T cells. (C) Expression of the eGFP-Crym protein in striatal neurons in primary culture. Images were obtained using confocal microscopy. Scale bars: 5 and 1 µm in left and right image, respectively; 10 µm in (C).
Finally, to study the subcellular localization of recombinant Crym in vivo, we generated an LV encoding Crym-HA (LV-Crym-HA) driven by the mouse PGK promoter. The LVs that we use almost exclusively transduce neurons in vivo and in vitro (21,34,35). Adult C57Bl/6 mice received intrastriatal injection of the LV-Crym-HA and 6 weeks later, brains were processed for histochemical evaluation. Immunohistochemical detection of the HA tag showed that the recombinant protein filled the entire soma and dendrites of neurons (Fig. 4). The nucleus was also stained. Confocal analysis of the striatum in mice injected with LV-Crym-HA confirmed that indeed, striatal neurons displayed high levels of Crym in the cytoplasm and dendrites branches and spines as well as in the nucleus (not shown).
Figure 4.

Recombinant Crym-HA expression in neurons after lentiviral infection of the mouse striatum. Photomicrographs showing the expression of Crym-HA 6 weeks after intrastriatal injection of LV-Crym-HA as revealed using anti-HA immunohistochemistry. (A) Low power magnification showing the expression in the anterior striatum. (B) Higher magnification showing densely labeled/compact neurons expressing Crym-HA. (C–E) Typical isolated striatal neurons expressing Crym-HA at distance from the core of the infection showing typical medium-size spiny neurons morphology. Note in (D) and (E) the dense labeling in the entire soma, and the presence of immunopositive dendritic spines (white arrowheads). Scale bars: 200 µm in (A), 20 µm in (B) and (C), 10 µm in (D) and (E).
Crym expression is reduced in HD models
Crym mRNA expression has been found to be reduced in HD models and patients (11,31,32). Here, we studied its expression in two additional genetic mouse models of HD. Analysis with qRT-polymerase chain reaction (PCR) showed that Crym mRNA levels were significantly reduced by 43% in the striatum of heterozygous BACHD mice at 6 months of age (mean mRNA expression ± S.E.M., wild-type littermates (n = 5), 5.96 ± 0.186; BACHD (n = 6), 3.38 ± 0.41; P < 0.0005, unpaired Student's t-test; not shown). Similarly, significant decrease in Crym mRNA levels was found in homozygous and to a lesser extent in heterozygous KI140CAG mice (13 months of age) when compared with wild-type mice (mean mRNA expression ± S.E.M., wild-type littermates (n = 8), 0.090 ± 0.009; heterozygous KI140CAG mice (n = 8), 0.080 ± 0.005; P < 0.05; homozygous KI140CAG mice (n = 8), 0.05 ± 0.005; P < 0.01. One-way ANOVA: P < 0.0001; F = 9.948; post hoc Bonferroni's multiple comparison test: wild-type littermates versus heterozygotes non-significant (n.s.); wild-type littermates versus homozygotes P < 0.001; heterozygotes versus homozygotes P < 0.05) (Fig. 5). Western blot analyses in KI140CAG mice showed reduced expression of Crym protein in the striatum when compared with age-matched wild-type littermates in homozygous mice with a trend to decrease in heterozygous mice (mean protein expression normalized with tubulin ± S.E.M., wild-type littermates (n = 3), 4.15 ± 0.34; heterozygotes (n = 8), 3.21 ± 0.32 n.s.; homozygotes (n = 8), 2.27 ± 0.13 ; P < 0.01. One-way ANOVA: P < 0.01, F = 11.13; post hoc Bonferroni's multiple comparison test: wild-type littermates versus heterozygotes n.s.; wild-type littermates versus homozygotes P < 0.01; heterozygotes versus homozygotes n.s.). Expression levels in the cortex were very low and could not be reliably quantified (not shown). Thus, these results show that Crym expression is down regulated in HD models at both the mRNA and protein levels.
Figure 5.

Crym expression in the KI140CAG mouse model of HD. Endogenous Crym expression was characterized by qRT-PCR (A) in the striatum and by western blot (B–D) in the striatum and cerebral cortex of knock in heterozygous (+/KI) and homozygous (KI/KI) 140CAG mice and wild-type littermates (+/+). Results are expressed as mean ± S.E.M. (n = 3–8). *P < 0.05; **P < 0.01; one-way ANOVA and post hoc Bonferroni test.
Crym overexpression reduces the toxicity of an N-terminal fragment of mHtt
We next examined whether overexpressing Crym could modify the toxicity of an N-terminal fragment of mHtt. We first designed and characterized an LV to increase Crym expression in vivo (LV-Crym-HA). For the quantitative analysis of Crym-HA mRNA expression when compared with endogenous Crym, LV-Crym-HA was mixed with an LV expressing GFP (LV-GFP) to allow dissection of the infected region under fluorescence binoculars. A lentivirus encoding β-galactosidase (LV-LacZ) was used as a control of viral load. The fluorescent region expressing GFP (and thus Crym-HA) was dissected out for mRNA expression analysis. Results of the quantitative RT-PCR (qRT-PCR) showed that LV-Crym-HA produced a 14.6-fold increase of Crym expression compared with control (LV-LacZ) [mean mRNA expression ± S.E.M.; LacZ (n = 4), 0.030 ± 0.005; Crym-HA (n = 4), 0.437 ± 0.049; P < 0.0002, unpaired Student's t-test] (Supplementary Material, Fig. S1). Thus, the LV-Crym-HA was efficacious to increase Crym expression in the mouse striatum.
To study the effect of Crym overexpression on the neurotoxicity of mHtt in striatal neurons in vivo, we used a lentiviral model of HD in which stereotaxic injection of an LV encoding a short fragment of mHtt (LV-Htt171-82Q) produces a progressive loco-regional cell dysfunction and degeneration characterized by mHtt- and ubiquitin-containing inclusions, loss of markers linked to neuronal integrity and astrogliosis within 6 weeks (33,36–38). This versatile model is particularly suitable to assess in vivo how mHtt toxicity can be modified by different factors that can be co-expressed with the mutant protein using injection of a mixture of LVs in mouse striatum (for a review 33).
