

Abstract
We examined the characteristics of interferon alpha/beta (IFN-α/β) induction after alphavirus or control Sendai virus (SeV) infection of murine fibroblasts (MEFs). As expected, SeV infection of wild-type (wt) MEFs resulted in strong dimerization of IRF3 and the production of high levels of IFN-α/β. In contrast, infection of MEFs with multiple alphaviruses failed to elicit detectable IFN-α/β. In more detailed studies, Sindbis virus (SINV) infection caused dimerization and nuclear migration of IRF3, but minimal IFN-β promoter activity although surprisingly, the infected cells were competent for IFN production by other stimuli early after infection. A SINV mutant defective in host macromolecular synthesis shut-off induced IFN-α/β in the MEF cultures dependent upon the activities of the TBK1 IRF-3 activating kinase and host pattern recognition receptors (PRRs) PKR and MDA5 but not RIG-I. These results suggest that wild type alphaviruses antagonize IFN induction after IRF-3 activation but also may avoid detection by host PRRs early after infection.
Keywords: Interferon, alphavirus, PKR, MDA5, TBK1, RIG-I, antagonism
Introduction
Data collected using mice deficient in type I interferon (interferon alpha/beta [IFN-α/β]) responses have shown this innate immune system to be critical for host survival of infections with many viruses, including alphaviruses (Ryman et al., 2000; White et al., 2001; Couderc et al., 2008; Fragkoudis et al., 2007; Gardner et al., 2009). The IFN-α/β response is induced by interaction of particular virus structures or replicative intermediates (known as pathogen–associated molecular patterns [PAMPs]) with host pattern recognition receptors (PRRs) that initiate signaling cascades within infected cells. However, virtually nothing is known about the particular PAMPs or host PRRs involved in IFN-α/β induction by the alphaviruses or how activation of these pathways differs between alphavirus types. Potential PRRs include extracellular and endosomal receptors such as toll-like receptors (TLRs; (Iwasaki & Medzhitov, 2004; Uematsu & Akira, 2008), C-type lectin scavenger receptors (McGreal et al., 2004; Geijtenbeek & Gringhuis, 2009), and intracellular receptors that appear to recognize intermediates of RNA virus replication such as PKR (Jiang et al., 2003; Williams, 2001) and the RIG-I, MDA5 and Lgp2 RNA helicases (Yoneyama et al., 2004; Kang et al., 2002; Kato et al., 2006; Venkataraman et al., 2007; Meylan et al., 2006). Potential viral PAMPS include glycoproteins (C-type lectins; TLR2) and viral ssRNA (TLR7) or dsRNA (TLR3, PKR, and RIG-I) (Meylan et al., 2006; Uematsu & Akira, 2008; Geijtenbeek & Gringhuis, 2009; Williams, 2001).
A distinction is generally made between the cell-surface/endosomal PRRs (C-type lectins, TLR3, TLR7 and others) and the cytoplasmic PRRs (PKR, RIG-I and MDA5) in that the cell-surface PRRs are expressed only by subsets of dendritic cells (DCs) or macrophages, whereas cytoplasmic PRRs are expressed by most cells (Meylan & Tschopp, 2006; Pollara et al., 2005; Uematsu & Akira, 2008). Therefore, viral induction of IFN-α/β may exhibit cell-type specificity, depending upon the pathway(s) triggered. Several signaling pathways resulting in IFN-α/β induction have been elucidated for the various PRRs. The canonical induction pathway was originally defined as dsRNA replicative intermediates in the cytoplasm activating the IRF3 transcription factor, which is then translocated to the nucleus where it activates IFN-β gene transcription (Schafer et al., 1998). This involves detection of viral RNAs by the RIG-I or MDA5 RNA helicases, followed by recruitment of the IPS-1 adapter molecule and activation of tank binding kinase 1 (TBK1 also IKKε in lymphoid cells), which may directly phosphorylate IRF3 (Lee & Kim, 2007; Paz et al., 2006; McWhirter et al., 2004; Fitzgerald et al., 2003). Interestingly, recent studies indicate that, while MDA5 may interact with dsRNA, RIG-I is activated by interaction with uncapped RNA molecules containing 5′-triphosphates (Kang et al., 2002; Gitlin et al., 2006; Pichlmair et al., 2006; Hornung et al., 2006). PKR, a dsRNA-binding protein, appears to augment the induction of IFN-α/β by signaling through the NFκB pathway and may be a downstream modulator of TLR3 signaling (Jiang et al., 2003). With the exception of TLR3, TLRs signal through the canonical TLR pathway including MyD88-IRAK1 adaptor proteins to activate NFκB and induce the IFN-β promoter. In contrast, TLR3 signals through the TRIF/TRAF6 pathway to activate both NFκB and TBK1-IRF3 (Honda et al., 2006).
For alphaviruses, it is known that type I IFN is induced in vitro by infection of some cell types (Griffin, 2001) but that infection of established murine fibroblast cell lines with the prototypic alphavirus, Sindbis virus (SINV), does not result in IFN production (Frolova et al., 2002). This has led to the hypothesis that some alphaviruses antagonize IFN production in vitro by shutoff of host macromolecular synthesis (Frolova et al., 2002; Aguilar et al., 2007) but the specific points of blockage of IFN production have not been delineated. In contrast with cultured cells, high levels of serum IFN-α/β are associated with systemic replication of Sindbis virus (SINV) and Venezuelan equine encephalitis virus (VEEV) in vivo (Charles et al., 2001; Klimstra et al., 1999; Ryman et al., 2000; Ryman et al., 2002; Gardner et al., 2008). However, the cell types responsible for its production and the relationship of induction pathways in vivo to those in cultured cells are unknown.
Regarding PRRs involved in IFN induction by alphaviruses, one study concluded that induction of IFN-α/β by Semliki Forest virus (SFV) in myeloid DC cultures occurred independently of MyD88, but was dependent upon IRF3 (Hidmark et al., 2005). Furthermore, IFN-α induction was minimally impaired in MDA5−/− macrophages infected with SINV, suggesting that this molecule was not vital to responses against alphaviruses with this cell type (Gitlin et al., 2006). Aside from these two studies, the alphavirus-expressed PAMPs and particular PRRs and pathways associated with IFN-α/β induction remain essentially uncharacterized.
In the current studies, we have evaluated the IFN-α/β induction characteristics after infection of primary murine fibroblast (MEF) cells with SINV, VEEV, eastern equine encephalitis (EEEV) or chikungunya (CHIKV) viruses. None of the viruses induced substantial IFN-α/β in the MEFs at any time after infection. In contrast, infection with Sendai virus (SeV) induced IFN-α/β promoter activity, formation of IRF3 dimers and their nuclear translocation, and production of biological IFN-α/β by 12 hours post-infection (h p.i.). In detailed analyses, wild-type SINV also induced activation and nuclear translocation of IRF3 and an increase in its DNA binding but no transcription or protein synthesis was detected from genes under the control of the IFN-β promoter. Interestingly, IFN-α/β induction was detected early after infection in cells co-treated with wt SINV (TR339 strain) and IFN agonists SeV or poly(I:C), suggesting that the infected cells were competent for IFN production. When macromolecular shut-off was reduced by use of non-cytopathic version of SINV (39nc), IFN-α/β was induced later after infection, largely dependent upon PKR and MDA5 PRRs signaling through TBK1. Together, these data suggest that wild-type SINV, and likely other alphaviruses, antagonize IFN-α/β induction from infected MEFs through shut-off of host macromolecular synthesis but also appear to avoid induction prior to the time at which shutoff is complete.
Results
Wild-type alphaviruses do not trigger IFN-α/β production from infected MEF cultures
Many previous studies examining IFN-α/β induction by RNA viruses have used murine fibroblasts because of the availability of mice deficient in specific components of the IFN-α/β induction pathways and ease of MEF cell line generation. To test the role of TBK1 in alphavirus IFN-α/β induction, we began by comparing wild-type (wt) MEFs to those deficient in TBK1. Using a biological IFN-α/β assay we found that infection of wt MEFs with SeV, used as a positive control for RIG-I-dependent IFN-α/β induction (Kato et al., 2005; Yoneyama et al., 2004), resulted in detectable production of IFN-α/β by 12 h p.i., whereas in the absence of TBK1, SeV failed to induce detectable IFN-α/β((Figure 1), consistent with previous reports (e.g., McWhirter et al., 2004). Furthermore, the wt MEFs responded to transfection with poly(I:C), a MDA5/PKR agonist (Gitlin et al., 2006; Diebold et al., 2003) with production of IFN-α/β (data not shown). These data suggest that RIG-I and MDA5/PKR virus detection pathways were functional in the MEF cultures.
Figure 1. Interferon production from MEFs after infection with different alphaviruses.
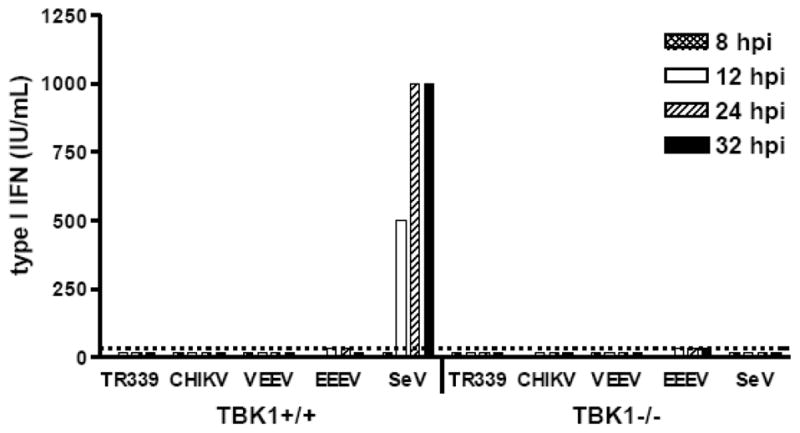
Control and TBK1−/− MEFs were infected with each virus for 1 h at 37°C at an MOI of 3. Supernatants were harvested for biological interferon assay at the times indicated. Each bar represents the average of triplicate samples. Error bars are standard deviations but are too small to be seen. Some of these data were published previously (Ryman & Klimstra, 2009).
