

Abstract
Fragile X syndrome, a common cause of intellectual disability and autism, is due to mutational silencing of the FMR1 gene leading to the absence of its gene product, fragile X mental retardation protein (FMRP). FMRP is a selective RNA binding protein owing to two central K-homology domains and a C-terminal arginine-glycine-glycine (RGG) box. However, several properties of the FMRP amino terminus are unresolved. It has been documented for over a decade that the amino terminus has the ability to bind RNA despite having no recognizable functional motifs. Moreover, the amino terminus has recently been shown to bind chromatin and influence the DNA damage response as well as function in the presynaptic space, modulating action potential duration. We report here the amino terminal crystal structures of wild-type FMRP, and a mutant (R138Q) that disrupts the amino terminus function, containing an integral tandem Agenet and discover a novel KH motif.
Introduction
Fragile X mental retardation protein (FMRP) is an RNA binding protein that is responsible for regulating local protein synthesis within dendritic synapses (1). Loss of FMRP results in fragile X syndrome (FXS, OMIM: 300624), which is the most frequent inherited cause of intellectual disability (ID), and also one of the leading genetic causes of autism spectrum disorder (2).
Since the discovery of FMRP over 20 years ago, many insights about the functions of this protein have been gleaned by studying its various structural domains. For instance, FMRP has three RNA binding domains that include two centrally located KH domains (KH1 and KH2) and a C-terminal arginine-glycine-glycine (RGG) box (Fig. 1A) (3). These domains allow FMRP to bind and regulate the translation of a specific subset of mRNA targets involved in synaptic plasticity (4,5). Tightly controlled local protein synthesis is important for many forms of synaptic plasticity, and in the absence of FMRP, defects in these protein synthesis-dependent synaptic plasticity pathways contribute significantly to the pathophysiology of FXS (6–9). Indeed, the only two FMR1 missense mutations in patients presenting with FXS have been substitutions within the KH domains, illustrating the importance of FMRP's RNA binding domains in the pathophysiology of FXS (10,11).
Figure 1.

Structure of FMRP amino terminal domain. (A) Schematic representation of human FMRP structure. Tandem Agenet and KH0 domain, KH1 and KH2 domains and RGG box are shown. (B and C) Two views of FMRP amino terminal structure, with Agenet1 in blue, Agenet2 in green and KH0 in brown. (D) Intra-molecular polar interactions on the interface of Agenet1 (blue) and 2 (green). (E) Intra-molecular polar interactions between strand β5 of Agenet1 (blue) and Loop-2 (green). (F) Intra-molecular hydrophobic interactions on the interface of Agenet1 (blue) and KH0 (brown).
The amino terminus of FMRP (defined here as amino acids 1–215) has been largely understudied and there are still many pending questions about its exact in vivo functions. It is the site for most of FMRP's protein–protein interactions, including NUFIP1, 82-FIP and CYFIP1/2 (12–14), however the biological role of these protein interactions with FMRP is not well understood. Recently, an additional interaction between the FMRP amino terminus and the presynaptic large conductance calcium-activated potassium (BK) channel β4 subunit has been demonstrated to modulate action potential duration, potentially linking the amino terminal domain with FMRP's presynaptic function (15). Moreover, it had been known for many years that FMRP shuttles between the nucleus and cytoplasm through its nuclear localization and export sequences (NLS and NES, respectively) (16). However, a clear nuclear function for FMRP had not been able to be specified until recently when nuclear FMRP was found to interact with chromatin and modulate the DNA damage response via its two amino terminal Agenet domains, also known as tandem Tudor domains (17). Interestingly, a patient missense mutation within the amino terminal domain, c.413G>A (p.(Arg138Gln), referred to as R138Q throughout the remaining text), disrupts both BK channel and chromatin binding (17,18).