We stereotaxically injected a mixed suspension of LV-Htt171-82Q and LV-Crym-HA into the mouse striatum. At 6 weeks post-infection, LV-Htt171-82Q produced a loss of NeuN and DARPP32 labeling in vicinity of the injection site, indicating overt neurodegeneration (Fig. 6). In separate experiments, we checked that co-infection with LV-LacZ (control) did not change the size of the lesion seen using NeuN immunohistochemistry when compared with LV-Htt171-82Q alone [data not shown; mean ± S.E.M.; NeuN, LV-Htt171-82Q alone (n = 8), 0.440 ± 0.043 mm3; LV-Htt171-82Q+LV-LacZ (n = 6), 0.526 ± 0.088 mm3; unpaired Student's t-test, n.s.]. Quantitative histological evaluation using NeuN immunohistochemistry showed that the striatal lesions produced by a mixture of LV-Htt171-82Q and LV-Crym-HA were significantly smaller than those produced by LV-Htt171-82Q mixed with LV-LacZ (Fig. 6) [mean NeuN-depleted volume ± S.E.M.; LV-Htt171-82Q+LV-Crym-HA (n = 10), 0.229 ± 0.033 mm3; LV-Htt171-82Q+LV-LacZ (n = 7), 0.442 ± 0.095 mm3; Student's t-test, P < 0.03]. In line with this, analysis of striatal degeneration using COX histochemistry also showed that the overexpression of Crym reduced the lesions produced by LV-Htt171-82Q (Fig. 6) [mean COX-depleted volume ± S.E.M.; LV-Htt171-82Q+LV-Crym-HA (n = 10), 0.236 ± 0.024 mm3; LV-Htt171-82Q+LacZ (n = 7), 0.360 ± 0.057 mm3; unpaired Student's t-test, P < 0.05]. In contrast, analysis of striatal degeneration using DARPP-32 immunohistochemistry did not show significant differences between groups (Fig. 6) [mean DARPP-32-depleted volume ± S.E.M.; LV-Htt171-82Q+LV-Crym-HA (n = 10), 0.450 ± 0.043 mm3; LV-Htt171-82Q + LV-LacZ (n = 7), 0.444 ± 0.023 mm3; unpaired Student's t-test, n.s.].
Figure 6.
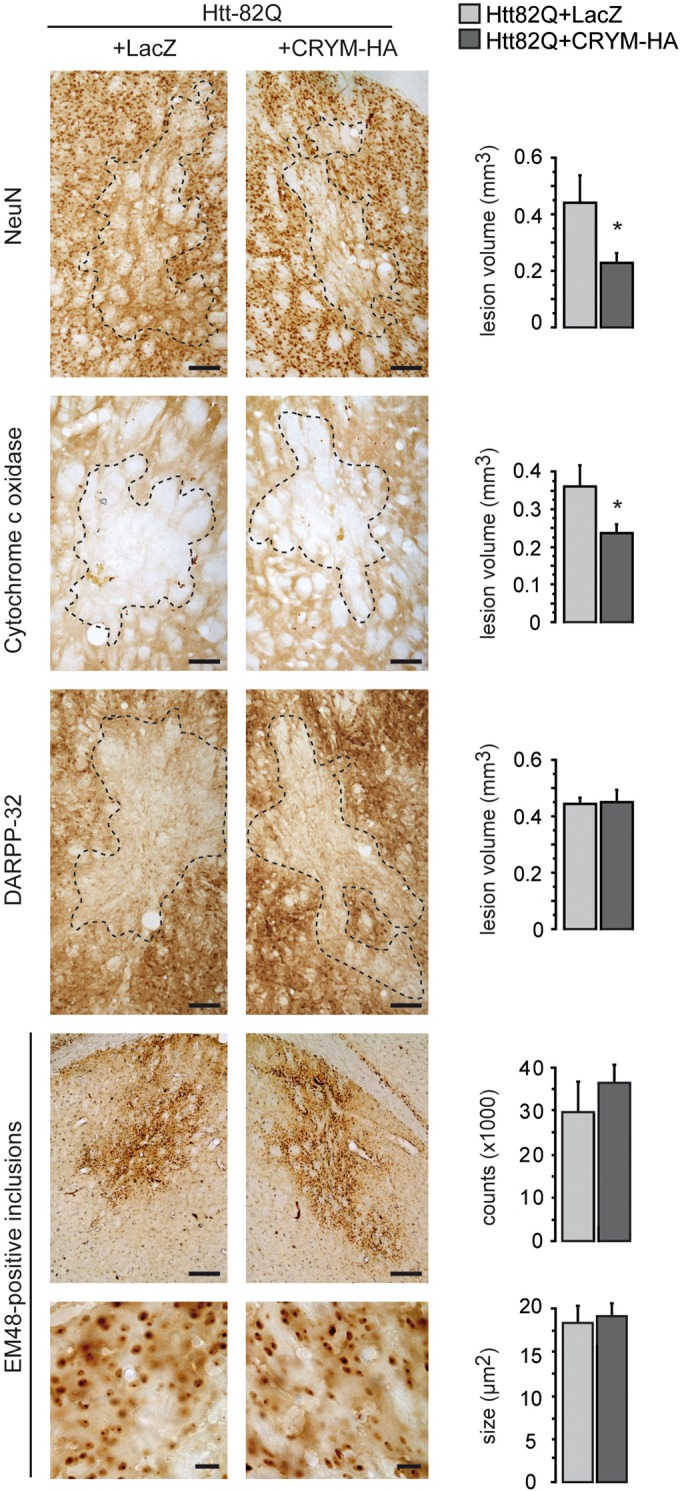
Effects of Crym-HA overexpresson in the striatum on the neurotoxicity of an N-terminal fragment of mHtt. Adult male mice received a bilateral intrastriatal injection of LV-Htt171-82Q mixed with either LV-LacZ (control) or an LV coding for Crym-HA (LV-Crym-HA). Six weeks after infection, brains were processed for histological evaluation using NeuN, DARPP-32 and Cytochrome oxidase (COX) histochemistry to assess the mHtt171-82Q toxicity. Left panel: typical coronal mouse brain sections where areas with depleted staining are delineated. Right panel: histograms representing quantitative determination of the volume of the striatum with depleted staining in the different groups and the number of ubiquitin-positive inclusions. Results are expressed as mean (n = 7–10/group) ± S.E.M. Scale bars: Neun, Cox, DARPP32, 100 µm; EM48 upper images, 200 µm; EM48 lower images, 20 µm. *P < 0.05; unpaired Student's t-test.