In contrast, infection (multiplicity of infection [MOI]=3) of either wt or TBK1-deficient MEFs with wild-type SINV strain TR339 or the more human-virulent alphaviruses VEEV, EEEV or CHIKV resulted in very low (<10 IU/ml) or no detectable IFN-α/β release prior to death of the cells (between 18 and 32 h p.i.) measured by bioassay (Figure 1) or IFN-β ELISA (data not shown). The failure of these cells to produce IFN-α/β was generally applicable to multiply passaged MEF-like cells as confirmed by infection of four other wt C57BL/6-derived MEF lines, murine Swiss 3t3 fibroblasts or murine L929 fibroblasts (data not shown).
Wild-type alphaviruses do not block IRF3 activation or translocation to the nucleus in infected MEF cultures
In order to determine the mechanism by which the alphaviruses antagonize IFN-α/β production from infected MEFs, we monitored cellular responses to virus infection at each sequential step in the IFN-α/β induction pathway. Constitutively-expressed transcription factor IRF3 is a master regulator of host antiviral defense activation (Hiscott et al., 1999; Hiscott, 2007). Upon virus detection, latent IRF3 is phosphorylated by TBK1 or IKKε kinases, causing IRF3 dimerization and translocation to the nucleus where it binds to the IFN-β promoter and stimulates transcription of the gene (Weaver et al., 1998; Lee & Kim, 2007; Taniguchi et al., 2001); (Fitzgerald et al., 2003). A number of viruses antagonize/evade these early steps in the IFN-α/β induction pathway in order to avoid the IFN-α/β response (Randall & Goodbourn, 2008; Haller et al., 2007).
To determine if the lack of IFN-α/β production from SINV TR339-infected MEF cultures was due to the failure or inhibition of IRF3 activation, we first used native-PAGE analysis to examine the differential migration of activated IRF3 dimers versus inactive monomers. We confirmed the formation of activated IRF3 dimers in wild-type MEFs at 12 h p.i. with SeV and demonstrated similar activation in SINV TR339-infected cells; however, IRF3 dimerization was not detected after infection of TBK1-deficient MEFs with either SeV or SINV TR339 (Figure 2A). These data indicate that the activation of IRF3 is not blocked in SINV-infected MEFs and is triggered primarily via a TBK1-dependent detection mechanism. Subcellular fractionation analyses indicated that translocation of dimerized IRF3 to the nucleus was also not blocked after infection of wt MEFs with either SeV or SINV TR339 viruses (Figure 2B). In this assay, staining for tubulin (cytoplasmic protein) and lamin A/C (nuclear protein) in control samples confirmed that the fractionation was successful.
Figure 2. Activation and nuclear translocation of IRF3 after infection of MEFs.

A) Control or TBK1−/−MEFs were mock infected or infected with each virus at an MOI of 3 and lysates were harvested at 12 h p.i. for Native-PAGE separation of proteins and western blot for IRF3. B) Cells were infected as in A and processed for fractionation as described in Materials and Methods. Proteins transferred to membranes were stained with either IRF3 or fractionation control proteins tubulin or lamin A/C. C) Cells were infected and harvested as in A and processed for oligonucleotide pull down assay and IRF3 western blots as described in Materials and Methods. Numbers in the figure indicate relative levels of bound IRF3 with values for mock-infected cells set to 1.0. Values from virus-infected cells are presented as a ratio to mock for each cell type. D) Control MEFS were transfected with a plasmid encoding an IRF3/GFP fusion protein as described in Materials and Methods followed by mock infection or infection with each virus 18 h later at an MOI of 3. At 8 h p.i., appropriate cells were stained for either SINV or VEEV structural proteins. Then the subcellular location of GFP signal was scored in triplicate using fluorescence microscopy for at least 50 cells in which virus proteins and GFP were co-localized. Error bars are standard deviations and some are too small to be seen.
As a functional measure of IRF3 activation, the DNA-binding capacity of IRF3 was examined with an oligonucleotide pull-down assay performed on lysates from SINV TR339 or SeV-infected MEFs (Figure 2C). Samples from wt, but not TBK1-deficient, MEFs infected with either SeV or SINV TR339 resulted in a reproducible increase in IRF3-DNA binding compared to mock-infected cells, suggesting that some portion of IRF3 is activated by both viruses via a TBK1-dependent pathway to bind DNA after infection of MEFs.
These assays give only a measurement of total IRF3 activation in the cultures and no information regarding the extent of IRF3 activation within individual cells. To examine the latter, we transfected wt or TBK1-deficient MEFs with a plasmid expressing an IRF3/GFP fusion protein (Mibayashi et al., 2007) and evaluated the cytoplasmic versus nuclear localization of GFP signal at 8 h p.i. with SINV TR339 or VEEV to represent the Old and New World alphaviruses, respectively (Figure 2D). While anti-alphavirus staining revealed that virtually all of the cells were infected, those cells that were successfully transfected with the plasmid were rendered refractory to alphavirus infection, by an IFN-α/β signaling-dependent mechanism. Therefore, we used MEFs derived from IFN-α/β receptor-null mice (IFNAR−/−) and included in the analyses only those cells positive for anti-alphavirus staining and expressing IRF3/GFP. As expected, significant nuclear translocation of IRF3/GFP was observed following SeV infection (p<0.01 versus mock). Similarly, infection with SINV TR339 or VEEV resulted in a significant translocation of IRF3/GFP to the nucleus (p<0.01 for each virus versus mock). Under these conditions, VEEV appeared to cause more consistent nuclear translocation in infected cells, similar to SeV. Direct antibody staining of nucleus-translocated IRF3 could not be achieved in the MEF culture system; however, we confirmed the IRF3/GFP plasmid results by costaining for IRF3 and alphavirus structural protein in infected 293 human kidney cells followed by confocal microscopy (data not shown).
Taken together, these data suggest that at least some of the Old and New World alphaviruses behave similarly in that they do not antagonize IFN-α/β inductive signaling pathways upstream of the nuclear translocation of IRF3 dimers in infected MEFs. Examination of IRF3 binding to DNA after SINV infection suggests this step is also not antagonized.
Synthesis of IFN-β is inhibited in SINV TR339-infected MEF cultures
The absence of detectable IFN-α/β production from alphavirus-infected MEFs, in spite of the successful activation, nuclear translocation and DNA binding of the IRF3 master transcription factor, led us to examine whether the IFN-β promoter was activated after SINV TR339 infection of wt or TBK1-deficient MEFs. Activity of the IFN-β promoter, as well as the transcription factor-specific positive regulatory domains (PRDI/III) of the promoter, were examined (Figure 3A and B). In keeping with the results in Figure 1, SeV infection of wt MEFs resulted in IFN-β promoter and IRF3-specific PRDI/III activity; however, promoter activity was abrogated in the absence of TBK1. In contrast, SINV TR339 infection of neither wt nor TBK1-deficient MEFs resulted in IFN-β promoter or PRDI/III activity greater than detected in mock infected cells, measured at the level of mRNA accumulation (data not shown) or luciferase production (Figure 3A and 3B). From the data presented thus far, we infer that the antagonism of IFN-α/β production from SINV TR339-infected MEFs occurs due to arrest of host transcription/translation of IRF3-dependent genes including the type I IFNs, not because upstream signaling pathways are inhibited.
Figure 3. Interferon promoter activity after SINV TR339 infection of MEFs.

Control and TBK1−/− MEFs were co-transfected with the A) IFN-β promoter or B) PRDI/III and a renilla control reporter constructs as described in Materials and Methods. Approximately 18 h later, the cells were mock infected or infected with each virus at an MOI of 3. At 12 h p.i., lysates were harvested for dual luciferase assay. Data are presented as the ratio of relative light units (RLU) from a control renilla luciferase-expressing plasmid to RLU from each inducible reporter. Error bars represent standard deviations and some are too small to be seen.
Amplification of IFN-α/β induction pathway components is perturbed in SINV TR339-infected cells
To confirm that the MEFs expressed mRNA for various components of the IFN-α/β induction pathways, we performed RT-PCR on RNA derived from uninfected or infected wt or TBK1−/− MEFs, evaluating the abundance of mRNAs specific for TLR-3, TLR-7, PKR, MDA5, RIG-I, TBK1, IκKε, IRF3 and IRF7. With the exception of IRF7, mRNAs for each gene product were present in mock-treated cells (Figure 4), consistent with IFN-α/β production being induced by both RIG-I and MDA5/PKR agonist (Figure 1 and data not shown). SeV infection induced TBK1-dependent upregulation of mRNAs for RIG-I, MDA5, PKR, IRF7 and TLR-3 at 8 h p.i. suggestive of direct TBK1-dependent, IRF3-mediated upregulation of these mRNAs by the virus infection and/or the amplifying activity of signaling by early IFN-α/β release, consistent with previous findings (Yount et al., 2007). In contrast, no transcriptional upregulation was observed in SINV TR339-infected cells for any of these PRRs or cell signal mediator genes, consistent with the demonstrated failure of these cells to secrete IFN-α/β and the arrest of host macromolecular synthesis in SINV-infected MEFs (Frolova et al., 2002). These data indicate that, in addition to blocking the synthesis of type I IFN, wild-type SINV infection inhibits the IRF3 and/or IFN-α/β-dependent amplification of proteins that allow the host cell to detect the presence of the invading pathogen.
Figure 4. RT-PCR for mRNAs encoding IFN induction pathway components.