Of particular interest is how the FMRP amino terminal domain binds RNA. As early as 15 years ago, the FMRP amino terminus was demonstrated capable of binding to RNA homopolymers, even though this region did not contain any recognizable RNA-binding motifs (19–21). Furthermore, brain cytoplasmic RNA BC1, and its primate analog BC200, have been shown to specifically bind the FMRP amino terminus and this interaction requires residues 180–217 (22,23). The subject of FMRP–BC1 RNA interaction has been quite controversial over the past 10 years and still remains unresolved. Several studies were unable to replicate BC1 and FMRP interaction (24,25), while others have successfully replicated the interaction (21,26–28) but not the requirement of the amino terminus (21). Some insight comes from a recent attempt to model the BC1 RNA interaction with the FMRP amino terminal NMR structure of the first 134 residues (28). BC1 RNA was shown to interact with specific residues within Agenet2 on FMRP, and while these interactions were necessary, they were not sufficient for FMRP–BC1 RNA interaction. It was proposed that the complete RNA binding surface for BC1 interaction would require the downstream residues adjacent to the tandem Agenet domain, however structural data for FMRP between residues 135 and 215 were unavailable. Here we show the wild-type (WT) and R138Q mutant crystal structures of FMRP amino terminal fragment (residues 1–202). In addition to the expected Agenet domains, we surprisingly discovered a novel KH motif immediately preceding the KH1 and KH2 domains, which we have termed KH0. This previously unrecognized KH fold might contribute to this regions ability to bind RNA and could potentially resolve some unanswered questions about the functions of the FMRP amino terminal domain.
Results
FMRP amino terminal region forms an integral domain containing two tandem Agenet modules and a novel KH module
We expressed, purified and crystallized WT and R138Q mutant of human FMRP residues 1–213 in two different space groups (P43212 and P41212) and determined the structures to the resolution of 3.2 and 3.0 Å, respectively (Supplementary Material, Table S1). For the structures of both space groups, the crystallographic asymmetric unit contains two molecules. The four monomeric structures are highly similar with a root-mean-square deviation (r.m.s.d.) of <0.6 when pairwise comparing two monomers. Thus, we will describe the monomeric structure of slightly higher resolution (3.0 Å) R138Q mutant structure.
The FMRP amino terminal domain consists of three structural modules, a tandem array of two Agenet folds and a novel KH fold (Fig. 1B and C). Each Agenet module contains a twisted β-sheet of five strands (β1–β5 for Agenet1 and β6–β10 for Agenet2). A 13-residue Loop-1 (residues 50–62) connects strand β5 of Agenet1 to strand β6 of Agenet2. The interface of the two Agenet modules is mediated by two ion pairs between side chains of E7 of strand β1 with R113 of strand β10, and E66 of strand β6 with R48 of strand β5 (Fig. 1D). In addition, aromatic residues, F15 of strand β2 and W79 of strand β7, are packed against the aliphatic carbons of R48 and R113, respectively. These two ion pairs are conserved among the FMRP, FXR1, FXR2 and Drosophila FMR proteins (Supplementary Material, Fig. S1), and mutations in residues R48C, E68K, R113C and R113H from a Drosophila forward genetic screen were unable to rescue FMRP overexpression-induced lethality (29) illustrating their importance for FMRP amino terminal function.
The KH0 module, residues 126–202, is composed of three antiparallel β strands of β11–β13 with three helices (αA–αC) packed on one side of the sheet (Fig. 1B and C). The KH0 module is located on the right side of Agenet1, opposite of Agenet2. A 12-residue Loop-2 (residues 114–125) connects strand β10 of Agenet2 to strand β11 of KH0 and appears to be critical to the structural integrity of the molecule. The loop extends through the entire length of Agenet1 and provides hydrogen bonds via two asparagine residues, N116 and N118, connecting E7 of strand β1, main-chain carbonyl oxygen atoms of F49 of strand β5 and P50 of Loop-1 (Fig. 1E).
The interface between Agenet1 and KH0 involves a large number of aromatic and hydrophobic interactions (Fig. 1F). These interactions include F126, F157, L171 and I173 of KH0 and L4, V20, I28, F44 and F49 of Agenet1. Emphasizing the importance of these interactions is the observation that the three structural modules are integral parts of the entire structure, glued together by the interactions mediated by strand β5 in the center of the structure. To the left, R48 of strand β5 stabilizes a network of polar interactions involving Agenet2 (Fig. 1D). To the right, F49 of strand β5 is part of the hydrophobic core involving KH0 (Fig. 1F). In addition, the main-chain carbonyl oxygen atom of F49 interacts with residues in the middle of Loop-2 (Fig. 1E). These intra-molecule interactions likely confer stability to the molecule.
