

Abstract
Thrombocytopenia absent radius (TAR) syndrome is a very rare and infrequently seen congenital disorder with an approximate frequency of 0.42/100,000 live births. It is associated with bilateral absence of radii, hypo-megakaryocytic thrombocytopenia, and presence of both thumbs. The other systems which are affected by TAR syndrome include skeletal, hematologic, and cardiac systems. Intracranial hemorrhages due to thrombocytopenia and cardiac disorders are a common association usually seen with this syndrome and are usual cause of death. We describe a 3-month-old infant who was diagnosed with TAR syndrome on the basis of clinical features (thrombocytopenia and bilateral absent radius bone and confirmed by genetic analysis). The patient was diagnosed to have Tetralogy of Fallot, for which the infant was managed with definitive repair and thrombocytopenia was managed with platelet transfusion. Infants with TAR syndrome should be assessed for other associated malformations of various systems and followed up regularly and parents should be counseled for associated expected complications in these patients. We report an infant with TAR syndrome with Tetralogy of Fallot, which has not been reported in medical literature until now and this is the first case of its type.
Keywords: thrombocytopenia absent radius syndrome, Tetralogy of Fallot, microdeletion 1q21.1, RBM8A
Introduction
Thrombocytopenia absent radius (TAR) is a rare syndrome associated with bilateral absence of radii, hypo-megakaryocytic thrombocytopenia, and presence of both thumbs.1 TAR syndrome abnormalities include various systems, including skeletal, hematologic, and cardiac system abnormalities. This association of simultaneous involvement of various systems has been postulated due to the simultaneous development of the heart, radii, and megakaryocytes at 6–8 weeks’ gestation. The reported frequency of TAR syndrome in medical literature has been expected to be around 0.42/100,000 population with unknown genetic inheritance pattern with few case reports showing both autosomal recessive and dominant pattern of inheritance. TAR syndrome is the result of noncoding single nucleotide polymorphisms located in the 5′UTR region or the first intron of the gene RBM8A and also because of microdeletion of chromosome 1q21.1 region.2 This case report will help pediatricians to be aware of the associated problems with TAR syndrome other than simply the functional disability associated with absent radius. We report an infant with TAR syndrome with Tetralogy of Fallot which has not been reported in medical literature until now and this is the first case of its type.
Case report
A 3-month old male infant who was the first child of a non-consanguineous couple and born at term with a birth weight of 2,480 grams was brought to the outpatient department with repeated episodes of bluish discoloration of lips on excessive crying. In the prenatal history the mother was 25 years old, primigravida, with unsupervised antenatal checkup. There was no history in the mother of any drug or radiation exposure. The infant was delivered at 39 weeks by normal vaginal delivery with normal Apgar score of 8/8/9 at 1, 5, and 10 minutes. The infant was exclusively breastfed and the couple did not consult any doctor for abnormalities before coming to our hospital. On physical examination the child was noted to be pink with 89%–90% saturation on room air, bilateral absent radius, radial club hand and flexion against the palm (Figures 1 and 2), and a grade 2/6 murmur in the left parasternal border, depressed. He was provisionally diagnosed to have TAR syndrome on the basis of the physical findings. X-ray of upper limbs suggested absence of bilateral radius bone (Figure 3). Two-dimensional echocardiography revealed Tetralogy of Fallot physiology with ventricular septal defect (VSD), infundibular pulmonary stenosis with overriding of the aorta. Complete blood count revealed thrombocytopenia and anemia with a platelet count of 53,000/mm3 and hemoglobin value of 9.2 gm/dL. Peripheral smear showed normal shaped red blood cells and thrombocytopenia. The platelets were normal in size. A bone marrow study revealed a decreased number of megakaryocytes. Head ultrasound was normal. Genetic analysis showed interstitial microdeletion in 1q21.1 and a hypomorphic allele in RBM8A chromosomes, which confirmed our provisional diagnosis of TAR syndrome.
Figure 1.