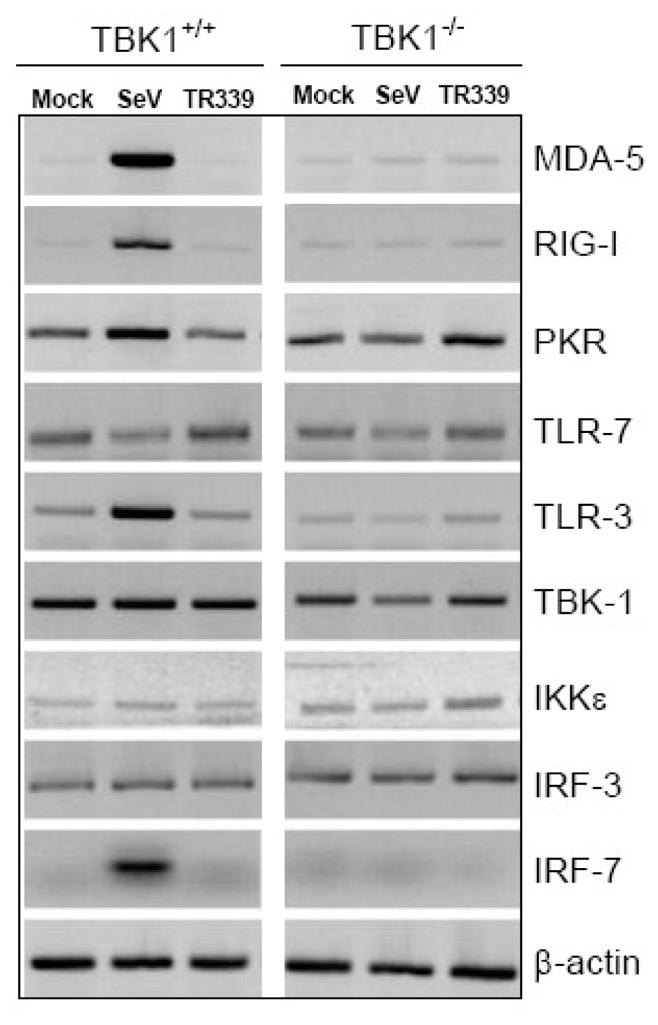
Control or TBK1−/−MEFS were mock-infected or infected with each virus at an MOI of 3 followed by harvesting of total RNA at 8 h p.i. RT-PCR for eachmRNA and agarose gel separation of products was performed as described in Materials and Methods.
A SINV mutant that does not inhibit host macromolecular synthesis induces IFN-α/β after infection of MEFs, dependent upon the presence of TBK1
The nsP2 protein of Old World alphaviruses such as SINV is a multifunctional protein that contains protease activity required for nonstructural protein processing, as well as RNA helicase, NTPase and RNA triphosphatase activities important in RNA replication (Gomez et al., 1999; Rikkonen et al., 1994; Vasiljeva et al., 2000). In addition, several studies have implicated the nsP2 protein in the shut-off of host cell transcription and translation (Breakwell et al., 2007; Garmashova et al., 2006) and it has been proposed that host shutoff functions to antagonize stress responses in infected cells, including the induction of IFN-α/β (Frolova et al., 2002; Breakwell et al., 2007). Single amino acid changes in nsP2 such as the glycine to proline mutation at amino acid position 726 reduce SINV-mediated shut-off of host transcription and translation, abrogate cytopathogenicity and promote IFN-α/β induction (Frolova et al., 2002; Garmashova et al., 2006). Therefore, we utilized a version of SINV TR339 that encodes this mutation to confirm the production of IFN-α/β by infected cells in the absence of transcription/translation arrest and to identify PRRs involved in recognition of SINV by host cells resulting in IFN-α/β production. Furthermore, use of the mutant virus allowed quantitative measurement of IFN-α/β which would be especially important if induction was dependent upon multiple PRRs.
We first confirmed that the nsP726 gly-pro mutant of SINV TR339 (designated SINV 39nc) replicated normally and was non-cytopathic when compared to SINV TR339. Growth curve analyses of SINV TR339 and SINV 39nc demonstrated that the mutant was not defective in replication in the MEFs; although growth was somewhat slower (Figure 5A). Consistent with previous reports of another SINV strain containing this mutation (Frolova et al., 2002), both gross manifestations of cytopathic effect (CPE) examined by light microscopy and shut-off of host protein synthesis evaluated with radioactive label pulse chase analysis were minimal through 36 h p.i. (data not shown). In comparison, CPE was extensive within SINV TR339-infected cultures by 18 h p.i. with 100% CPE occurring by 24 h p.i. and a significant reduction in host protein synthesis was observed by 8 h p.i. (data not shown). We then analyzed the ability of SINV 39nc mutant-infected MEF cultures to produce IFN-α/β. Wt or TBK1-deficient MEFs were infected with SINV 39nc at an MOI of 3 and supernatants were collected and used in biological IFN-α/β assays (Figure 5B). Using a GFP-expressing virus, we determined that this MOI resulted in 100% of the cells being infected at 24 h p.i. Unlike the wild-type SINV TR339, the non-cytopathic virus induced IFN-α/β in wt, but not TBK1−/−, MEFs, suggesting that TBK1 is critical for induction of IFN-α/β by SINV 39nc. Surprisingly, although progeny SINV 39nc virion release could be detected within 6–12 h p.i., IFN-α/β was detected only at later times p.i. (>30 h p.i.), a time at which complete CPE was observed for MEFs infected at the same MOI with SINV TR339.
Figure 5. Virus replication, IFN-α/β induction and IRF3 activation after SINV 39nc infection of MEFs.

A) Control or TBK1−/− MEFs were infected with SINV TR339 (squares) or SINV 39nc (triangles) at an MOI of 3 and supernatants were harvested at various intervals for plaque titration of virus. B) Control or TBK1−/− MEFs were infected with each virus at an MOI of 3 and supernatants were collected for biological interferon assay at the times indicated. Some of these data were published previously (Ryman & Klimstra, 2009). C) Control cells were mock-infected or infected with each virus at an MOI of three and processed for IRF3 dimerization analysis as described in Materials and Methods at 12 h p.i. (SeV) or 24 h p.i. (SINV 39nc). Error bars are standard deviations and some are too small to be seen.
We examined the dimerization of IRF3 and its translocation to the nucleus after SINV 39nc infection of the MEFs. Altered migration of IRF3 consistent with dimerization was observed after 24 h p.i. with SINV 39nc and it appeared similar to that observed with SeV at 12 h p.i. (Figure 5C). In contrast with SINV TR339, dimerization was not detected at 12 h p.i. with SINV 39nc (data not shown) which may reflect the reduced replication rate of SINV 39nc (e.g., Figure 5A).
The effects of IFN-α/β induction in SINV 39nc-infected fibroblasts and their dependence upon the presence of TBK1 were further explored by semi-quantitative RT-PCR for IFN-α/β induction pathway components (Figure 6). At 8 h p.i., consistent with the lack of IFN-α/β production by SINV 39nc-infected cells at this time and similar to SINV TR339, no upregulation of mRNAs for IFN-inducible gene products RIG-I, MDA5, PKR, IRF7 and TLR-3 was observed (Figure 6A). However, both SeV infection and IFN-α/β treatment resulted in upregulation within 8 h. By 24 h p.i., transcription of these mRNAs was clearly upregulated after SINV 39nc infection of wt but not TBK1-deficient cells, while IFN-α/β treatment upregulated these mRNAs in both cell types (Figure 6B).
Figure 6. Effects of SINV 39nc infection upon abundance of mRNAs for interferon induction pathway components.
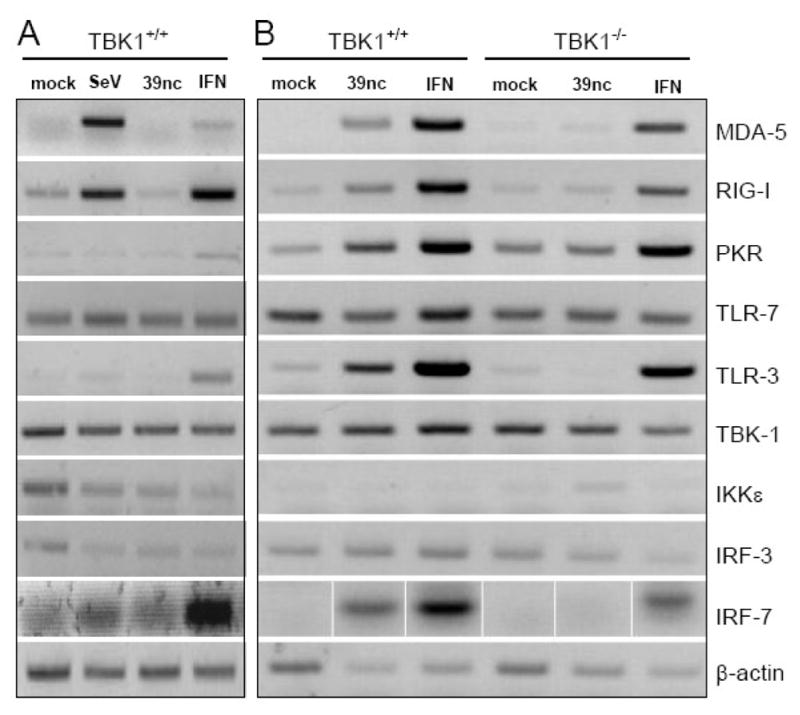
Control or TBK1−/−MEFS were mock-infected, infected with SeV or SINV 39nc at an MOI of 3, or treated with 1000 IU/ml of IFN-α/β followed by harvesting of total RNA at either 8 h p.i. (A), or 24 h p.i. (B). RT-PCR for each mRNA and agarose gel separation of products was performed as described in Materials and Methods.
Taken together with the lack stimulation of IFN-β promoter activity by SINV TR339, these data suggest that wt alphavirus infection precludes IFN-α/β induction in MEFs by blocking gene expression events after migration of activated IRF-3 to the nucleus. The mutation in SINV 39nc overcomes this blockade most likely through failure to arrest host macromolecular synthesis. However, it should be noted that activation of the IFN-α/β induction pathway by the mutant virus is evident only relatively late after infection.