We also assessed the number and size of Em48-positive nuclear inclusions in mice (Fig. 6) injected with a mixture of LV-Htt171-82Q and LV-Crym-HA or a mixture of LV-Htt171-82Q and LV-LacZ. Microscopic quantitative analysis of the sections processed by Em48 antibody-immunohistochemistry showed that overexpression of Crym has a tendency to increase the number of Em48-containing inclusions [mean inclusion number ± S.E.M.; LV-Htt171-82Q+LV-Crym-HA (n = 8), 36 306 ± 4091; LV-Htt171-82Q+LV-LacZ (n = 6), 29 525 ± 7112; Student's t-test, n.s.]. The mean size of Em48-positive inclusions did not show differences between groups [inclusion size, LV-Htt171-82Q+ LV-Crym-HA (n = 8), 19.09 ± 1.48 µm2; LV-Htt171-82Q+LV-LacZ (n = 6), 18.39 ± 1.86 µm2; unpaired Student's t-test, n.s.]. The mean density of Em48-positive inclusions per millimeter squared shows a higher tendency in the LV-Crym-HA injected mice [density inclusions/mm², LV-Htt171-82Q+ LV-Crym-HA (n = 8), 935.42 ± 62.48 µm2; LV-Htt171-82Q+LV-LacZ (n = 6), 831.63 ± 48.15 µm2; unpaired Student's t-test, n.s.]. The number and size of ubiquitin-positive nuclear inclusions were not different from what was observed with Em48 staining (data not shown).
Discussion
The mechanisms underlying the vulnerability of the striatum in HD remain unclear. In the present study, we tested the hypothesis that the protein Crym may play a role in the susceptibility of adult striatal neurons to mHtt.
Our results show that expression of Crym mRNA is reduced in KI140 mice and BACHD mice. This is in agreement with the down regulation of its mRNA we and others previously found in different HD mouse models and HD patients (11,31,32,39). In addition, we also show that the levels of the protein Crym is markedly reduced in KI140 mice, indicating that down regulation that occurs at the mRNA levels leads to loss of the protein, likely impairing its function in the striatum. We reasoned that this reduction in Crym expression could participate in the striatal vulnerability to mHtt toxicity. Results of our study indicated that Crym overexpression could reduce the susceptibility of striatal neurons in vivo toward the toxicity of an N-terminal fragment of human mHtt. The effect of the continuous expression of mHtt in lentiviral models has been characterized by the loss of expression of three markers, DARPP-32, NeuN and COX (33). The loss of NeuN immunoreactivity and the loss of COX histochemical labeling are associated with actual neuronal degeneration and dysfunction, as assessed using markers of cell death (40) and stereological cell counts (34). Thus, the present results indicate that overexpressing Crym reduces cell death induced by the mHtt fragment.
The number and size of Em48- and ubiquitin-positive inclusions in the striatum, a neuropathological hallmark of HD, were not significantly changed by the overexpression of Crym. This suggests that the neuroprotective effects produced by the increased expression of Crym in striatal neurons are likely independent from a direct effect on mHtt expression or aggregation. If Crym had reduced the levels of cellular mHtt, we should have observed a lower number of inclusions. Neuroprotective effects can be produced without major change in inclusions and aggregates size or numbers. For example, expression of the Ip and Fp subunits of mitochondrial complex II protects striatal neurons against mHtt in vitro and in vivo, while the number and size of inclusions remain unchanged (36).
The neuroprotective effects of Crym overexpression using a lentiviral approach in our mouse model are unlikely linked to methodological bias. Indeed, we have tested different transgenes using the very same methods. Recently, we studied the striatal marker Capucin (Syndig1l) whose expression is markedly reduced in HD patients and mouse models (38). Overexpression of Capucin, using a similar LV-mediated approach produced no significant change in the toxicity of LV-Htt171-82Q in the striatum. Using similar co-infection approaches in rats, we have also identified the neuroprotective roles of some proteins against mHtt fragment, including the transcription factor CA150 (41), chaperone proteins (42), signaling proteins of the JNK pathway (43), the MSK-1 (30) and the subunits of mitochondrial complex II (36).
The mechanisms underlying reduced expression of Crym mRNA (and, as shown here, of Crym protein as well) are unknown. The down regulation of striatal transcripts in HD may result from the functional disruption of striatum-specific transcriptional activators such as Bcl11b (44). Depending on the transcript, this phenomenon can be protective, detrimental or have no effect on striatal neurons (20). The down regulated expression of several proteins enriched in the striatum and involved in different neuronal functions such as neurotransmission, intracellular signaling and transcription have been reported to play a role in striatal vulnerability in HD. For example, compelling experimental evidence shows that the loss of A2A-R, CB1-R, MSK-1, STEP61, Bcl11b and Fox1b likely renders striatal cells more prone to degeneration in HD models (for a review 20,45,).