FMRP tandem Agenet domain is structurally similar to other tandem Tudor domains that bind methylated lysine
Distance matrix alignment (DALI) search (30) against structures in the Protein Data Bank (PDB) showed that the FMRP tandem Agenet domain is structurally similar to the mammalian UHRF1 tandem Tudor domain and Arabidopsis SHH1 tandem Tudor-like domain (z score of 9.9 and 9.7, respectively) (Fig. 2), both of which bind methylated lysine 9 of histone H3 (H3K9me) peptides via an aromatic cage (31,32).
Figure 2.

Structural similarity of FMRP tandem Agenet with other tandem Tudor domains. (A) FMRP amino terminal structure with aromatic cage residues in Agenet1 (blue) and Agenet2 (green) indicated. (B–D) Tandem Tudor domains of UHRF1, SHH1 and 53BP1 bind methylated lysine in the Tudor1 aromatic cage (corresponds to Agenet1 of FMRP). (E) Tandem Tudor domain of JMJD2A binds methylated lysine in the Tudor2 aromatic cage (corresponds to Agenet2 of FMRP).
The tandem Agenet domain of FMRP, particularly the second Agenet motif, was reported as a potential methylated lysine binder and has been shown to bind H3K79me2 both in vivo and in vitro (17,33). Additionally, the structurally related FXR1/2 proteins have also been shown to recognize methylated lysine and bind H4K20me1/2/3 in vitro via their Agenet domains (34). In support of a histone binding function for the FMRP amino terminal domain, we found that each Agenet fold contains an aromatic cage with potential for binding methylated lysine: Y16, F32 and W36 within Agenet1 and W80, Y96 and Y103 within Agenet2 (Fig. 2A). Indeed, solution NMR spectrometry and in vitro binding assays have identified Y103 of Agenet2 (whose side chain is disordered in the absence of binding substrate) important for the recognition of trimethylated lysine (17,33). Among the structurally characterized tandem Tudor domains, UHRF1, SHH1 and 53BP1 bind methylated lysine with their first Tudor domain (Fig. 2B–D), whereas JMJD2A binds methylated lysine with its second Tudor domain (Fig. 2E). For FMRP, both Agenet motifs contain a recognizable aromatic cage (Fig. 2A), although only Agenet2 has been shown to interact with methylated lysine (17,33).
FMRP has a KH0 module that may participate in binding of nucleic acids
A surprising structural motif was found at residues 126–202, for which the DALI server recognized as a KH fold (we named it as KH0). KH folds are well studied as DNA/RNA binding domains (35), and FMRP already has two other tandem KH folds (KH1 and KH2) that follow immediately after KH0 (36). Among the three FMRP KH folds, KH0 and KH1 (or KH2) share 20% (or 18%) sequence identity and an r.m.s.d. of 2.4 Å (or 2.6 Å). Both KH1 and KH2 have the canonical G-X-X-G motif between helices αA and αB, but KH0 does not have such a motif (Fig. 3A). Instead, KH0 has an A-K-E-A between helices αA and αB (Fig. 3A), and the K143-E144 is in corresponding positions as that of K299-N300 of KH2, as seen by the positively charged surface of KH0 and KH2 (Fig. 3C and D). The Agenet1 and the KH0 together form a continuous basic surface patch, while Agenet2 has an acidic surface (Fig. 3C). If the aromatic cage of Agenet2 is indeed involved in binding positively charged histone lysine residues, then the basic surface patch of Agenet1 and KH0 could be involved in binding nucleosomal DNA. Alternatively, the basic surface patch of Agenet1 and KH0 could be involved in binding to RNA instead, which would potentially resolve some reports of the amino terminal domain's RNA binding capability despite the absence of any recognizable RNA binding motifs.
Figure 3.