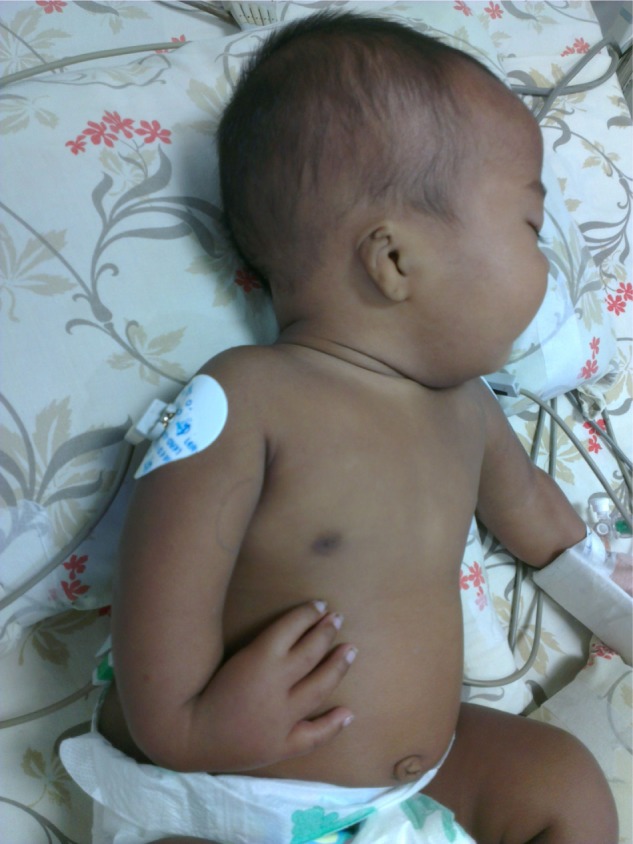
Bilateral absent radius, radial club hand, and flexion against the palm.
Figure 2.
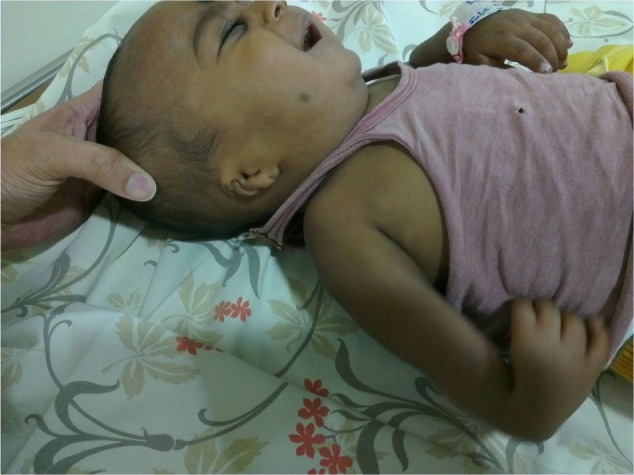
Bilateral absent radius, radial club hand, and flexion against the palm.
Note: Note thumb of opposite hand is visible in the photo.
Figure 3.
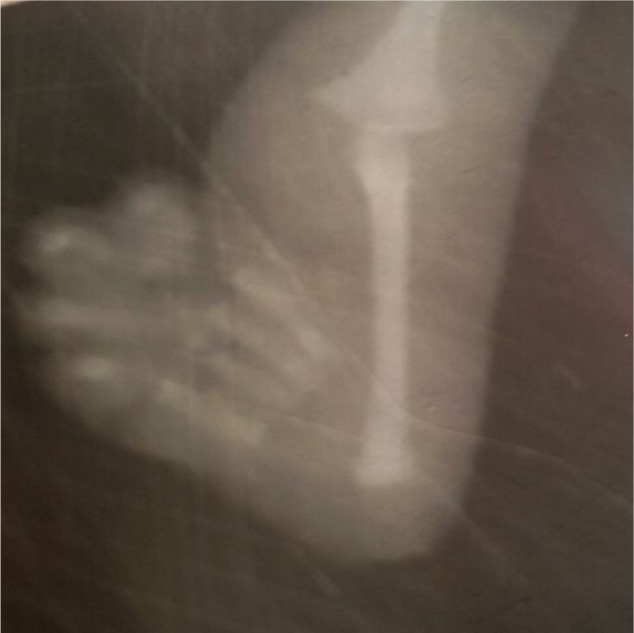
X-ray of upper limb showing absence of radius bone in the patient.