The mechanisms of whereby Crym may reduce the vulnerability of striatal neurons to mHtt are not yet elucidated. However, this may have to be associated with the possible alteration in the levels of the THs in HD patients, given that THs regulate multiple cellular events, including energy expenditure and cellular differentiation (46). In their pioneering studies, Aziz and collaborators showed that levels of T3, which is a ligand of Crym, are increased in a small cohort of HD patients when compared with age-matched controls (47). In a larger cohort, no significant increase in T3 was shown but the hormone levels were found to be negatively correlated with severity of clinical impairments, supporting the hypothesis that the thyrotropic axis is altered in HD patients (48). In line with this, a functional interaction of mHtt with thyroid hormone receptor (TR) and the nuclear co-repressor NCor, which represses TR-mediated transcription, has been reported, suggesting that mHtt may lead to abnormal transcription of TR targets in the HD striatum (49). In the striatum, such mechanism would be exacerbated by the loss of Crym, which is normally expressed at high levels, likely facilitating the transport of T3 from the cytoplasm into the nucleus where it interacts with TR. Further studies are awaited to experimentally address these hypothetical T3-dependent mechanisms.
In summary, the present results indicate that the expression of the protein Crym in striatal neurons may be one of the important molecular determinants of the preferential vulnerability of the striatum in HD and further links HD pathogenesis with possible alterations of TH-mediated regulation of transcription in the striatum.
Materials and Methods
Animals
Mice were housed in a temperature-controlled room maintained on a 12 h light–dark cycle. Food and water were available ad libitum. All animal studies were conducted according to the French regulation (EU Directive 86/609—French Act Rural Code R 214-87 to 131). The animal facility was approved by veterinarian inspectors (authorization n°A 92-032-02) and complies with Standards for Humane Care and Use of Laboratory Animals of the Office of Laboratory Animal Welfare (OLAW—n°#A5826-01). All procedures received approval from the ethical committee. Adult male C57BL/6J mice (25 g each; Charles River, Saint Germain sur l'Arbresle, France) were used for lentiviral infections and preparation of brain extracts for biochemical studies. For biochemical studies on endogenous Crym, brains of male Sprague-Dawleys rats (350 g) were also used. We also had access to coronal brain slices from a control adult macaque fascicularis.
For the study of endogenous Crym mRNA levels in the context of HD, we used the transgenic mouse model of HD generated and maintained in the FvB inbred background, the BACHD mice, that express full-length human mHtt from its own regulatory elements on a 240 kb BAC, which contains the intact 170 kb human htt locus plus ∼20 kb of 5′ flanking genomic sequence and 50 kb of 3′ (50). We used 6-month-old male BACHD mice for the present study, a time point where there is no detectable striatal atrophy (51).
We also studied 13-month-old knock-in mice expressing chimeric mouse/human exon 1 containing 140 CAG repeats inserted in the murine Htt gene (KI140) and their littermate controls. KI140 colony was maintained by breeding heterozygotes KI140 males and females (52). Mice were N3 (B6) on a 129 Sv × C57BL/6 J background. The resulting different genotypes were used for the present study. At this age, homozygous and heterozygous KI140 mice showed no major striatal atrophy, although these animals had motor deficits (Rotarod, CAtWalk and open field) (data not shown).
Genotyping was determined from PCR of tail snips taken at 10–15 days of age for BACHD and KI140 mice.
Lentiviral vector construction, production and infection
DNA sequences coding for GFP and mouse Crym were cloned into the SIN-W-PGK LV to generate LV-GFP and LV-Crym, respectively (40). The LVs expressing an mHtt fragment (LV-Htt171 82Q) or β-galactosidase (LV-LacZ) have been described previously (34,37). Viral particles were produced as described elsewhere (53). The particle content of the viral batches was determined by ELISA for the p24 antigen (Gentaur, Paris, France). LV-Htt171-82Q was used at a concentration of 150 ng/µl of p24, LV-Crym and LV-LacZ at a concentration of 100 ng/µl of p24. In experiments performed for PCR analysis, LV-GFP was mixed with LV-Crym or LV-LacZ at a concentration of 50 ng/µl of p24. After being anesthetized (ketamine/xylazine), mice received a total volume of 2 µl of lentiviral suspension into the mouse striatum as previously reported (37,38), using the following stereotaxic coordinates: 1.0 mm anterior and 2.0 mm lateral to the bregma, at a depth of 2.7 mm from the dura, with the tooth bar set at 0.0 mm.
Histological and cytological analyses
Animal studies
After deep anesthesia by intraperitoneal injection of a sodium pentobarbital solution (50 µg/g of body weight), mice were transcardially perfused with 100 ml of phosphate buffer containing 4% paraformaldehyde at 8 ml/min. The brains were removed, post-fixed overnight in the same solution, then cryoprotected by immersion in a 15% sucrose solution for 24 h followed by immersion in a 30% sucrose solution for another 24 h. Free-floating 40 µm-thick serial coronal sections throughout the striatum were collected using a freezing sliding microtome (SM2400; Leica Microsystems, Wetzlar, Germany).
Immunohistochemistry
For immunohistochemistry, sections were treated with 0.3% hydrogen peroxide for 20 min, washed three times in phosphate-buffered saline (PBS), blocked in PBS containing 4.5% normal goat serum for 1 h, then incubated 48 h at 4°C in PBS containing 3% normal goat serum and one of the following antibodies: rabbit anti-DARPP 32 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1 : 1000), mouse anti-NeuN (Millipore, Molsheim, France; 1 : 1000), mouse anti-Em48 (Chemicon, MAB5374, Temecula, CA, USA; 1 : 1000), rabbit anti-Ubiquitin (Wako Chemicals, Neuss, Germany; 1 : 1000) or mouse anti-HA (Covance, Princeton, NJ, USA; 1 : 1000). Sections were rinsed three times in PBS before incubation with the appropriate anti-IgG biotinylated antibody (Vector Laboratories, Burlingame, CA, USA) at a 1 : 1000 dilution for 1 h. Staining was visualized by the addition of avidin-biotinylated peroxidase and incubation with DAB substrate (Vector Laboratories) for 30 s to 1 min. Stained sections were mounted on microscopic slides.
