

Abstract
Nicotinamide adenine dinucleotide phosphate (NADPH) biogenesis is an essential mechanism by which both normal and cancer cells maintain redox balance. While antitumor approaches to treat cancers through elevated reactive oxygen species (ROS) are not new ideas, depleting specific NADPH-biogenesis pathways that control recovery and repair pathways are novel, viable approaches to enhance cancer therapy. However, to elicit efficacious therapies exploiting NADPH-biogenic pathways, it is crucial to understand and specifically define the roles of NADPH-biogenesis pathways used by cancer cells for survival or recovery from cell stress. It is equally important to select NADPH-biogenic pathways that are expendable or not utilized in normal tissue to avoid unwanted toxicity. Here, we address recent literature that demonstrates specific tumor-selective NADPH-biogenesis pathways that can be exploited using agents that target specific cancer cell pathways normally not utilized in normal cells. Defining NADPH-biogenesis profiles of specific cancer-types should enable novel strategies to exploit these therapeutic windows for increased efficacy against recalcitrant neoplastic disease, such as pancreatic cancers. Accomplishing the goal of using ROS as a weapon against cancer cells will also require agents, such as NQO1 bioactivatable drugs, that selectively induce elevated ROS levels in cancer cells, while normal cells are protected.
Keywords: reactive oxygen species (ROS), NQO1-bioactivatable drugs, nicotinamide adenine dinucleotide phosphate (NADPH), glutathione (GSH), biogenic pathways, antioxidant
Introduction
Reduced nicotinamide adenine dinucleotide phosphate (NADPH) is a necessary cofactor for anabolic reactions, such as lipid and nucleic acid biosynthesis. Additionally, NADPH provides reducing power to oxidation–reduction reactions necessary for protecting cancer cells against the accumulation of reactive oxygen species (ROS) produced during rapid cellular proliferation.1
While increased ROS in cancer cells may be an important initiating event in carcinogenesis, excessive levels of ROS can be toxic and lead to cell death by causing irreversible damage to DNA, lipids, and proteins.1–3 Many chemotherapeutic agents act by inducing excessive ROS damage in cancer cells, but lack the ability to differentiate between normal and tumor tissues, leading to a narrow therapeutic window.4,5 In addition, some cancers in advanced stages may become resistant to intrinsic oxidative stress and can up-regulate canonical antioxidant defenses to protect against ROS-inducing agents. Reduced glutathione (GSH) and thioredoxin (TRX) are essential ROS scavenging molecules in cancer and in normal cells.6 GSH and TRX are necessary for peroxidases, thioreductases, and peroxiredoxins to detoxify ROS. GSH and TRX rely on continuous reduction from NADPH to sustain their function as ROS scavengers.6 Therefore, the strategies to inhibit NADPH-biogenesis may dramatically alter the ROS scavenging abilities of cancer cells and sensitize them to oxidative damage. However, to achieve therapeutic selectivity, NADPH must be modulated through tumor-specific NADPH-biogenesis pathways that are necessary for cancer cells, but expendable in normal cells. To this end, this review describes cancer-selective alterations in NADPH biogenesis, defines potential therapies that exploit these pathways to sensitize cancer to ROS damage, and provides a method to predict cancer-specific NADPH-biogenesis profiles. We will not focus on pharmacological modulation of de novo GSH and/or TRX pathways, as these topics have been comprehensively reviewed elsewhere.7–9
NADPH-biogenesis pathways in normal vs cancer cells
Oxidative pentose phosphate pathway (PPP)
A key mechanism of NADPH generation in normal cells is through the oxidative arm of the PPP. The PPP consists of two phases: the oxidative phase and the non-oxidative phase. The non-oxidative phase produces ribose from glucose, while the oxidative phase generates two NADPH molecules for every glucose entering the pathway (Figure 1).10 NADPH produced from the oxidative PPP is essential for protection against ROS damage arising from mitochondrial respiration, ionizing radiation, and various xenobiotic agents.11 In this pathway, glucose 6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6PGD) reduce NADP+ to NADPH while oxidizing glucose-6-phosphate (G6P) and carboxylating 6-phosphogluconate (6PG), respectively (Figure 1).12,13
Figure 1.