Following admission the baby received three aliquots of single donor platelet transfusion and this increased the repeat platelet count to 142,000/mm3. The central lines were established and the patient was monitored for any arrhythmias. The infant underwent definitive repair of Tetralogy of Fallot (definitive repair included VSD closure and relief of RVOT obstruction through a combination of infundibular muscle resection and pulmonary valvotomy). After the operation the baby was given post-operative care in the form of invasive ventilation for the following 48 hours and there was improvement in the neonate’s saturation with a maximum of 96% and the infant was extubated successfully. The feeds were started on post-operative day 3 and gradually increased and breast feeding was started 7 days after the operation. The baby was discharged 2 weeks later and was to be followed up for the intermittent screening for thrombocytopenia and platelet transfusion for approximately 2 months. Orthopedic/plastic surgeon consultation for the limb deformity was later planned, but the patient was later lost to follow-up. After enquiries it was discovered that the child had died, the grounds of which was unknown.
Discussion
TAR is a rare syndrome associated with bilateral absence of radii, hypo-megakaryocytic thrombocytopenia, and presence of both thumbs.1 It was first reported in medical literature in 1959. The incidence of TAR syndrome has been reported to be 0.42/100,000 population. The genetic inheritance pattern is uncertain with few documented reports mentioning autosomal recessive pattern, whereas others mention autosomal dominant pattern of inheritance. High-throughput sequencing studies suggest that TAR syndrome is biallelic inherited with noncoding single nucleotide polymorphisms located in the 5′UTR region or the first intron of the gene RBM8A (coding for Y14 protein, association to TAR syndrome not yet cleared up) and a chromosome 1q21.1 region microdeletion.2 The pathophysiology of this term is mostly unknown, but theories have run from a common (unknown) injury in 6–8 weeks of gestation when the heart, limbs, and megakaryocytes develop simultaneously or due to the “contiguous gene model” which is founded on the premise that phenotypic findings are related when genes responsible for each defect are geographically associated in a chromosome. Similarly, the pathophysiology for thrombocytopenia is largely unknown, but has been purported to be due to abnormal signal transduction to stimulator like thrombopoietin.3 Thrombocytopenia begins in the neonatal period,4 50% of affected infants present with thrombocytopenia in the first week of life and approximately 90% develop thrombocytopenia by 4 months of age. Other non-skeletal abnormalities commonly associated include gastroenteritis and cow’s milk intolerance (47%), renal malformations (23%), cardiac defects like atrial septal defects, VSD, and very rarely Tetralogy of Fallot (15%), facial dysmorphism (53%), short stature (95%), macrocephaly (76%), and capillary hemangioma (24%).5 The predominant skeletal abnormality includes bilateral radial aplasia with presence of bilateral thumb. Nevertheless, additional skeletal abnormalities are often observed, those including rarely more extensive upper limb malformations, phocomelia, and lower limb malformations in as many as 50% of the patients.5 TAR syndrome is commonly associated with a combination of physical findings. TAR syndrome is commonly labelled when there is a constellation of various physical findings:
Fanconi anemia is characterized by congenital anomalies, progressive bone marrow failure, and predisposition to cancers like myelodysplastic syndrome and acute myelogenous leukemia. The skeletal abnormalities include radial bone and thumb abnormalities, short stature, skin hyperpigmentation, and/or café au lait macules. The presence of thumbs in our patient helped us to rule out Fanconi anemia and genetic analysis confirmed our diagnosis.6
Roberts syndrome is inherited in autosomal recessive pattern and the affected patient has various problems which include severe growth retardation, cleft lip and palate, nose and ears anomalies, facial hemangioma, hypertelorism, microcephaly, oligodactyly or tetraphocomelia, renal malformations like polycystic and dysplastic kidneys, congenital heart defects, and enlarged male genitalia. The karyotype analysis shows typical “railroad track” appearance because of chromatic and premature centromere separation in metaphase spreads. The gene, which has been postulated in Roberts syndrome as ESCO2 genetic, is located on 8p21.1 locus. The genetic analysis of our patient ruled out Robert syndrome in the index case.7