Quantitative histological evaluation
The area of the striatal lesions resulting from LV-Htt171-82Q infection was delineated manually by identifying the border of the lesion (loss of DARPP-32 and NeuN immunolabeling). Lesion area was delineated using ×5 and ×10 objectives. Depending on the antero-posterior extension of the lesions, 3–8 coronal sections were analyzed for each mouse. Observation of sections and calculation of the surface of lesioned area were performed using a Leica DM6000 equipped with a motorized stage and an automated image acquisition and analysis system (Mercator software, Explora Nova, La Rochelle, France). The volume of the striatal lesion (V) was determined using the Cavalieri method (34,36,38). The number of ubiquitin-positive inclusions was quantified as previously described (34,36,38) with the following modifications: the inter-section distance was 320 µm (i.e. one in every seven sections was used) and observations were performed using a ×10 objective on an Axioplan 2 Imaging microscope (Carl Zeiss, Le Pecq, France) equipped with a motorized stage and an automated image acquisition and analysis system (Mercator software, Explora Nova). With this set-up, objects with an apparent cross-sectional area >3 µm2 (i.e. diameter ) could be reliably detected.
Real-time quantitative PCR
Adult mice were deeply anesthetized by intraperitoneal injection of a sodium pentobarbital solution (50 µg/g of body weight) before decapitation. The brains were immediately removed and positioned in a coronal brain matrix (Ted Pella, Redding, CA, USA).
For the quantification of overexpressed Crym mRNA levels, mice were infected with a mixture of LV-Crym-HA and LV-GFP. Injection of LV-LacZ was used as controls for viral load. The striatal region displaying fluorescence was dissected out using a circular punch (1.5 mm diameter) from 1 mm-thick fresh coronal brain sections visualized under a fluorescence binocular (Leica). Total RNA extraction and real-time qRT-PCR were performed as previously described (38,54), using the following primer sequences for Crym mRNA Crym-U CTATGAGGGCCACAGCAACA and Crym-L ATGACCGCCAGCAGGGAG. Primers recognized sequence in mouse (endogenous) Crym and recombinant Crym-HA after viral infection.
Cell studies
The primary culture of striatal neurons has been performed as previously described (21). These neurons were electroporated with the mouse striatal neuron Nucleofector® kit according to the supplier's manual (Amaxa, Biosystem, Köln, Germany) for detection of a chimeric eGFP-Crym construct.
Human embryonic kidney 293T (HEK293T) cells were grown at 37°C in 5% CO2 in Dulbeco's modified Eagle's medium supplemented with 10% bovine calf serum, 1% l-glutamine and antibiotics (50 units/ml penicillin and 50 μg/ml streptomycin). For the study of recombinant Crym, cultured cells were seeded on glass coverslips, and transfected with recombinant Crym-HA vector or equivalent amount of empty vector for HEK293T cells, and 48 h later fixed with cold-ice methanol and EGTA. Cells were rinsed with TBST, and blocked with 2% bovine serum albumin (BSA), 0.1% Tween-20 in TBS. Primary polyclonal antibody rabbit anti-HA (H6908, Sigma, St Louis, MO, USA; 1 : 800) was used at 2 μg/ml in 2% BSA, 0.1% Triton X-100, 0.1% azide in PBS and incubated for 1 h at room temperature. Similar attempt to detect native recombinant Crym protein using the affinity purified IgY chicken anti-Crym (Michel—see below for western blot) were also used unsuccessfully. Cells were washed with 0.05% Tween-20 in PBS and incubated with the appropriate fluorescent secondary antibody for 1 h. Nuclei were counterstained with Hoechst (DAPI). Coverslips were mounted in FluorSafe and examined with a Leika microscope fitted with the appropriate fluorescence filters, Retiga camera and Morphostrider imaging software prior to analysis in Confocal Zeiss LSM for eGFP-Crym in primary cultures and the SPE TCS Leica confocal with Leica LAS AF interface for the HEK experiments.
Biochemical analysis
Cell preparation
Cells were harvested 48 h after transfection and lyzed in modified RIPA buffer: 50 mm Tris, pH 8.0, 50 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, 1% NP40 and protease inhibitor cocktail (Roche). Cell lysates were centrifuged at 15 000 g for 20 min at 4°C.
Brain tissue preparation
Mice and rats were euthanized by rapid decapitation and brains were removed, blocked and cut into 1 mm-thick coronal slices using brain matrices (Ted Pella, Redding, CA, USA). On one coronal slice (+1 mm from bregma), tissue punches were taken from the striatum using a tissue corer (1.5 mm in diameter) and the cerebral cortex was dissected out using scalpel blades under binoculars.
Cortical and striatal (caudate and putamen) tissue samples were prepared from a control non-human primate (macaca fascicularis, #2055, male ∼7 years old). Briefly, the animal received a lethal dose of pentobarbital, brain was rapidly removed from the skull and cut into 5 mm-thick coronal slabs. Slabs were put on ice-cooled Petri dish and the pre-commissural putamen, caudate and a piece of cerebral cortex (gray matter ribbon) in the somato-sensori cortex were dissected out using a scalpel blade.
For determination of total level of Crym expression in brain tissue, punches were lysed in 50 mm Tris, pH 8, 150 mm NaCl, 1 mm EDTA, 0.5% Triton X-100 and 1% NP40, with protease inhibitor cocktail (Complete, Roche) and phosphatase inhibitor cocktail 2 (Sigma) using glass–glass pestle potters.
Differential centrifugation experiments were performed to determine the cytosolic levels of Crym in comparison with cell membranes, tissue punches were homogenized in solution A containing 50 mm Tris, pH 7.4, 100 mm NaCl, 0.25 m sucrose with protease inhibitor cocktail (Complete, Roche) using a Teflon pestle and Thomas potter. After 10 min centrifugation at 10 000 g, the supernatant S2 was collected and centrifuged at 135 000 g, 30 min 4°C. The corresponding supernatant (S0) contained the cytosolic fraction, which was mixed in Laemli. The pellet P2 (containing membranes) was re-suspended to further remove cytosolic contaminants, placed on a 1.7 m sucrose cushion and centrifuged at 135 000 g for 30 min at 4°C. The material (membranes) blocked at the 0.25/1.7 m sucrose interface was collected and re-suspended in solution A, then centrifuged again 30 min at 135 000 g, 4°C to remove sucrose; pellet was re-suspended in Laemli before electrophoresis.