PKR and MDA5, but not RIG-I, are important for detection of the non-cytopathic SINV in MEFs
It has been established that the PRRs for RNA viruses include the TLRs 3 and 7, and the DexD/H box helicases, RIG-I and MDA5 and that these PRRs function in a cell type-specific manner (see Introduction). In addition to these canonical PRRs for RNA viruses, PKR may detect poly(I:C) and activate the transcription factor NFκB (Bonnet et al., 2000; Gil et al., 2000; Zamanian-Daryoush et al., 2000) and has been implicated in IFN-α/β induction by West Nile virus, another positive-sense RNA virus (Gilfoy & Mason, 2007). Previously, we described a role for PKR in IFN-α/β induction by bone marrow-derived dendritic cells (DCs), demonstrated by a lag in IFN production from cells lacking PKR and RNase L, but not RNase L alone (Ryman et al., 2002). Recently, a role for PKR in IFN-α/β induction by MEFs was shown during SFV infection (Barry et al., 2009). Therefore, PKR may also be involved in alphavirus-induced IFN-α/β production. We have excluded TLR-3 and TLR-7 from these analyses because preliminary experiments revealed that the MEFs did not produce IFN-α/β in response to their agonists (cell-surface poly(I:C) for TLR-3 or imiquimod for TLR-7) suggesting that the TLR pathways were nonfunctional (data not shown). To assess the role of RIG-I, MDA5 and PKR in IFN-α/β induction in the MEF model system, we first used siRNA technology to specifically silence expression of each mRNA, followed by infection with SeV or SINV 39nc. Reduction in the abundance of appropriate mRNAs was confirmed by semi-quantitative PCR (Figure 7B). As expected, SeV-mediated IFN-α/β induction was greatly reduced (p<0.001) in the presence of RIG-I siRNA; however, no change in the level of supernatant IFN-α/β was observed in the presence of MDA5 siRNA (p>0.05; Figure 7A). With SINV 39nc, a significant (p<0.001) reduction in supernatant IFN-α/β of approximately 50% was consistently observed from MDA5 siRNA-treated MEFs whereas, surprisingly, knockdown of RIG-I mRNA reproducibly increased IFN-α/β production in response to SINV 39nc (p<0.05). Finally, we made numerous attempts to reduce PKR mRNA using the siRNA approach; however, knockdown of PKR mRNA and protein was minimal at the time of IFN-α/β measurement after SINV 39nc infection (>30 h p.i.) perhaps due to induction of PKR gene transcription by the virus infection (Figure 6).
Figure 7. siRNA-mediated knockdown of MDA5 or RIG-I.

A) Control MEFs were transfected with siRNAs specific for cyclophilin B, RIG-I, MDA5, a non-targeting (negative) control or treated with transfection reagent (DF3). At 48 h post-treatment, cells were infected with SeV or SINV 39nc at an MOI of 3 and supernatants were collected for biological IFN-α/β assay at 12 h p.i. (SeV; open bars) or 48 h p.i. (SINV 39nc; solid bars). Results for other treatments are normalized to the cyclophilin B siRNA treatment which is set to 100%. Error bars are standard deviations. B) Semi-quantitative RT-PCR for mRNA of the target genes encoding cyclophilin B, RIG-I, MDA5 or β-actin which was used as a loading control. Samples are duplicates of the treatments described above. At 96 h post siRNA transfection, total RNA was harvested from mock-infected cells and processed for RT-PCR and agarose gel separation as described in Materials and Methods.
To further assess the role of PKR and MDA5 in the alphavirus IFN-α/β induction pathway, we utilized two additional strategies: infection of MEFs deficient in either PKR or MDA5 (Figure 8) or pharmacological inhibition of PKR (data not shown). As expected, neither the MDA5−/− nor PKR−/− cells produced IFN-α/β after SINV TR339 infection (data not shown), while SeV infection of either cell type induced ~500 IU/mL IFN-α/β, similar to wt cells. After SINV 39nc infection, a decrease in the amount of detectable IFN-α/β was observed in PKR-deficient MEFs (125 IU/mL) when compared to wt MEFs (1667 IU/mL); however, IFN-α/β was still produced in the absence of PKR suggesting the presence of another PRR capable of detecting SINV 39nc infection. Similar results were observed with PKR/RNase L−/− MEFs (data not shown). Consistent with the siRNA results, MDA5−/− cells produced ~80% less IFN-α/β versus controls after SINV 39nc infection (p<0.01).
Figure 8. Interferon induction after infection of MDA5−/− or PKR−/− MEFs.
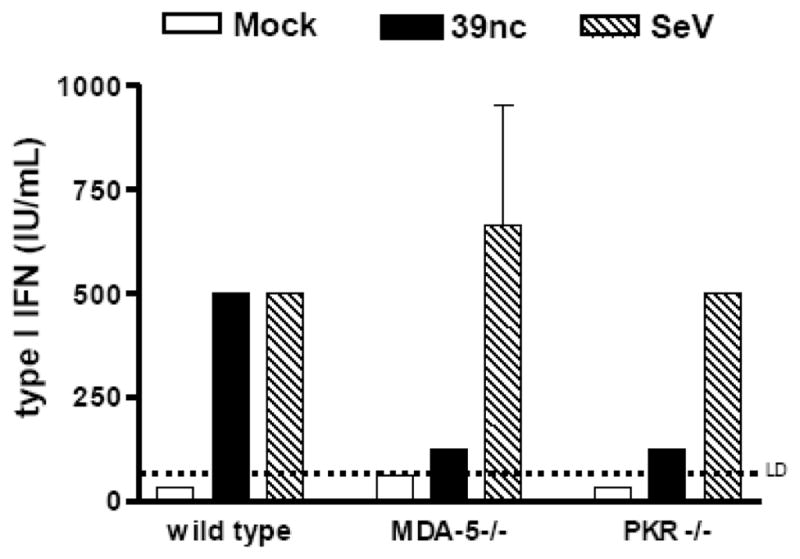
Control MEFs (a different line for each knockout) or MDA5-deficient or PKR-deficient MEFs were infected with each virus at an MOI of 3. Supernatants were harvested at 24 h p.i. (SeV) or 48 h p.i. (SINV 39nc) for biological IFN-α/β assay. Each bar represents the average of triplicate samples. Error bars are standard deviations and some are too small to be seen.
Treatment of four independently-derived wt C57BL/6 MEF cell lines with the PKR inhibitor 50μM 2-aminopurine (Zinn et al., 1988), had no effect upon IFN-α/β induction by SeV while it reduced SINV 39nc-mediated induction in each cell line to varying degrees. However the reduction was significant (p<0.05) versus a DMSO control in only one cell line (data not shown). We also attempted to use another commercially available PKR inhibitor (Calbiochem) but this drug appeared to lack specificity for PKR in that, while IFN-α/β induction by SINV 39nc was consistently reduced in a dose-dependent manner, early IFN-α/β induction by SeV (<12 h p.i.) was inhibited if the drug was maintained in the cultures longer than 1 h and, even when cells were exposed to the drug for 1 h only (as reported (Gilfoy & Mason, 2007), IFN-α/β produced in response to SeV was reduced when measured after 12 h p.i.
Finally, since a number of reports have implicated activation of the NFκB pathway in PKR-mediated IFN-α/β induction (Kumar et al., 1994; Kumar et al., 1997; Uetani et al., 2000), we examined the stimulation of a transfected NFκB-dependent PRDII promoter-driven luciferase reporter (McWhirter et al., 2004) after infection with SeV or SINV 39nc. In contrast to SINV TR339, which did not stimulate reporter activity over mock infected cells (data not shown), both SeV (consistent with the results of others; e.g., (Zhou & Perlman, 2007) and SINV 39nc resulted in significant promoter activity after infection (Figure 9; p<0.001 versus mock for both). Taken together, our results indicate that MDA5 and PKR contribute to alphavirus IFN-α/β induction in fibroblasts; however, we cannot exclude the activity of additional PRRs that may also detect SINV 39nc or that detection of wild-type viruses may not be identical.
Figure 9. Activation of the NFκB pathway after SINV 39nc infection.
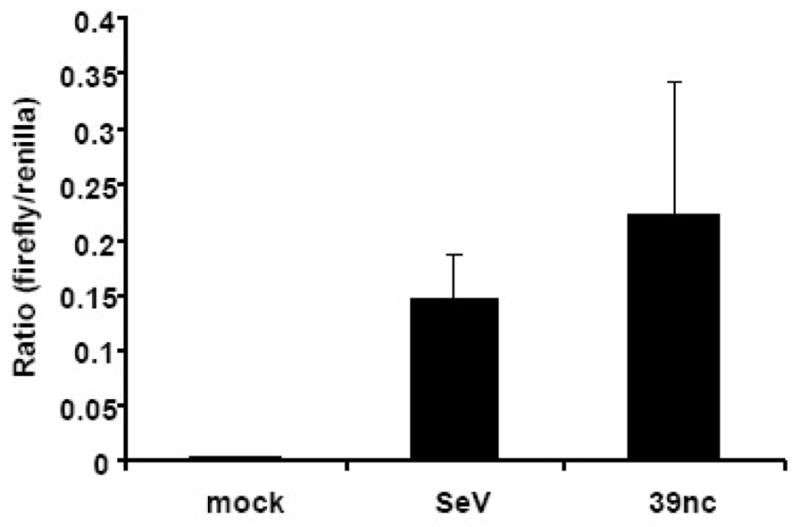
Wild type C57BL/6 MEFs were co-transfected with PRDII-dependent firefly luciferase and constitutive renilla reporters. At 24 h post-transfection, cells were mock-infected or infected with either SeV or SINV 39nc and lysates were collected for a dual luciferase assay at 12 h p.i. (SeV) or 48 h p.i. (mock and SINV 39nc) hpi. Data are presented as the ratio of relative light units (RLU) from a control renilla luciferase-expressing plasmid to RLU from each inducible reporter. Error bars are standard deviations and some are too small to be seen.