FMRP KH0 motif is an integral part of the amino terminal structure. (A) Sequence and structural alignments of FMRP KH0, KH1 and KH2. White letters on black are identical or conserved residues among all three KH modules, and white on gray are identical or conserved in at least two. The G-X-X-G motifs of KH1 and KH2 and A-X-X-A motif of KH0 are depicted in white on red background. The position of the patient mutation (R138Q) and hydrophobic residues involved in the hydrophobic core (Fig. 1F) unique to KH0 are shown in red. Conserved residues are as defined by the following groupings: V, L, I and M; F, Y and W; K and R, E and D; Q and N; E and Q; D and N; S and T, and A, G and P. (B) Cartoon structure of Agenet1-Agenet2-KH0 (PDB: 4QW2) connected to KH1-KH2 (PDB: 2QND) by a flexible linker. (C and D) The surface charge distribution of amino terminal Agenet1-Agenet2-KH0 (C) and KH1-KH2 (D) at neutral pH, displayed as blue for positive, red for negative and white for neutral, in a similar orientation as shown in (B). The orange circles indicate the position of A-X-X-A (KH0) or G-X-X-G (KH1 and KH2).
Functionally relevant mutation of the FMRP amino terminal domain
A FMRP missense mutation at arginine 138 to glutamine (R138Q), reported in a patient with ID and seizures (37), is located within KH0 helix αA. However, the WT and R138Q crystal structures were very similar with an r.m.s.d. of 0.8 Å (Fig. 4). The R138Q missense is a partial loss-of-function mutation that impairs FMRP's ability to interact with BK channels and modulate action potential duration in hippocampal and cortical neurons (18) as well as impairs chromatin binding (17). This mutation did not affect the ability of full length FMRP to bind RNA in vivo, at least not with the well-known FMRP targets: Map1b, PSD95 or CamKII (18). We were able to express and purify the R138Q mutant 1–213 protein with similar purity and amount as that of WT protein. The one difference noted between WT and R138Q purification is that R138Q protein eluted slightly earlier from anion exchange Q column (data not shown). This is evidence that the mutation may affect the protein–protein interactions of FMRP, which is also supported by biochemical data indicating R138Q disrupts FMRP's interaction with BK channels (18). Thus, we were unable to determine any gross structural changes that would cause R138Q mutation to lose chromatin or BK channel binding, however this positively charged arginine residue may be critical for specific protein–protein interactions that are unable to form when mutated to a polar glutamine.
Figure 4.
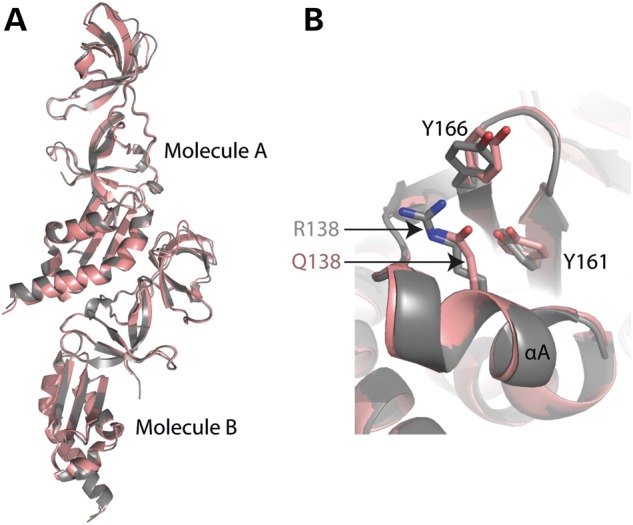
Comparison of WT and R138Q structures. (A) WT (gray, space group P43212) and R138Q (brown, space group P41212) crystal structures are highly similar with an r.m.s.d. of 0.8 Å. (B) Superposition of WT (gray) and R138Q (brown). Residues R138 and Q138 of helix αA are indicated.
Discussion
We determined the crystal structure of the WT and R138Q mutant FMRP amino terminal domains (residue 1–202). We found that the amino terminal fragment forms a stable domain that contains two tandem Agenet modules and a novel KH module. The three structural modules are arranged with Agenet1 flanked by Agenet2 and KH0 forming an integral structure, stabilized by intra-molecular polar interactions (Agenet2–Agenet1) and intra-molecular hydrophobic interactions (Agenet1–KH0). This is the first crystal structure reported for the amino terminal domain of FMRP and our work helps provide structural evidence for many of the proposed functions of the amino terminal domain including chromatin binding and RNA binding.
The Agenet domains within FMRP were previously identified based on structural alignment (38) and the NMR solution structure of FMRP residues 1–134 (33). Our crystal structure analysis further confirms their existence and also supports a chromatin binding function for these domains. We found that both Agenet modules contain an aromatic cage, similar to other Tudor/Agenet domains, and have potential for binding methylated histone lysines. However, further structural studies of the FMRP amino terminal domain with bound substrate(s) will help clarify exactly how the Agenet domains recognize and bind to chromatin.