Western blotting
Total protein concentrations were determined using the BCA kit (Pierce). Equal amounts of total protein extract were subjected to SDS–PAGE in 4–12% Bis–Tris gels (NuPAGE® Novex Bis-tris midi gel 15 wells, Invitrogen) and transferred to nitrocellulose membranes. Blocked membranes (5% milk in TBS–0.1% Tween-20) were incubated with primary antibodies: Crym [1 : 500, chicken IgY directed against AVGASRPDWRELDDE and affinity purified (AgroBio)], actin (1 : 2000, mouse, Sigma), HA (1 : 3000, mouse, Covance), Dopamine- and cAMP-regulated PhosphoProtein 32 kDa (DARPP-32, 1 : 3000, rabbit, Cell Signaling), α-tubulin (1 : 3000, mouse, Sigma); and washed three times with TBS–0.1% Tween-20 (TBST) for 10 min. Membranes were then labeled with secondary IgG-HRP antibodies raised against each corresponding primary antibody. After three washes with TBST, the membranes were incubated with ECL chimioluminescent reagent (Clarity Western ECL substrate; Bio-Rad or Immun-Star WesternC kit) according to the instructions of the supplier. Peroxydase activity was detected with camera system Fusion TX7 (Fisher Scientific). Normalization was done by densitometry analysis with Bio1D software.
Supplementary Material
Funding
This work was supported by the ‘Commissariat à l'Energie Atomique et aux Energies Alternatives’ (CEA) and ‘Centre National de la Recherche Scientifique’ (CNRS). L.G. was supported by the Neuropôle de Recherche Francilien and the Fondation pour la Recherche Médicale. L.F. was supported by the French Research Ministry. The research leading to these results has received funding from the European Community's Seventh Framework Program FP7/2007-2013 under grant agreement no. HEALTH-F5-2008-222925. We also received financial support from the IMAGEN European integrated project (EUR617037286). Funding to pay the Open Access publication charges for this article was provided by the Centre National de la Recherche Scientifique (CNRS).
Supplementary Material
Acknowledgements
We thank Dr William Yang for his help to set up the BACHD mice colony. We are also grateful to Scott Zeitlin and Sandrine Humbert for their help to start the KI140CAG mice colony.
Conflict of Interest statement. None declared.
References
- 1.Wistow G., Kim H. Lens protein expression in mammals: taxon-specificity and the recruitment of crystallins. J. Mol. Evol. 1991;32:262–269. doi: 10.1007/BF02342749. [DOI] [PubMed] [Google Scholar]
- 2.Vie M.P., Evrard C., Osty J., Breton-Gilet A., Blanchet P., Pomerance M., Rouget P., Francon J., Blondeau J.P. Purification, molecular cloning, and functional expression of the human nicodinamide-adenine dinucleotide phosphate-regulated thyroid hormone-binding protein. Mol. Endocrinol. 1997;11:1728–1736. doi: 10.1210/mend.11.11.9915. [DOI] [PubMed] [Google Scholar]
- 3.Ortiga-Carvalho T.M., Sidhaye A.R., Wondisford F.E. Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat. Rev. Endocrinol. 2014;10:582–591. doi: 10.1038/nrendo.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki S., Suzuki N., Mori J., Oshima A., Usami S., Hashizume K. Micro-crystallin as an intracellular 3,5,3′-triiodothyronine holder in vivo. Mol. Endocrinol. 2007;21:885–894. doi: 10.1210/me.2006-0403. [DOI] [PubMed] [Google Scholar]
- 5.Umeda S., Suzuki M.T., Okamoto H., Ono F., Mizota A., Terao K., Yoshikawa Y., Tanaka Y., Iwata T. Molecular composition of drusen and possible involvement of anti-retinal autoimmunity in two different forms of macular degeneration in cynomolgus monkey (Macaca fascicularis) FASEB J. 2005;19:1683–1685. doi: 10.1096/fj.04-3525fje. [DOI] [PubMed] [Google Scholar]
- 6.Abe S., Katagiri T., Saito-Hisaminato A., Usami S., Inoue Y., Tsunoda T., Nakamura Y. Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. Am. J. Hum. Genet. 2003;72:73–82. doi: 10.1086/345398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oshima A., Suzuki S., Takumi Y., Hashizume K., Abe S., Usami S. CRYM mutations cause deafness through thyroid hormone binding properties in the fibrocytes of the cochlea. J. Med. Genet. 2006;43:e25. doi: 10.1136/jmg.2005.034397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daoud H., Valdmanis P.N., Gros-Louis F., Belzil V., Spiegelman D., Henrion E., Diallo O., Desjarlais A., Gauthier J., Camu W., et al. Resequencing of 29 candidate genes in patients with familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011;68:587–593. doi: 10.1001/archneurol.2010.351. [DOI] [PubMed] [Google Scholar]
- 9.Fukada Y., Yasui K., Kitayama M., Doi K., Nakano T., Watanabe Y., Nakashima K. Gene expression analysis of the murine model of amyotrophic lateral sclerosis: studies of the Leu126delTT mutation in SOD1. Brain Res. 2007;1160:1–10. doi: 10.1016/j.brainres.2007.05.044. [DOI] [PubMed] [Google Scholar]
- 10.Hallen A., Cooper A.J., Jamie J.F., Haynes P.A., Willows R.D. Mammalian forebrain ketimine reductase identified as mu-crystallin: potential regulation by thyroid hormones. J. Neurochem. 2011;118:379–387. doi: 10.1111/j.1471-4159.2011.07220.x. [DOI] [PubMed] [Google Scholar]
- 11.Brochier C., Gaillard M.C., Diguet E., Caudy N., Dossat C., Segurens B., Wincker P., Roze E., Caboche J., Hantraye P., et al. Quantitative gene expression profiling of mouse brain regions reveals differential transcripts conserved in human and affected in disease models. Physiol. Genomics. 2008;33:170–179. doi: 10.1152/physiolgenomics.00125.2007. [DOI] [PubMed] [Google Scholar]