MEFs are competent for IFN-α/β induction early after alphavirus infection
The data described above suggest that SINV and likely other alphaviurses antagonize IFN-α/β induction in MEFs primarily through arrest of host macromolecular synthesis. However, since the activation of inductive pathways appears to be delayed compared with SeV-infected cells and IFN-α/β production is not observed at any time p.i. with wt SINV, we sought to determine if infected cells were competent for IFN-α/β production early after infection prior to the time at which shut-off occurred. Failure of cells to produce IFN-α/β at early times would imply additional mechanisms of antagonism. Therefore, we infected wt MEFs with SINV TR339 or SINV 39nc and co-treated with RIG-I or PKR/MDA-5 agonists, SeV or poly(I:C) respectively (Kato et al., 2005; Yoneyama et al., 2004; Gitlin et al., 2006; Diebold et al., 2003), either simultaneously or at 6 h p.i. with SINV. Supernatants were collected and used in biological IFN assays to compare the amount of IFN-α/β produced by SINV TR339-infected cells to mock-infected cells (Figure 10). Simultaneous infection of MEFs with SINV TR339 and SeV resulted in ~20% reduction in IFN-α/β production compared to mock/SeV co-treatment. When SeV infection occurred 6 h p.i. with wild-type SINV, IFN-α/β production was reduced by ~80% when compared to mock-infected; although IFN-α/β was still reproducibly detected (Figure 10A). After poly(I:C) transfection of SINV TR339-infected cells, IFN-α/β induction was attenuated similar to that for SeV when delivered simultaneously and slightly greater than for SeV (2-fold) when the poly(I:C) was given 6 h p.i. with SINV TR339. Therefore, provided that PAMPs are detected by host PRRs, both the RIG-I and MDA-5/PKR induction pathways are functional early after wt SINV infection in a time-frame in which the cells can produce IFN-α/β. We also tested if the reduction in SeV-initiated IFN induction observed with wild-type SINV TR339 was associated with ability of this virus to shut-off host processes. Accordingly, treatment of SINV 39nc-infected cells with SeV or poly(I:C), either simultaneous with or 6 h p.i., did not result in any reduction of IFN-α/β produced when compared to mock treated cells (Figure 10B). From these data we infer that the PRRs that detect SINV in the MEFs do not do so until relatively late in infection, by which time arrest of host macromolecular synthesis prevents the production of IFN-α/β.
Figure 10. Effects of SINV infection upon IFN-α/β induction by IFN pathway agonists.

WT MEFs were infected with SINV TR339 (A) or SINV 39nc (B) andtreated with SeV (infection) or poly(I:C) (transfection) either simultaneously with (open bars) or 6 hours after (black bars) SINV infection as described in Materials and Methods. IFN-α/β levels were measured at 24 hours post SINV infection and are presented as a percentage of those in control wells that were treated with SeV or poly(I:C) but not infected with SINV. Interferon was measured with either the IFN-α/β bioassay or an IFN-β ELISA (PBL). Error bars are standard deviations and some are too small to be seen.
Discussion
We initiated these studies to examine the PRRs responsible for IFN-α/β induction after alphavirus infection of cultured cells choosing the MEF model system due to the abundance of genetically-modified mice with deletions of components of the innate immune response and the availability of MEF cell lines derived from them. However, in initial studies, wt MEFs infected with alphaviruses including SINV, VEEV, EEEV and CHIKV failed to secrete significant IFN-α/β. This was in contrast to the well-studied negative-sense virus SeV which, we and others have observed, induces high levels of IFN-α/β in a TBK1-dependent manner by 8–12 h p.i. (e.g., (McWhirter et al., 2004). With SINV, we demonstrated that this phenotype reflected minimal upregulation of transcription or protein production from genes driven by the IFN-β promoter implying an antagonism at, or upstream of, transcription of the IFN gene mRNAs; although, other activities that alter mRNA abundance may also be involved. Increased IRF3 dimerization, nuclear translocation and DNA binding activity could be demonstrated in the MEFs after SINV TR339 infection. Similarly, but perhaps more efficiently than SINV TR339, VEEV caused an increase of IRF3 nuclear translocation after infection. In contrast to SINV TR339, the SINV 39nc virus which fails to rapidly arrest host macromolecular synthesis after infection induced TBK1-dependent IRF3 activation and the release of detectable IFN-α/β into supernatants. However, in comparison with SeV this was observed later after infection (30 h p.i.). Examination of the PRRs responsible for IFN-α/β induction by the noncytopathic virus implicated PKR and MDA5, but not RIG-I, as host detectors of replicative intermediates in the MEFs.
Our previous results revealed a role for PKR in the efficiency of induction of IFN-α/β early after infection of bone marrow-derived conventional DC cultures (Ryman et al., 2002). The current results confirm these data and suggest that PKR may also play a role in IFN-α/β(induction in MEFs. Several recent publications have implicated PKR in IFN-α/β induction by positive-sense RNA viruses and/or poly(I:C) (Gilfoy & Mason, 2007; Diebold et al., 2003; Ryman et al., 2002; Balachandran et al., 2000; Carpentier et al., 2007; Yang et al., 1995)}(Kalali et al., 2008). The primary mechanism through which PKR is thought to stimulate IFN-α/β production is by inducing phosphorylation of IκB and activation of the NFκB transcription pathway (Kumar et al., 1994; Kumar et al., 1997; Uetani et al., 2000); although evidence has also been presented that PKR is involved in signaling downstream of TLRs (Oganesyan et al., 2006). In our studies, IFN-α/β induction by SINV 39nc was absolutely dependent upon the presence of TBK1 and appeared to be associated with activation of the NFκB-dependent PRDII promoters.
Our data also indicate that MDA5 is partially responsible for IFN-α/β induction by SINV 39nc in MEF cultures. Recognition of alphavirus infection by MDA5 is logical in that dsRNA is a strong activator of this RNA helicase (Gitlin et al., 2006; Kato et al., 2006) and high levels of dsRNA are produced as a consequence of alphavirus replication (Griffin, 2001). The fact that inhibition or deletion of PKR also reduced IFN-α/β induction may suggest that the MDA5 and PKR induction pathways or expression levels are interdependent. PKR associates with TRAF3, an essential component of the intracellular IFN-α/β induction pathway and binding partner of TBK1 (Oganesyan et al., 2006), perhaps accounting for this phenotype. This also may explain reports in the literature in which either MDA5 or PKR were independently shown to be essential for intracellular detection of poly(I:C) in cells of myeloid lineage (Kato et al., 2006; Gitlin et al., 2006; Diebold et al., 2003).
Interestingly, reduction of RIG-I mRNA abundance actually increased IFN-α/β induction by SINV 39nc which may be secondary to increased virus replication in the cultures. Increased IFN-α/β induction was also observed from SINV 39nc-infected wt MEFs expressing a tetracycline-inducible dominant-negative RIG-I mutant (data not shown). These results suggest several interpretations with respect to the role of RIG-I: i) this PRR exerts a direct antiviral activity versus SINV; ii) constitutive RIG-I activity is responsible for induction of other antiviral activities that suppress SINV replication in cells; or iii) RIG-I is a minor PRR for SINV and induces amounts of IFN-α/β that are below the limit of detection of our assays but, nonetheless, inhibit SINV replication. In each case, it is possible that when RIG-I activity is suppressed by siRNA-mediated knockdown or dominant negative mutant expression, SINV replication and signaling through other PRRs is increased. Overall, our data implicate the involvement of MDA5 and PKR in IFN induction by alphaviruses. However other PRRs, for example the LGP2 helicase (Venkataraman et al., 2007), may also be involved.
In contrast with our results, Gorchakov and coworkers (Gorchakov et al., 2004) indicated that IFN-α/β induction was only slightly reduced in supernatants of PKR−/− cell cultures infected with a nsP2 726G mutant SINV compared with Swiss 3t3 MEFs used as controls; however, the magnitude of induction was not shown and wt MEFs analogous to the PKR-deficient cells were not tested. It is possible that the biological assay employed by these investigators was more sensitive than our assay or there might be differences in IFN-α/β induction between the SINV Toto1101 strain used by that group as a backbone for mutagenesis and the SINV TR339 strain we have used. Occasionally we detected higher IFN-α/β induction in the PKR−/− cell cultures; however, this was found primarily when the cells were very confluent (data not shown). It is possible that these conditions affected the expression level or activity of other compensatory PRRs.
Studies with SFV support our results that IRF3 translocation is not inhibited after infection in cells that fail to induce IFN-α/β (Breakwell et al., 2007). Interestingly, studies examining the activities of alphavirus proteins that promote host shut-off have indicated that the capsid protein of New World alphaviruses (but not Old World alphaviruses) may arrest host cell transcription by disruption of nuclear translocation of some proteins (Atasheva et al., 2008). A similar transcription inhibitory function has been attributed to the nsP2 protein of Old World alphaviruses (but not New World alphaviruses); however, the mechanism through which nsP2 promotes shut-off remains uncharacterized (Garmashova et al., 2007; Breakwell et al., 2007). Our data suggest that type I IFN induction is blocked by Old World and at least some New World alphaviruses by interference with accumulation of IFN-α/β gene mRNAs. However, we do not observe IFN-α/β production by MEFs infected with non-propagative VEEV replicon particles that do not express the capsid protein (data not shown), suggesting that another host process is also blocked (e.g., translation; (J. Yin C. Gardner, C. Burke, K. Ryman and W. Klimstra, J. Virol. in press).
The results we describe may indicate a mechanism of antagonism of the IFN-α/β response of MEFs by alphaviruses in addition to virus-mediated host macromolecular shut-off as it appears that MEFs fail to detect SINV replicative intermediates early after infection. In our system, production of IFN-α/β is not detected in response to SINV TR339 infection prior to the time at which arrest of host transcription/translation occurs although co-treatment with RIG-I or PKR/MDA-5 agonists during this time induces the release of IFN-α/β from SINV-infected cells. With the shut-off-defective SINV 39nc, detectable IFN-α/β is induced only > 30 h p.i., a time at which virus titer has increased multiple orders of magnitude over input and replicative intermediates must be abundant. Therefore, it appears that virus replicative intermediates remain hidden from cellular PRRs during the earlier stages of infection. We (Ryman & Klimstra, 2009) and others (Gorchakov et al., 2008) have suggested this hypothesis previously for SINV and a similar mechanism has also been proposed to explain the failure of mouse hepatitis virus to induce an IFN-α/β response in MEFs (Zhou & Perlman, 2007) and delayed induction by West Nile virus infection (Fredericksen et al., 2004). However the observation that SINV 39nc replication was delayed versus SINV TR339 may suggest that production or accumulation of replicative intermediates that trigger IFN-α/β induction may simply be delayed such that PRRs are triggered only by 30 h p.i. with the noncytopathic virus. Also, as yet uncharacterized differences between non-cytopathic and cytopathic alphaviruses must be considered. Understanding of the particular alphavirus replicative intermediates that stimulate PRRs and the timing of their production will be required to clarify this issue.