Holt–Oram syndrome (OMIM 142900) is associated with upper limb extremity abnormalities which include abnormality of radius, metacarpals, or carpal bones. There is usually associated congenital heart defects like atrial septal defect and VSD. The inheritance usually seen is autosomal dominant manner. The only gene which has been known to cause Holt–Oram syndrome, TBX5 gene, is located on chromosome 12 (12q24.1). Our patient did not have any abnormality of metacarpal and carpal bones and genetic analysis showed interstitial microdeletion in 1q21.1 and a hypomorphic allele in RBM8A chromosomes which confirmed TAR syndrome.8
Diamond-Blackfan anemia/Aase syndrome is characterized by erythroid hypoplasia, macrocytic anemia, and normal white blood cell and platelet counts. The affected patient usually has other congenital anomalies, such as triphalangeal, bifid, or subluxed thumbs, flattening of the thenar eminence, with a normal radius bone. The other systems also involved include genitourinary and cardiovascular. These patients usually have webbed neck, Klippel-Feil anomaly or Sprengel deformity. The absence of radius and reduced platelet count helped us to differentiate Diamond-Blackfan anemia from TAR syndrome and our suspicion was confirmed by genetic analysis.9
In TAR syndrome, significant laboratory findings include thrombocytopenia with normal platelet morphology in peripheral smear and hypo-megakaryocytic thrombocytopenia with small, basophilic, vacuolated megakaryocytes on bone marrow examination with normal or elevated plasma thrombopoietin levels.10 Radial abnormality can be detected as early as 13 weeks by prenatal ultrasonography.11 Affected infants have an increased risk of bleeding manifestations especially intracranial hemorrhage which is more common during the first 2 years of life. We suspect that cause of death in our index case could be because of intracranial hemorrhage as these infants are more prone to it.
Hence, these babies should follow general thrombocytopenic precautions like avoidance of trauma and avoidance of antiplatelet drugs. The mainstay of treatment includes platelet transfusions. Prophylactic platelet transfusions to raise the platelet count above 40,000/mm3 (extrapolated from thrombocytopenia with leukemia) is currently pursued in patients with high risk of clinically important hemorrhage. Other recent treatment modalities include treatment with erythropoietin,12 IL-6,13 and hematopoietic stem cell transplantation in refractory thrombocytopenic patients.14 Splenectomy in adults has shown to improve thrombocytopenia. Deformity-splinting of the limbs during infancy improves future function. Later, adaptive devices can be applied as they have functioning fingers. The risk of bleeding, thrombocytopenia is significant particularly in the first 2 years of life, subsequently platelet count increases with age. Yet there is a reported increased risk of leukemia with TAR syndrome.15
Similar events have been described in the past, some of them have included additional findings. In a serial publication published by Greenhalgh et al in 2002, all patients had documented thrombocytopenia and bilateral radial aplasia, 47% had lower limb anomalies, 47% intolerance to cow’s milk, 23% renal anomalies, and 15% cardiac anomalies. Congenital anomalies also included facial capillary hemangiomata, intracranial vascular malformation, sensorineural hearing loss, and scoliosis.5 Menghsol et al reported a patient with TAR syndrome who also had coarctation of the aorta and axial rotation of the kidney. Other findings included adducted thumbs, radial aplasia, hypoplasia of the cerebellar vermis, and axial malrotation of the kidney.16
Conclusion
TAR is a rare syndrome and is associated with bilateral absence of radii, hypo-megakaryocytic thrombocytopenia, and presence of both thumbs. TAR syndrome abnormalities include various systems including skeletal, hematologic, and cardiac system abnormalities. Babies with suspected TAR syndrome should be assessed for other associated malformations of various systems like renal malformations, cardiac defects like atrial septal defects, VSD, and very rarely Tetralogy of Fallot, facial dysmorphism, short stature, macrocephaly, capillary hemangioma, and hematological malignancies in later life, requiring regular follow-up. Antenatal diagnosis of TAR syndrome is possible in the first trimester but that requires thorough antenatal checkup and regular follow-up. The risk of bleeding is highest during the first 2 years of life span after which the platelet count may improve. Close surveillance for thrombocytopenia with regular follow-ups and platelet transfusion to raise platelet count above 40,000/mm3 are the mainstay of treatment. Treatment of associated deformities requires a multidisciplinary approach that contributes to the successful management of such cases.
Footnotes
Disclosure
The authors have no conflicts of interest to report.