- 12.de Chaldee M., Gaillard M.C., Bizat N., Buhler J.M., Manzoni O., Bockaert J., Hantraye P., Brouillet E., Elalouf J.M. Quantitative assessment of transcriptome differences between brain territories. Genome. Res. 2003;13:1646–1653. doi: 10.1101/gr.1173403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker F.O. Huntington's disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 14.The-Huntington's-Disease-Collaborative-Research-Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 15.Zuccato C., Cattaneo E. Huntington's Disease. Handb. Exp. Pharmacol. 2014;220:357–409. doi: 10.1007/978-3-642-45106-5_14. [DOI] [PubMed] [Google Scholar]
- 16.Damiano M., Galvan L., Deglon N., Brouillet E. Mitochondria in Huntington's disease. Biochim. Biophys. Acta. 2010;1802:52–61. doi: 10.1016/j.bbadis.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 17.Fan M.M., Raymond L.A. N-Methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Prog. Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Hinckelmann M.V., Zala D., Saudou F. Releasing the brake: restoring fast axonal transport in neurodegenerative disorders. Trends Cell Biol. 2013;23:634–643. doi: 10.1016/j.tcb.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Roze E., Saudou F., Caboche J. Pathophysiology of Huntington's disease: From huntingtin functions to potential treatments. Curr. Opin. Neurol. 2008;21:497–503. doi: 10.1097/WCO.0b013e328304b692. [DOI] [PubMed] [Google Scholar]
- 20.Francelle L., Galvan L., Brouillet E. Possible involvement of self-defense mechanisms in the preferential vulnerability of the striatum in Huntington's disease. Front. Cell. Neurosci. 2014;8:295. doi: 10.3389/fncel.2014.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benchoua A., Trioulier Y., Diguet E., Malgorn C., Gaillard M.C., Dufour N., Elalouf J.M., Krajewski S., Hantraye P., Deglon N., et al. Dopamine determines the vulnerability of striatal neurons to the N-terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II. Hum. Mol. Genet. 2008;17:1446–1456. doi: 10.1093/hmg/ddn033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charvin D., Roze E., Perrin V., Deyts C., Betuing S., Pages C., Regulier E., Luthi-Carter R., Brouillet E., Deglon N., et al. Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo. Neurobiol. Dis. 2008;29:22–29. doi: 10.1016/j.nbd.2007.07.028. [DOI] [PubMed] [Google Scholar]
- 23.Charvin D., Vanhoutte P., Pages C., Borrelli E., Caboche J. Unraveling a role for dopamine in Huntington's disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc. Natl Acad. Sci. USA. 2005;102:12218–12223. doi: 10.1073/pnas.0502698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramaniam S., Sixt K.M., Barrow R., Snyder S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science (New York, NY) 2009;324:1327–1330. doi: 10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seredenina T., Gokce O., Luthi-Carter R. Decreased striatal RGS2 expression is neuroprotective in Huntington's disease (HD) and exemplifies a compensatory aspect of HD-induced gene regulation. PLoS ONE. 2011;6:e22231. doi: 10.1371/journal.pone.0022231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blum D., Hourez R., Galas M.C., Popoli P., Schiffmann S.N. Adenosine receptors and Huntington's disease: implications for pathogenesis and therapeutics. Lancet Neurol. 2003;2:366–374. doi: 10.1016/s1474-4422(03)00411-3. [DOI] [PubMed] [Google Scholar]
- 27.Mievis S., Blum D., Ledent C. Worsening of Huntington disease phenotype in CB1 receptor knockout mice. Neurobiol. Dis. 2011;42:524–529. doi: 10.1016/j.nbd.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 28.Chiarlone A., Bellocchio L., Blazquez C., Resel E., Soria-Gomez E., Cannich A., Ferrero J.J., Sagredo O., Benito C., Romero J., et al. A restricted population of CB1 cannabinoid receptors with neuroprotective activity. Proc. Natl Acad. Sci. USA. 2014;111:8257–8262. doi: 10.1073/pnas.1400988111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roze E., Betuing S., Deyts C., Marcon E., Brami-Cherrier K., Pages C., Humbert S., Merienne K., Caboche J. Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. 2008;22:1083–1093. doi: 10.1096/fj.07-9814. [DOI] [PubMed] [Google Scholar]
- 30.Martin E., Betuing S., Pages C., Cambon K., Auregan G., Deglon N., Roze E., Caboche J. Mitogen- and stress-activated protein kinase 1-induced neuroprotection in Huntington's disease: role on chromatin remodeling at the PGC-1-alpha promoter. Hum. Mol. Genet. 2011;20:2422–2434. doi: 10.1093/hmg/ddr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodges A., Strand A.D., Aragaki A.K., Kuhn A., Sengstag T., Hughes G., Elliston L.A., Hartog C., Goldstein D.R., Thu D., et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum. Mol. Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn A., Goldstein D.R., Hodges A., Strand A.D., Sengstag T., Kooperberg C., Becanovic K., Pouladi M.A., Sathasivam K., Cha J.H., et al. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- 33.Ruiz M., Deglon N. Viral-mediated overexpression of mutant huntingtin to model HD in various species. Neurobiol. Dis. 2012;48:202–211. doi: 10.1016/j.nbd.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 34.Diguet E., Petit F., Escartin C., Cambon K., Bizat N., Dufour N., Hantraye P., Deglon N., Brouillet E. Normal aging modulates the neurotoxicity of mutant huntingtin. PLoS ONE. 2009;4:e4637. doi: 10.1371/journal.pone.0004637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Regulier E., Pereira de Almeida L., Sommer B., Aebischer P., Deglon N. Dose-dependent neuroprotective effect of ciliary neurotrophic factor delivered via tetracycline-regulated lentiviral vectors in the quinolinic acid rat model of Huntington's disease. Hum. Gene. Ther. 2002;13:1981–1990. doi: 10.1089/10430340260355383. [DOI] [PubMed] [Google Scholar]