Finally, any discussion of antagonism or evasion of host innate immunity should be considered in light of the documented and expected responses of intact vertebrate hosts to infection with alphaviruses. Upon subcutaneous inoculation, arthropod-borne viruses such as alphaviruses interact with cells in interstitial tissue spaces such as DCs and macrophages and virions are also likely introduced into the bloodstream resulting in infection of similar cells in the spleen and elsewhere (Ryman et al., 2000; MacDonald & Johnston, 2000). Subsequently, depending upon the virus, pancreatic cells, osteoblasts, skeletal muscle, neurons and other cells of the central nervous system are targeted (Griffin, 2001). In contrast with the failure of all alphaviruses tested to induce substantial IFN-α/β in MEF cultures, multiple studies have shown robust IFN-α/β responses after infection of animals with VEEV and SINV (Klimstra et al., 1999; Ryman et al., 2000; Griffin, 2001; Charles et al., 2001; Gardner et al., 2008) and our global transcription analysis of SINV-infected DC cultures indicated upregulation of the transcription of IFN-α/β and other innate response genes followed by accumulation of high levels of IFN-α/β in supernatants (Ryman et al., 2005; Ryman et al., 2002). Conversely, we have found that primary murine osteoblast and cortical neuron cultures, like MEFs, fail to produce substantial IFN-α/β after infection with SINV, VEEV, EEEV or CHIKV (C.W. Burke, C.L. Gardner, J. Yin, K.D. Ryman and W.B. Klimstra unpublished observations and J. Yin, C. Gardner, C. Burke, K. Ryman and W. Klimstra, J. Virol. in press). Therefore, the capacity to respond to infection by production of antiviral cytokines may be highly dependent upon the characteristics of interaction of the virus with a particular cell-type, with MEFs more closely resembling other non-myeloid cells. These observations underscore the importance of examining the inductive phase of the innate immune response in cells and tissues relevant to virus replication and disease pathogenesis in vivo.
Materials and Methods
Cell lines
TBK1 −/− (Perry et al., 2004) PKR−/−(Yang et al., 1995), MDA5−/−(Gitlin et al., 2006), PKR/RNaseL−/− (Zhou et al., 1999)or wild type C57BL/6 mouse embryonic fibroblasts (MEFs) and 293 human kidney cells were maintained in filtered Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco), 0.29 mg of L-glutamine/ml (mediatech), 1% nonessential amino acids (Mediatech), and 1X β-mercaptoethanol (Gibco). L929 and Baby Hamster Kidney (BHK-21) cells (ATCC) were maintained in alpha minimal essential medium (AMEM) supplemented with 10% donor calf serum (Gibco), 2.9 mg of tryptose phosphate/ml, 0.29 mg of L-glutamine/ml, and 100U of penicillin/ml and 0.05mg of streptomycin/ml (Mediatech). All cells were cultured at 37°C and 5% CO2.
Viruses and antibodies
The SINV TR339 strain of SINV has been described (Klimstra et al., 1998). The FL93-939 cDNA clone of North American EEEV, ZPC738 cDNA clone of VEEV (a gift from Dr. Scott Weaver, UTMB, Galveston) and the La Reunion cDNA clone of CHIKV (a gift from Dr. Stephen Higgs, UTMB, Galveston) have been described (Aguilar et al., 2007; Anishchenko et al., 2004; Tsetsarkin et al., 2006). SeV was obtained from ATCC. The SINV TR339-nsP2 726-G virus (designated SINV 39nc) was constructed by overlap PCR using a mutagenic primer that introduced a C–G mutation at nucleotides 3855 and 3856 of the SINV nsP2 gene resulting in a proline-glycine coding change previously shown to greatly reduce the cytopathogeniciy and transcription/translation shut-off of the Toto1101 strain of SINV (Frolova et al., 2002).
Antisera reactive with PKR, IRF3, tubulin and lamin A/C were obtained from Santa Cruz Biotechnology and an additional antiserum reactive with IRF3 was obtained from Zymed.
Virus stocks, infections and drug treatments
To prepare the alphavirus stocks, the cDNA clone was linearized and used in an in vitro transcription reaction. The resulting RNA was electroporated into BHK-21 cells (2 pulses at 0.22kV, 1.0mF of capacitance). Virus containing culture supernatants were collected 24 hours post electroporation, clarified by centrifugation and used as viral stocks. Alphavirus stocks were titered by standard plaque assay on BHK-21 cells or on each of the corresponding cell types and expressed as PFU per ml. Sendai virus Cantell strain (ATCC) titer was calculated by plating serial dilutions on BHK-21 cells and visualizing the infectious dose that caused 50% CPE. Encephalomyocarditis virus stocks were amplified in and titered on BHK cells. Unless otherwise stated MEFs were infected at an MOI of 3 with alphaviruses or 3 TCID50 of SeV for one hour followed by two washes with Dulbecco’s phosphate-buffered saline with calcium and magnesium (PBS; Mediatech) supplemented with 1% donor calf serum followed by medium replacement. For co-treatment experiments, cells were either simultaneously infected with SeV and SINV or simultaneously infected with SINV and transfected with poly(I:C) (75μg/ml; Dharmafect 4 reagent) or infected with SINV and infected/transfected with SeV or poly (I:C) six hours after SINV.
Cells were treated with several doses of a PKR inhibitor (Calbiochem #527450; ranging from 1μM to 100μM) prior to infection with the drug either replaced or not replaced after infection depending upon the experiment. Cells were treated with 50μM 2-aminopurine (dimethyl sulfoxide [DMSO] diluent) for 1 hour prior to infection. Cells were treated with the TLR-7 agonist imiquimod or TLR-3 agonist poly(I:C) (75μg/ml) (Invivogen) and IFN induction was measured at 12 hours post treatment. Poly(I:C) was also transfected into cells (Dharmafect 4 reagent) either alone or after SINV infection as above.
Interferon alpha/beta bioassay
The concentration of biologically active IFN-α/β was measured by standard IFN assay on L929 murine fibroblast cells with Encephalomyocarditis virus (EMCV) as the challenge virus (described previously (Trgovcich et al., 1996). Briefly, L929 cells were seeded in 96-well plates at a concentration of 3x104 cells/well and incubated for 24 hours. Experimental samples were adjusted to a volume of 250 μl and acidified to pH 2 with 1N HCl. Acidified samples were incubated 24 hours at 4°C. The samples were returned to pH 7 with 2N NaOH and 100 μl of each were put into the first well of the 96-well plates in duplicate. The samples were two-fold serially diluted down the length of the plate and the plates were incubated 24 hours at 37°C. Commercially available IFN-α/β (Access Biomedical) of a known concentration was used on each plate to serve as a standard and was treated in the same manner as the samples. EMCV was added to all wells and incubated for 24 hours at 37°C and then cells were stained with crystal violet. The end point, defined as the amount of IFN-α/β necessary to protect 50% of the indicator cells from EMCV CPE, was then used to calculate the IFN-α/β concentration by comparing the experimental samples to the IFN-α/β standard.
Reporter transfection
Lipofectamine2000 (Invitrogen), or DharmaFECT 3 (Dharmacon) reagent was used to transfect cells one day post plating following the manufacturer’s protocol. The following concentrations of DNA were used unless otherwise stated: pβLUX, 0.5 μg; pRL-SV40, 0.02 μg; and PRDI/III and PRDII, 0.5 μg. Transfected cells were incubated at 37°C with 5% CO2 for 18 hours prior to infection. The IFN-β promoter, pβLUX, and control, pRL-SV40, plasmids were gifts from Dr. Barbara Sherry, North Carolina State University (Noah et al., 1999)) and the PRDI/III and PRDII plasmids were gifts from Dr. Tom Maniatis, Harvard University (McWhirter et al., 2004)).
RT-PCR detecting mRNA for IFN induction pathway components
Total cellular RNA was harvested from MEFs using the RNeasy kit (Qiagen). Random hexamers were used for cDNA synthesis using Superscript III reverse transcriptase and equal volumes of total cellular RNA. Equal concentrations of cDNA were then used in PCR reactions using primers specific to each target mRNA. Amplicons were then separated on agarose gels followed by ethidium bromide staining. Control reactions in which no reverse transcriptase was added indicated that amplified products were derived from mRNA.
Dual luciferase assay
Dual luciferase assays were performed following the manufacturers protocol (Promega). Briefly, the surface of the cells was washed twice with PBS. Passive lysis buffer was added and the plates were incubated at room temperature on an orbital shaker for 15 minutes. Homogeneous lysates were made by scraping the wells with a rubber policeman, were collected into 1.5 ml tubes and were then stored at −80°C. Measurements were made using the POLARstar OPTIMA (BMG LABTECH) and results were normalized by dividing firefly luciferase activity by renilla luciferase activity.