References
- 1.de Ybarrondo L, Barratt MS. Thrombocytopenia Absent Radius Syndrome. Pediatr Rev. 2011;32(9):399–400. doi: 10.1542/pir.32-9-399. [DOI] [PubMed] [Google Scholar]
- 2.Albers CA, Newbury-Ecob R, Ouwehand WH, Ghevaert C. New insights into the genetic basis of TAR (thrombocytopenia-absent radii) syndrome. Curr Opin Genet Dev. 2013;23(3):316–323. doi: 10.1016/j.gde.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Sekine I, Hagiwara T, Miyazaki H, et al. Thrombocytopenia with absent radii syndrome: studies on serum thrombopoietin levels and megakaryopoiesis in vitro. J Pediatr Hematol Oncol. 1998;20(1):74–78. doi: 10.1097/00043426-199801000-00012. [DOI] [PubMed] [Google Scholar]
- 4.Toriello HV. Thrombocytopenia-absent radius syndrome. Semin Thromb Hemost. 2011;37(6):707–12. doi: 10.1055/s-0031-1291381. [DOI] [PubMed] [Google Scholar]
- 5.Greenhalgh KL, Howell RT, Bottani A, et al. Thrombocytopenia-absent radius syndrome: a clinical genetic study. J Med Genet. 2002;39(12):876–881. doi: 10.1136/jmg.39.12.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khincha PP, Savage SA. Genomic characterization of the inherited bone marrow failure syndromes. Semin Hematol. 2013;50(4):333–347. doi: 10.1053/j.seminhematol.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dupont C, Bucourt M, Guimiot F, et al. 3D-FISH analysis reveals chromatid cohesion defect during interphase in Roberts syndrome. Mol Cytogenet. 2014;7(1):59. doi: 10.1186/s13039-014-0059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garavelli L, De Brasi D, Verri R, et al. Holt-Oram syndrome associated with anomalies of the feet. Am J Med Genet A. 2008;146A(9):1185–1189. doi: 10.1002/ajmg.a.32170. [DOI] [PubMed] [Google Scholar]
- 9.Chirnomas SD, Kupfer GM. The inherited bone marrow failure syndromes. Pediatr Clin North Am. 2013;60(6):1291–1310. doi: 10.1016/j.pcl.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ballmaier M, Schulze H, Strauss G, et al. Thrombopoietin in patients with congenital thrombocytopenia and absent radii: elevated serum levels, normal receptor expression, but defective reactivity to thrombopoietin. Blood. 1997;90(2):612–619. [PubMed] [Google Scholar]
- 11.Luthy DA, Mack L, Hirsch J, Cheng E. Prenatal ultrasound diagnosis of thrombocytopenia with absent radii. Am J Obstet Gynecol. 1981;141(3):350–351. doi: 10.1016/s0002-9378(16)32646-1. [DOI] [PubMed] [Google Scholar]
- 12.Dempfle CE, Burck C, Grutzmacher T, Wizenmann J, Heene DL. Increase in platelet count in response to rHuEpo in patient with thromboctopenia and absent radii syndrome. Blood. 2001;97(7):2189–2190. doi: 10.1182/blood.v97.7.2189. [DOI] [PubMed] [Google Scholar]
- 13.Aquino VM, Mustafa MM, Vackus L, Wooley R, Buchanan GR. Recombinant interleukin-6 in the treatment of congenital thrombocytopenia associated with absent radii. J Pediatr Hematol Oncol. 1998;20(5):474–476. doi: 10.1097/00043426-199809000-00013. [DOI] [PubMed] [Google Scholar]
- 14.Brochstein JA, Shank B, Kernan NA, Terwilliger JW, O’Reilly RJ. Marrow transplantation for thrombocytopenia-absent radii syndrome. J Pediatr. 1992;121(4):587–589. doi: 10.1016/s0022-3476(05)81152-7. [DOI] [PubMed] [Google Scholar]
- 15.Go RS, Johnston KL. Acute myelogenous leukemia in an adult with thrombocytopenia with absent radii syndrome. Eur J Haematol. 2003;70(4):246–248. doi: 10.1034/j.1600-0609.2003.00054.x. [DOI] [PubMed] [Google Scholar]
- 16.Menghsol SC, Harris RD, Ornvold K. Thrombocytopenia and absent radii, TAR syndrome: report of cerebellar dysgenesis and newly identified cardiac and renal anomalies. (Letter) Am J Med Genet A. 2003;123A(2):193–196. doi: 10.1002/ajmg.a.20251. [DOI] [PubMed] [Google Scholar]