In contrast to the expected Agenet domains, the discovery of the novel KH0 was quite surprising and remained elusive until now because KH0 shares low sequence homology with the other FMRP KH domains (20 and 18% for KH1 and KH2, respectively) and does not contain the canonical G-X-X-G motif. Subsequently, this fourth FMRP RNA binding motif could not be identified until structural data became available for residues 135–215. Even though KH0 has not been directly tested for RNA binding, several lines of evidence do suggest that it is capable of binding RNA. (1) The Agenet1 and KH0 possess a continuous basic surface patch with potential for binding nucleic acids. (2) Previous studies have shown that the amino terminal domain binds to RNA homopolymers (FMRP fragment 1–214) (19,20) and has a specific RNA interaction with BC1 RNA (FMRP fragment 1–217) (22,28). (3) Two shorter FMRP fragments consisting of residues 1–134 or 1–180 do not bind BC1 RNA, indicating that the region necessary for RNA binding requires the residues between 180 and 217, which is where KH0 lies (residues 126–202). Thus, the discovery of the KH0 module helps to finally resolve the issue of how the amino terminal domain of FMRP could participate in RNA binding despite the absence of any discernible RNA binding motifs. Further studies will be needed to determine whether the KH0 module alone is capable of binding RNA, if it works in conjunction with the KH1 and KH2 domains to enhance RNA binding specificity, and what, if any, are the biological targets of KH0.
Materials and Methods
Protein expression and purification
We initially generated hexahistidine-SUMO-tagged constructs of human FMRP residues 1-234 and residues 1–298, for both WT and R138Q mutant. We performed proteolytic digestion on purified 1-234 fragment and found that trypsin generated a stable ∼30 kDa protein (see below). The molecular mass was determined by MALDI-TOF-MS, which corresponds to an FMRP protein fragment of residues 1-213, which was the construct used for crystallization.
The proteins were expressed in E. coli BL21 (DE3)-Gold cells with the RIL-Codon plus plasmid (Stratagene). Cultures were grown at 37°C until the OD 600 nm reached 0.5; the temperature was then shifted to 16°C, and isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to 0.4 mm to induce expression. After 16 h, cells were re-suspended with four volumes of 300 mM NaCl, 20 mm sodium phosphate, pH 7.4, 20 mm imidazole, 1 mm dithiothreitol (DTT) for WT or 0.5 mm tris(2-carboxyethyl)phosphine (TCEP) for R138Q and 0.3 mm phenylmethyl-sulphonyl fluoride. The cells were sonicated for 5 min (1 s on and 2 s off) and the lysates clarified by centrifugation at 38 000 g for 1 h. Hexahistidine fusion proteins were isolated on a nickel-charged HiTrap chelating column (GE Healthcare) and the His-SUMO tag was removed by incubating with Ulp1 for 16 h at 4°C. The cleaved protein was further purified by a HiTrap Q column (GE Healthcare) and eluted by increasing NaCl concentration from 0.1 to 1 m. The proteins were then loaded onto a Superdex 75 (16/60) column (equilibrated with 200 mm NaCl, 20 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.0, 1 mm DTT or 0.5 mm TCEP) where it eluted at ∼75 ml as a single peak corresponding to a monomeric protein. We increased NaCl concentration up to 500 mm during protein concentration.
Protease digestion and mass spectrometry
FMRP 1-234 protein (4 μg) in 20 mm HEPES, pH 7.0, 200 mm NaCl and 1 mm DTT was treated with serial dilutions of trypsin, chimotrypsin and elastase for final protease conditions of 0, 0.1, 1, 10 and 100 ng/μl. Digestion occurred for 30 min at room temperature before being separated on a 12% sodium dodecyl sulfate gel. Trypsin was found to form stable cleavage product of the FMRP 1-234 protein and was used for further analysis by MALDI-TOF-MS. To prepare samples for MALDI, FMRP 1-234 protein was again digested with trypsin at 0, 1 and 10 ng/μl, for 30 min at room temperature. One microliter of sinapic acid (dissolved in equal volumes of 0.1% trifluoroacetic acid and acetonitrile) and 1 μl of digested protein were mixed together and spotted onto a MALDI plate and allowed to air dry overnight before determining the respective masses of the uncut and cut proteins by MALDI-TOF-MS using a Burker Ultra FlexII TOF/TOF instrument (Biochemistry Department, Emory University).