Oligonucleotide pull down Assay
ONPD assays were performed similarly to methods previously described (Melchjorsen et al., 2005). Briefly, virus treated cells were washed one time with PBS, collected, and lysed in a buffer containing 1M Hepes pH 7.4, 1.5 M KCl, 1 M DTT, 50% glycerol, 0.5 M EDTA, 250 mM EGTA, 10% Triton-x-100, 100 mM Na3VO4, and protease inhibitors (Roche). Lysates were incubated sequentially with neutravidin-agarose beads coupled to 5′-biotinylated oligonucleotides of the IRF3 binding sites in the IFN-β promoter (Siren et al., 2005). Agarose bound proteins were disassociated by boiling in SDS sample buffer for 7 minutes and equal amounts of protein were run on 10% SDS-PAGE gels. Proteins were transferred to PVDF membrane using the Trans-Blot electrophoretic transfer system (BioRad) at 0.9 A for two hours. Membranes were blocked in TBST containing 5% milk overnight at 4°C. The following day, IRF3 was detected using the Zymed anti-IRF3 antibody (1:1000) in TBST containing 1% milk. IRF-3 binding was calculated by densitometric analysis of films using the VersaDoc instrument and software (BioRad).
Native PAGE and western blot
Native PAGE analysis was performed following a previously described method (Iwamura et al., 2001). Cells were lysed using a buffer containing 50mM Tris-Cl, pH 8.0, 1% NP40, 150 mM NaCl, phosphatase inhibitor cocktail (Sigma), and complete protease inhibitor (Roche). Collected cells were vortexed and the insoluble fraction was removed by centrifugation (15,000 rpm, 10 minutes). 4–15% precast acrylamide gels (BioRad) were pre-run at 4°C 40 mA for 30 min with 0.2% DOC in cathode chamber buffer. 50 μg of protein in 2X sample buffer without SDS and β-ME was loaded and run at 4°C. Proteins were transferred to PVDF membrane using the mini Trans-Blot electrophoretic transfer system (BioRad). Membranes were blocked in TBST containing 5% milk overnight at 4°C. IRF3 was detected using the Zymed anti-IRF3 antibody (1:1000) in TBST containing 1% milk.
siRNA transfection and gene knockdown confirmation
PKR (Cat# L-040807-00-0005), MDA5 (Cat# L-048303-00-0005), RIG-I (Cat# L-065328-00-0005), positive control Cyclophilin B (Cat# D-001820-20-05), and negative control non-targeting (Cat# D-001810-10-05) synthetic siRNA were purchased from Dharmacon RNA technologies. Transfection of siRNAs was carried out following manufacturer’s suggested protocol. Briefly, MEF/3T3 cells were plated into 24-well dishes 18–24 hours prior to transfection. 100nM siRNA was transfected into cells using DharmaFECT 3 (DF3) transfection reagent 48 hours prior to mock, SeV, or non-cytopathic SINV infection. Supernatants were collected 24 and 48 hours post infection and used in biological IFN assays. RNA was collected from mock-infected cells at 48 hours post infection using the RNeasy Kit (Qiagen) and used to confirm knockdown of gene expression by RT-PCR (methods explained in (Zhang et al., 2007)).
Cell fractionation
Mock or virus infected cells were trypsinized, collected, and washed with PBS. Cells were then washed with hypotonic buffer (10mM Hepes pH7.6, 1.5mM MgCl2, 10mM KCl) and split to collect a total cell lysate and a separated lysate. Cells used to collect a total cell lysate were pelleted, resuspended in RIPA buffer, and incubated on ice for 20 minutes. After incubation the total cell lysate was centrifuged to pellet cellular debris and the supernatant was collected. Cells used to collect cytoplasmic and nuclear fractions were pelleted, resuspended in 1mL hypotonic buffer, and incubated for 90 minutes on ice. After 60 minutes of incubation, 0.5% NP-40 was added to the cells in hypotonic buffer. At the end of the incubation, cells were disrupted using 30 strokes of a Dounce homogenizer then spun at 2800g for 5 minutes to pellet the nuclei. The supernatant was removed and kept as the cytoplasmic fraction. The pelleted nuclei were resuspended in 1mL of nuclei buffer (250mM sucrose, 5mM MgCl2, 25mM KCl, 20mM Tricine-NaOH pH7.8) and then layered over an Optiprep Discontinuous gradient (35%, 30%, and 25%). Nuclei were then centrifuged for 30 minutes 10,000g at 4°C. Clean nuclei were collected from the 30/35% interface and pelleted. Nuclei were lysed using RIPA buffer as described above. Equal protein concentrations of total, nuclear, and cytoplasmic fractions were loaded, lysates were separated on SDS PAGE gels and subjected to western blot to detect IRF3, and nucleus-associated proteins lamin A/C and tubulin.
Fluorescence microscopy and antibody staining of cells
An IRF3/GFP fusion plasmid (a gift from Dr. Adolfo Garcia-Sastre, Mount Sinai School of Medicine; (Mibayashi et al., 2007) was transfected into MEF/3T3 cells 18 hours prior to infection. Cells were mock-infected or infected with SINV TR339, VEEV or SeV at a MOI of 10 and GFP translocation was monitored using a Nikon inverted fluorescence microscope. At 16 hours post infection, cells were stained with virus-specific antisera. For cell expressing virus antigen and GFP, the percentage of cells counted with GFP in the nucleus versus cytoplasm were calculated for 50 cells at each sampling time.
Statistical analysis
Statistical significance was determined using a two-tailed Student’s t test with two sample equal variance.
Acknowledgments
We would like to thank Drs. Scott Weaver, Steven Higgs, Robert Johnston, Barbara Sherry, Thomas Maniatis and Adolfo Garcia-Sastre for generous gifts of reagents. DeAquinita McKinney, Tanya Debenport and Michael Farmer provided excellent technical assistance. This work was supported by NIH grants R21AI072350 (WBK) and R21AI069158 (KDR) and grants from NIAID/NIH through the Western Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (WRCE) U54 AI057156 (KDR and WBK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Aguilar PV, Weaver SC, Basler CF. Capsid protein of eastern equine encephalitis virus inhibits host cell gene expression. J Virol. 2007;81:3866–3876. doi: 10.1128/JVI.02075-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anishchenko M, Paessler S, Greene IP, Aguilar PV, Carrara AS, Weaver SC. Generation and characterization of closely related epizootic and enzootic infectious cDNA clones for studying interferon sensitivity and emergence mechanisms of Venezuelan equine encephalitis virus. J Virol. 2004;78:1–8. doi: 10.1128/JVI.78.1.1-8.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasheva S, Garmashova N, Frolov I, Frolova E. Venezuelan equine encephalitis virus capsid protein inhibits nuclear import in Mammalian but not in mosquito cells. J Virol. 2008;82:4028–4041. doi: 10.1128/JVI.02330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, Barber GN. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity. 2000;13:129–141. doi: 10.1016/s1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- Barry GM, Breakwell L, Fragkoudis R, Attarzadeh-Yazdi G, Rodriguez-Andres J, Kohl A, Fazakerley JK. PKR acts early in infection to suppress Semliki Forest virus production and strongly enhances the type-I interferon response. J Gen Virol. 2009 doi: 10.1099/vir.0.007336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol Cell Biol. 2000;20:4532–4542. doi: 10.1128/mcb.20.13.4532-4542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breakwell L, Dosenovic P, Karlsson Hedestam GB, D’Amato M, Liljestrom P, Fazakerley J, McInerney GM. Semliki Forest virus nonstructural protein 2 is involved in suppression of the type I interferon response. J Virol. 2007;81:8677–8684. doi: 10.1128/JVI.02411-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier PA, Williams BR, Miller SD. Distinct roles of protein kinase R and toll-like receptor 3 in the activation of astrocytes by viral stimuli. Glia. 2007;55:239–252. doi: 10.1002/glia.20450. [DOI] [PubMed] [Google Scholar]
- Charles PC, Trgovcich J, Davis NL, Johnston RE. Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology. 2001;284:190–202. doi: 10.1006/viro.2001.0878. [DOI] [PubMed] [Google Scholar]
- Couderc T, Chretien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, Touret Y, Barau G, Cayet N, Schuffenecker I, Despres P, Arenzana-Seisdedos F, Michault A, Albert ML, Lecuit M. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008;4:e29. doi: 10.1371/journal.ppat.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al Shamkhani A, Flavell R, Borrow P, Reis e Sousa Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Fragkoudis R, Breakwell L, McKimmie C, Boyd A, Barry G, Kohl A, Merits A, Fazakerley JK. The type I interferon system protects mice from Semliki Forest virus by preventing widespread virus dissemination in extraneural tissues, but does not mediate the restricted replication of avirulent virus in central nervous system neurons. J Gen Virol. 2007;88:3373–3384. doi: 10.1099/vir.0.83191-0. [DOI] [PubMed] [Google Scholar]
- Fredericksen BL, Smith M, Katze MG, Shi PY, Gale M., Jr The host response to West Nile Virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J Virol. 2004;78:7737–7747. doi: 10.1128/JVI.78.14.7737-7747.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova EI, Fayzulin RZ, Cook SH, Griffin DE, Rice CM, Frolov I. Roles of nonstructural protein nsP2 and Alpha/Beta interferons in determining the outcome of Sindbis virus infection. J Virol. 2002;76:11254–11264. doi: 10.1128/JVI.76.22.11254-11264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner CL, Burke CW, Tesfay MZ, Glass PJ, Klimstra WB, Ryman KD. Eastern and Venezuelan Equine Encephalitis Viruses Differ in Their Infectivity for Dendritic Cells and Macrophages: The Impact of Altered Cell Tropism on Pathogenesis. J Virol. 2008 doi: 10.1128/JVI.01323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner CL, Yin J, Burke CW, Klimstra WB, Ryman KD. Type I interferon induction is correlated with attenuation of a South American eastern equine encephalitis virus strain in mice. Virology. 2009;390:338–347. doi: 10.1016/j.virol.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmashova N, Gorchakov R, Frolova E, Frolov I. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J Virol. 2006;80:5686–5696. doi: 10.1128/JVI.02739-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmashova N, Gorchakov R, Volkova E, Paessler S, Frolova E, Frolov I. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J Virol. 2007;81:2472–2484. doi: 10.1128/JVI.02073-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009 doi: 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J, Alcami J, Esteban M. Activation of NF-kappa B by the dsRNA-dependent protein kinase, PKR involves the I kappa B kinase complex. Oncogene. 2000;19:1369–1378. doi: 10.1038/sj.onc.1203448. [DOI] [PubMed] [Google Scholar]