- 36.Damiano M., Diguet E., Malgorn C., D'Aurelio M., Galvan L., Petit F., Benhaim L., Guillermier M., Houitte D., Dufour N., et al. A role of mitochondrial complex II defects in genetic models of Huntington's disease expressing N-terminal fragments of mutant huntingtin. Hum. Mol. Genet. 2013;22:3869–3882. doi: 10.1093/hmg/ddt242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faideau M., Kim J., Cormier K., Gilmore R., Welch M., Auregan G., Dufour N., Guillermier M., Brouillet E., Hantraye P., et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum. Mol. Genet. 2010;19:3053–3067. doi: 10.1093/hmg/ddq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galvan L., Lepejova N., Gaillard M.C., Malgorn C., Guillermier M., Houitte D., Bonvento G., Petit F., Dufour N., Hery P., et al. Capucin does not modify the toxicity of a mutant Huntingtin fragment in vivo. Neurobiol. Aging. 2012;33:1845–1846. doi: 10.1016/j.neurobiolaging.2012.01.009. e1845. [DOI] [PubMed] [Google Scholar]
- 39.Desplats P.A., Kass K.E., Gilmartin T., Stanwood G.D., Woodward E.L., Head S.R., Sutcliffe J.G., Thomas E.A. Selective deficits in the expression of striatal-enriched mRNAs in Huntington's disease. J. Neurochem. 2006;96:743–757. doi: 10.1111/j.1471-4159.2005.03588.x. [DOI] [PubMed] [Google Scholar]
- 40.de Almeida L.P., Ross C.A., Zala D., Aebischer P., Deglon N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J. Neurosci. 2002;22:3473–3483. doi: 10.1523/JNEUROSCI.22-09-03473.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arango M., Holbert S., Zala D., Brouillet E., Pearson J., Regulier E., Thakur A.K., Aebischer P., Wetzel R., Deglon N., et al. CA150 expression delays striatal cell death in overexpression and knock-in conditions for mutant huntingtin neurotoxicity. J. Neurosci. 2006;26:4649–4659. doi: 10.1523/JNEUROSCI.5409-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perrin V., Regulier E., Abbas-Terki T., Hassig R., Brouillet E., Aebischer P., Luthi-Carter R., Deglon N. Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington's disease. Mol. Ther. 2007;15:903–911. doi: 10.1038/mt.sj.6300141. [DOI] [PubMed] [Google Scholar]
- 43.Perrin V., Dufour N., Raoul C., Hassig R., Brouillet E., Aebischer P., Luthi-Carter R., Deglon N. Implication of the JNK pathway in a rat model of Huntington's disease. Exp. Neurol. 2009;215:191–200. doi: 10.1016/j.expneurol.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Desplats P.A., Lambert J.R., Thomas E.A. Functional roles for the striatal-enriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington's disease. Neurobiol. Dis. 2008;31:298–308. doi: 10.1016/j.nbd.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seredenina T., Luthi-Carter R. What have we learned from gene expression profiles in Huntington's disease? Neurobiol. Dis. 2012;45:83–98. doi: 10.1016/j.nbd.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 46.Zhang C.Y., Kim S., Harney J.W., Larsen P.R. Further characterization of thyroid hormone response elements in the human type 1 iodothyronine deiodinase gene. Endocrinology. 1998;139:1156–1163. doi: 10.1210/endo.139.3.5849. [DOI] [PubMed] [Google Scholar]
- 47.Aziz N.A., Pijl H., Frolich M., Roelfsema F., Roos R.A. Altered thyrotropic and lactotropic axes regulation in Huntington's disease. Clin. Endocrinol. 2010;73:540–545. doi: 10.1111/j.1365-2265.2010.03836.x. [DOI] [PubMed] [Google Scholar]
- 48.Saleh N., Moutereau S., Durr A., Krystkowiak P., Azulay J.P., Tranchant C., Broussolle E., Morin F., Bachoud-Levi A.C., Maison P. Neuroendocrine disturbances in Huntington's disease. PLoS ONE. 2009;4:e4962. doi: 10.1371/journal.pone.0004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yohrling G.J., Farrell L.A., Hollenberg A.N., Cha J.H. Mutant huntingtin increases nuclear corepressor function and enhances ligand-dependent nuclear hormone receptor activation. Mol. Cell. Neurosci. 2003;23:28–38. doi: 10.1016/s1044-7431(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 50.Gray M., Shirasaki D.I., Cepeda C., Andre V.M., Wilburn B., Lu X.H., Tao J., Yamazaki I., Li S.H., Sun Y.E., et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boussicault L., Herard A.S., Calingasan N., Petit F., Malgorn C., Merienne N., Jan C., Gaillard M.C., Lerchundi R., Barros L.F., et al. Impaired brain energy metabolism in the BACHD mouse model of Huntington's disease: critical role of astrocyte-neuron interactions. J. Cereb. Blood Flow Metab. 2014;34:1500–1510. doi: 10.1038/jcbfm.2014.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menalled L.B., Sison J.D., Dragatsis I., Zeitlin S., Chesselet M.F. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J. Comp. Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- 53.Hottinger A.F., Azzouz M., Deglon N., Aebischer P., Zurn A.D. Complete and long-term rescue of lesioned adult motoneurons by lentiviral-mediated expression of glial cell line-derived neurotrophic factor in the facial nucleus. J. Neurosci. 2000;20:5587–5593. doi: 10.1523/JNEUROSCI.20-15-05587.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drouet V., Perrin V., Hassig R., Dufour N., Auregan G., Alves S., Bonvento G., Brouillet E., Luthi-Carter R., Hantraye P., et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. 2009;65:276–285. doi: 10.1002/ana.21569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.