Crystallography
Crystallization was carried out in a 2 μl sitting drop with equal volumes of protein solution (40–60 mg/ml) and well solution. Crystals appeared within 16 h at 16°C under the conditions of 27% (w/v) polyethylene glycol 3350, 0.2 m ammonium sulfate, 0.1 m HEPES, pH 7.0 (for WT protein) and 25–30% polyethylene glycol monomethyl ether 5000, 0.2 m ammonium sulfate, 0.1 m HEPES, pH 7.5 and 5 mm of (CH3)3PbOAc (for R138Q mutant). Crystals were cryoprotected by soaking in mother liquor supplemented with 20% (v/v) ethylene glycol and by plunging into liquid nitrogen. X-ray diffraction data sets were collected at the SER-CAT beamline at the Advanced Photon Source, Argonne National Laboratory and processed using HKL2000 (39). Because an appropriate choice of high-resolution cutoff is difficult, we followed the suggestion of Karpus and Diederichs (40) and used CC*, instead of Rmerge values, and I/σ(I) to guide the high-resolution limit (Supplementary Material, Table S1).
We combined molecular replacement (PDB: 3O8V) and single anomalous diffraction (SAD) to obtain crystallographic phases using Pb containing R138Q crystals. A data set was collected at 100 K at a wavelength of 0.94390 Å, at a slightly higher energy (100 eV) than the lead (Pb) absorption edge. Anomalous signal originally seemed to extend to only ∼7.4 Å, but after combination with the partial model from molecular replacement results, figure of merit and experimental phases were improved, and the resulting electron density map for KH0 domain was easily visible and the model was built using the program COOT (41). Finally, PHENIX scripts (42) were used for model refinement against the native WT data and R138Q mutant data (Supplementary Material, Table S1), respectively, with an optimized weight for the X-ray target and the stereochemistry of the atomic displacement parameters during the last refinement cycles.
The secondary structure matching (SSM) script in COOT-generated initial pairwise alignments, followed by visual inspections, between structures of FMRP 1–213 WT (PDB: 4QVZ), FMRP 1–213 R138Q (PDB: 4QW2), FMRP KH1-KH2 domain (PDB: 2QND) (36), FXR1/2 tandem Agenet domains (PDB: 3O8V, 3H8Z) (34), SHH1 Tudor domain with H3K9me3 peptide (PDB: 4IUR) (32) and UHRF1 tandem Tudor domain with H3K9me3 peptide (PDB: 3ASK) (31).
The X-ray structures (coordinates and structure factor files) of human FMRP 1-213 WT and R138Q 1-213 have been submitted to PDB under accession numbers 4QVZ (WT) and 4QW2 (R138Q), respectively.
Supplementary Material
Funding
This work was supported by grants from the National Institutes of Health (NS091859 from the National Institute of Neurological Disorders and Stroke and the Eunice Kennedy Shriver National Institute of Child Health & Human Development in support of the National Fragile X Research Center to S.T.W., GM049245-21 to H.H. and X.C.).
Supplementary Material
Acknowledgements
The Department of Biochemistry of Emory University School of Medicine supported the use of SER-CAT beamlines.
Conflicts of Interest statement. The authors have no conflicts of interest to report.