- Gilfoy FD, Mason PW. West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J Virol. 2007;81:11148–11158. doi: 10.1128/JVI.00446-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez dC, Ehsani N, Mikkola ML, Garcia JA, Kaariainen L. RNA helicase activity of Semliki Forest virus replicase protein NSP2. FEBS Lett. 1999;448:19–22. doi: 10.1016/s0014-5793(99)00321-x. [DOI] [PubMed] [Google Scholar]
- Gorchakov R, Frolova E, Williams BR, Rice CM, Frolov I. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J Virol. 2004;78:8455–8467. doi: 10.1128/JVI.78.16.8455-8467.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov R, Garmashova N, Frolova E, Frolov I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J Virol. 2008;82:10088–10101. doi: 10.1128/JVI.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DE. Alphaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott, Williams & Wilkins; Philadelphia: 2001. pp. 917–962. [Google Scholar]
- Haller O, Kochs G, Weber F. Interferon, Mx, and viral countermeasures. Cytokine Growth Factor Rev. 2007;18:425–433. doi: 10.1016/j.cytogfr.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidmark AS, McInerney GM, Nordstrom EK, Douagi I, Werner KM, Liljestrom P, Karlsson Hedestam GB. Early alpha/beta interferon production by myeloid dendritic cells in response to UV-inactivated virus requires viral entry and interferon regulatory factor 3 but not MyD88. J Virol. 2005;79:10376–10385. doi: 10.1128/JVI.79.16.10376-10385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- Hiscott J, Pitha P, Genin P, Nguyen H, Heylbroeck C, Mamane Y, Algarte M, Lin R. Triggering the interferon response: the role of IRF-3 transcription factor. J Interferon Cytokine Res. 1999;19:1–13. doi: 10.1089/107999099314360. [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Iwamura T, Yoneyama M, Yamaguchi K, Suhara W, Mori W, Shiota K, Okabe Y, Namiki H, Fujita T. Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells. 2001;6:375–388. doi: 10.1046/j.1365-2443.2001.00426.x. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Zamanian-Daryoush M, Nie H, Silva AM, Williams BR, Li X. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFkappa B and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J Biol Chem. 2003;278:16713–16719. doi: 10.1074/jbc.M300562200. [DOI] [PubMed] [Google Scholar]
- Kalali BN, Kollisch G, Mages J, Muller T, Bauer S, Wagner H, Ring J, Lang R, Mempel M, Ollert M. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling. J Immunol. 2008;181:2694–2704. doi: 10.4049/jimmunol.181.4.2694. [DOI] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A. 2002;99:637–642. doi: 10.1073/pnas.022637199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Klimstra WB, Ryman KD, Bernard KA, Nguyen KB, Biron CA, Johnston RE. Infection of neonatal mice with sindbis virus results in a systemic inflammatory response syndrome. J Virol. 1999;73:10387–10398. doi: 10.1128/jvi.73.12.10387-10398.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimstra WB, Ryman KD, Johnston RE. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J Virol. 1998;72:7357–7366. doi: 10.1128/jvi.72.9.7357-7366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc Natl Acad Sci U S A. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Yang YL, Flati V, Der S, Kadereit S, Deb A, Haque J, Reis L, Weissmann C, Williams BR. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. EMBO J. 1997;16:406–416. doi: 10.1093/emboj/16.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- MacDonald GH, Johnston RE. Role of dendritic cell targeting in Venezuelan equine encephalitis virus pathogenesis. J Virol. 2000;74:914–922. doi: 10.1128/jvi.74.2.914-922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGreal EP, Martinez-Pomares L, Gordon S. Divergent roles for C-type lectins expressed by cells of the innate immune system. Molecular Immunology. 2004;41:1109–1121. doi: 10.1016/j.molimm.2004.06.013. [DOI] [PubMed] [Google Scholar]
- McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A. 2004;101:233–238. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchjorsen J, Jensen SB, Malmgaard L, Rasmussen SB, Weber F, Bowie AG, Matikainen S, Paludan SR. Activation of innate defense against a paramyxovirus is mediated by RIG-I and TLR7 and TLR8 in a cell-type-specific manner. J Virol. 2005;79:12944–12951. doi: 10.1128/JVI.79.20.12944-12951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Tschopp J. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol Cell. 2006;22:561–569. doi: 10.1016/j.molcel.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M, Jr, Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noah DL, Blum MA, Sherry B. Interferon regulatory factor 3 is required for viral induction of beta interferon in primary cardiac myocyte cultures. J Virol. 1999;73:10208–10213. doi: 10.1128/jvi.73.12.10208-10213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- Paz S, Sun Q, Nakhaei P, Romieu-Mourez R, Goubau D, Julkunen I, Lin R, Hiscott J. Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway. Cell Mol Biol (Noisy -le-grand) 2006;52:17–28. [PubMed] [Google Scholar]
- Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Pollara G, Kwan A, Newton PJ, Handley ME, Chain BM, Katz DR. Dendritic cells in viral pathogenesis: protective or defective? Int J Exp Pathol. 2005;86:187–204. doi: 10.1111/j.0959-9673.2005.00440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Rikkonen M, Peranen J, Kaariainen L. ATPase and GTPase activities associated with Semliki Forest virus nonstructural protein nsP2. J Virol. 1994;68:5804–5810. doi: 10.1128/jvi.68.9.5804-5810.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman K, Klimstra W. Togaviruses. In: Brasier AR, Garcia-Sastre A, Lemon S, editors. Cellular signaling and innate immune responses to RNA virus infections. ASM Press; Washington, DC: 2009. pp. 353–372. [Google Scholar]
- Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol. 2000;74:3366–3378. doi: 10.1128/jvi.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman KD, Meier KC, Nangle EM, Ragsdale SL, Korneeva NL, Rhoads RE, Macdonald MR, Klimstra WB. Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells. J Virol. 2005;79:1487–1499. doi: 10.1128/JVI.79.3.1487-1499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman KD, White LJ, Johnston RE, Klimstra WB. Effects of PKR/RNase L-dependent and alternative antiviral pathways on alphavirus replication and pathogenesis. Viral Immunol. 2002;15:53–76. doi: 10.1089/088282402317340233. [DOI] [PubMed] [Google Scholar]
- Schafer SL, Lin R, Moore PA, Hiscott J, Pitha PM. Regulation of type I interferon gene expression by interferon regulatory factor-3. J Biol Chem. 1998;273:2714–2720. doi: 10.1074/jbc.273.5.2714. [DOI] [PubMed] [Google Scholar]
- Siren J, Pirhonen J, Julkunen I, Matikainen S. IFN-alpha regulates TLR-dependent gene expression of IFN-alpha, IFN-beta, IL-28, and IL-29. J Immunol. 2005;174:1932–1937. doi: 10.4049/jimmunol.174.4.1932. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- Trgovcich J, Aronson JF, Johnston RE. Fatal Sindbis virus infection of neonatal mice in the absence of encephalitis. Virology. 1996;224:73–83. doi: 10.1006/viro.1996.0508. [DOI] [PubMed] [Google Scholar]
- Tsetsarkin K, Higgs S, McGee CE, de LX, Charrel RN, Vanlandingham DL. Infectious clones of Chikungunya virus (La Reunion isolate) for vector competence studies. Vector Borne Zoonotic Dis. 2006;6:325–337. doi: 10.1089/vbz.2006.6.325. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Akira S. Toll-Like receptors (TLRs) and their ligands. Handb Exp Pharmacol. 2008:1–20. doi: 10.1007/978-3-540-72167-3_1. [DOI] [PubMed] [Google Scholar]
- Uetani K, Der SD, Zamanian-Daryoush M, de La MC, Lieberman BY, Williams BR, Erzurum SC. Central role of double-stranded RNA-activated protein kinase in microbial induction of nitric oxide synthase. J Immunol. 2000;165:988–996. doi: 10.4049/jimmunol.165.2.988. [DOI] [PubMed] [Google Scholar]
- Vasiljeva L, Merits A, Auvinen P, Kaariainen L. Identification of a novel function of the alphavirus capping apparatus. RNA 5′-triphosphatase activity of Nsp2. J Biol Chem. 2000;275:17281–17287. doi: 10.1074/jbc.M910340199. [DOI] [PubMed] [Google Scholar]
- Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, Barber GN. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007;178:6444–6455. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- Weaver BK, Kumar KP, Reich NC. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol Cell Biol. 1998;18:1359–1368. doi: 10.1128/mcb.18.3.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LJ, Wang JG, Davis NL, Johnston RE. Role of alpha/beta interferon in Venezuelan equine encephalitis virus pathogenesis: effect of an attenuating mutation in the 5′ untranslated region. J Virol. 2001;75:3706–3718. doi: 10.1128/JVI.75.8.3706-3718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR. Signal integration via PKR. Sci STKE. 2001;2001:RE2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, Williams BR, Aguet M, Weissmann C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995;14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Yount JS, Moran TM, Lopez CB. Cytokine-independent upregulation of MDA5 in viral infection. J Virol. 2007;81:7316–7319. doi: 10.1128/JVI.00545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20:1278–1290. doi: 10.1128/mcb.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Burke CW, Ryman KD, Klimstra WB. Identification and Characterization of Interferon-induced Proteins that Inhibit Alphavirus Replication. J Virol. 2007;81:11246–11255. doi: 10.1128/JVI.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, Paranjape JM, Der SD, Williams BR, Silverman RH. Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology. 1999;258:435–440. doi: 10.1006/viro.1999.9738. [DOI] [PubMed] [Google Scholar]
- Zhou H, Perlman S. Mouse hepatitis virus does not induce Beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J Virol. 2007;81:568–574. doi: 10.1128/JVI.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinn K, Keller A, Whittemore LA, Maniatis T. 2-Aminopurine selectively inhibits the induction of beta-interferon, c-fos, and c-myc gene expression. Science. 1988;240:210–213. doi: 10.1126/science.3281258. [DOI] [PubMed] [Google Scholar]