References
- 1.Bassell G.J., Warren S.T. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santoro M.R., Bray S.M., Warren S.T. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 3.Ashley C.T., Jr, Wilkinson K.D., Reines D., Warren S.T. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 4.Brown V., Jin P., Ceman S., Darnell J.C., O'Donnell W.T., Tenenbaum S.A., Jin X., Feng Y., Wilkinson K.D., Keene J.D., et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 5.Darnell J.C., Van Driesche S.J., Zhang C., Hung K.Y., Mele A., Fraser C.E., Stone E.F., Chen C., Fak J.J., Chi S.W., et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber K.M., Gallagher S.M., Warren S.T., Bear M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osterweil E.K., Krueger D.D., Reinhold K., Bear M.F. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Volk L.J., Pfeiffer B.E., Gibson J.R., Huber K.M. Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J. Neurosci. 2007;27:11624–11634. doi: 10.1523/JNEUROSCI.2266-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H., Wu L.J., Kim S.S., Lee F.J., Gong B., Toyoda H., Ren M., Shang Y.Z., Xu H., Liu F., et al. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron. 2008;59:634–647. doi: 10.1016/j.neuron.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 10.De Boulle K., Verkerk A.J., Reyniers E., Vits L., Hendrickx J., Van Roy B., Van den Bos F., de Graaff E., Oostra B.A., Willems P.J. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet. 1993;3:31–35. doi: 10.1038/ng0193-31. [DOI] [PubMed] [Google Scholar]
- 11.Myrick L.K., Nakamoto-Kinoshita M., Lindor N.M., Kirmani S., Cheng X., Warren S.T. Fragile X syndrome due to a missense mutation. Eur. J. Hum. Genet. 2014;22:1185–1189. doi: 10.1038/ejhg.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bardoni B., Schenck A., Mandel J.L. A novel RNA-binding nuclear protein that interacts with the fragile X mental retardation (FMR1) protein. Hum. Mol. Genet. 1999;8:2557–2566. doi: 10.1093/hmg/8.13.2557. [DOI] [PubMed] [Google Scholar]
- 13.Schenck A., Bardoni B., Moro A., Bagni C., Mandel J.L. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. USA. 2001;98:8844–8849. doi: 10.1073/pnas.151231598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bardoni B., Castets M., Huot M.E., Schenck A., Adinolfi S., Corbin F., Pastore A., Khandjian E.W., Mandel J.L. 82-FIP, a novel FMRP (fragile X mental retardation protein) interacting protein, shows a cell cycle-dependent intracellular localization. Hum. Mol. Genet. 2003;12:1689–1698. doi: 10.1093/hmg/ddg181. [DOI] [PubMed] [Google Scholar]
- 15.Deng P.Y., Rotman Z., Blundon J.A., Cho Y., Cui J., Cavalli V., Zakharenko S.S., Klyachko V.A. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron. 2013;77:696–711. doi: 10.1016/j.neuron.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eberhart D.E., Malter H.E., Feng Y., Warren S.T. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 1996;5:1083–1091. doi: 10.1093/hmg/5.8.1083. [DOI] [PubMed] [Google Scholar]
- 17.Alpatov R., Lesch B.J., Nakamoto-Kinoshita M., Blanco A., Chen S., Stutzer A., Armache K.J., Simon M.D., Xu C., Ali M., et al. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell. 2014;157:869–881. doi: 10.1016/j.cell.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Myrick L.K., Deng P.Y., Hashimoto H., Oh Y., Cho Y., Poidevin M.J., Suhl J.A., Visootsak J., Cavalli V., Jin P., et al. An independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. PNAS. 2014 doi: 10.1073/pnas.1423094112. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adinolfi S., Ramos A., Martin S.R., Dal Piaz F., Pucci P., Bardoni B., Mandel J.L., Pastore A. The N-terminus of the fragile X mental retardation protein contains a novel domain involved in dimerization and RNA binding. Biochemistry. 2003;42:10437–10444. doi: 10.1021/bi034909g. [DOI] [PubMed] [Google Scholar]
- 20.Adinolfi S., Bagni C., Musco G., Gibson T., Mazzarella L., Pastore A. Dissecting FMR1, the protein responsible for fragile X syndrome, in its structural and functional domains. RNA. 1999;5:1248–1258. doi: 10.1017/s1355838299990647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan X., Denman R.B. Conformational-dependent and independent RNA binding to the fragile X mental retardation protein. J Nucleic Acids. 2011;2011:246127. doi: 10.4061/2011/246127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zalfa F., Adinolfi S., Napoli I., Kuhn-Holsken E., Urlaub H., Achsel T., Pastore A., Bagni C. Fragile X mental retardation protein (FMRP) binds specifically to the brain cytoplasmic RNAs BC1/BC200 via a novel RNA-binding motif. J. Biol. Chem. 2005;280:33403–33410. doi: 10.1074/jbc.M504286200. [DOI] [PubMed] [Google Scholar]
- 23.Zalfa F., Giorgi M., Primerano B., Moro A., Di Penta A., Reis S., Oostra B., Bagni C. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]
- 24.Wang H., Iacoangeli A., Lin D., Williams K., Denman R.B., Hellen C.U., Tiedge H. Dendritic BC1 RNA in translational control mechanisms. J. Cell Biol. 2005;171:811–821. doi: 10.1083/jcb.200506006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iacoangeli A., Rozhdestvensky T.S., Dolzhanskaya N., Tournier B., Schutt J., Brosius J., Denman R.B., Khandjian E.W., Kindler S., Tiedge H. On BC1 RNA and the fragile X mental retardation protein. Proc. Natl. Acad. Sci. USA. 2008;105:734–739. doi: 10.1073/pnas.0710991105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabus C., Mazroui R., Tremblay S., Khandjian E.W., Darlix J.L. The fragile X mental retardation protein has nucleic acid chaperone properties. Nucleic Acids Res. 2004;32:2129–2137. doi: 10.1093/nar/gkh535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson E.M., Kinoshita Y., Weinreb D.B., Wortman M.J., Simon R., Khalili K., Winckler B., Gordon J. Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J. Neurosci. Res. 2006;83:929–943. doi: 10.1002/jnr.20806. [DOI] [PubMed] [Google Scholar]
- 28.Lacoux C., Di Marino D., Boyl P.P., Zalfa F., Yan B., Ciotti M.T., Falconi M., Urlaub H., Achsel T., Mougin A., et al. BC1-FMRP interaction is modulated by 2′-O-methylation: RNA-binding activity of the tudor domain and translational regulation at synapses. Nucleic Acids Res. 2012;40:4086–4096. doi: 10.1093/nar/gkr1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reeve S.P., Lin X., Sahin B.H., Jiang F., Yao A., Liu Z., Zhi H., Broadie K., Li W., Giangrande A., et al. Mutational analysis establishes a critical role for the N terminus of fragile X mental retardation protein FMRP. J. Neurosci. 2008;28:3221–3226. doi: 10.1523/JNEUROSCI.5528-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holm L., Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arita K., Isogai S., Oda T., Unoki M., Sugita K., Sekiyama N., Kuwata K., Hamamoto R., Tochio H., Sato M., et al. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc. Natl. Acad. Sci. USA. 2012;109:12950–12955. doi: 10.1073/pnas.1203701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Law J.A., Du J., Hale C.J., Feng S., Krajewski K., Palanca A.M., Strahl B.D., Patel D.J., Jacobsen S.E. Polymerase IV occupancy at RNA-directed DNA methylation sites requires SHH1. Nature. 2013;498:385–389. doi: 10.1038/nature12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramos A., Hollingworth D., Adinolfi S., Castets M., Kelly G., Frenkiel T.A., Bardoni B., Pastore A. The structure of the N-terminal domain of the fragile X mental retardation protein: a platform for protein–protein interaction. Structure. 2006;14:21–31. doi: 10.1016/j.str.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 34.Adams-Cioaba M.A., Guo Y., Bian C., Amaya M.F., Lam R., Wasney G.A., Vedadi M., Xu C., Min J. Structural studies of the tandem Tudor domains of fragile X mental retardation related proteins FXR1 and FXR2. PLoS One. 2010;5:e13559. doi: 10.1371/journal.pone.0013559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valverde R., Edwards L., Regan L. Structure and function of KH domains. FEBS J. 2008;275:2712–2726. doi: 10.1111/j.1742-4658.2008.06411.x. [DOI] [PubMed] [Google Scholar]
- 36.Valverde R., Pozdnyakova I., Kajander T., Venkatraman J., Regan L. Fragile X mental retardation syndrome: structure of the KH1-KH2 domains of fragile X mental retardation protein. Structure. 2007;15:1090–1098. doi: 10.1016/j.str.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 37.Collins S.C., Bray S.M., Suhl J.A., Cutler D.J., Coffee B., Zwick M.E., Warren S.T. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am. J. Med. Genet. A. 2010;152A:2512–2520. doi: 10.1002/ajmg.a.33626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maurer-Stroh S., Dickens N.J., Hughes-Davies L., Kouzarides T., Eisenhaber F., Ponting C.P. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 2003;28:69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z., Borek D., Majewski W., Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr. A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- 40.Karplus P.A., Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.