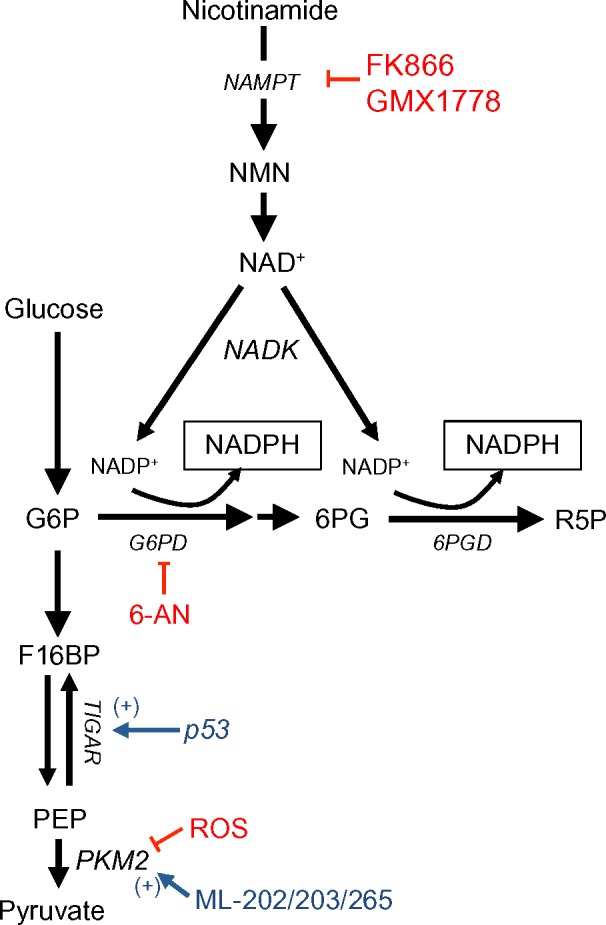
NADPH production from the oxidative PPP and one-carbon serine catabolism pathway.
Notes: Oxidative PPP uses glucose to generate NADPH via G6PD and 6PGD. G6PD is inhibited by then FDA-approved drug, 6-AN. NADP+ is generated through the NAD+ salvage pathway, where nicotinamide is converted to NMN via NAMPT. NADP+ is then formed by NADK. FK866 and GMX1778 inhibit NAMPT to block the production of NADP+, and therefore NADPH. During ROS stress, p53 positively regulates TIGAR to shunt glycolytic flux into the oxidative PPP. PKM2, which is overexpressed in many cancers, is inhibited by ROS, allowing glycolytic flux to be shuttled into the oxidative PPP for NADPH generation. The small-molecule compounds, ML-202/203/265, can positively modulate PKM2, thereby decreasing glycolytic flux into the oxidative PPP and blunting NADPH biogenesis during ROS.
Abbreviations: PPP, pentose phosphate pathway; NADPH, nicotinamide adenine dinucleotide phosphate; G6PD, glucose-6-phosphate dehydrogenase; 6PGD, 6-phosphogluconate dehydrogenase; 6-AN, 6-aminonicotinamide; NMN, nicotinamide mononucleotide; NAMPT, nicotinamide phosphoribosyltransferase; NADK, NAD+-kinase; ROS, reactive oxygen species; TIGAR, TP53-induced glycolysis and apoptosis regulator; PKM2, pyruvate kinase 2; G6P, glucose-6-phosphate; 6PG, 6-phosphogluconate; R5P, ribulose-5-phosphate; F16BP, fructose-1,6-bisphosphate; PEP, phosphoenolpyruvate; FDA, food and drug administration; NAD, nicotine adenine dinucleotide.
Pyruvate kinase (PK) is an essential glycolytic enzyme for conversion of phosphoenolpyruvate (PEP) to pyruvate (Figure 1). The M2 isoform of PK (PKM2) is found in many cancer cells and self-renewing cells, but is expressed in an inactive state in normal adult tissues.14 In many human cancers, PKM2 can be inactivated by ROS, which diverts glycolytic flux back into the oxidative PPP to generate NADPH and detoxify ROS (Figure 1).15 After ROS stress, PKM2 is an essential in cancer, but not normal, cells to maintain cell viability via redox scavenging. It could provide a potential efficacious antitumor therapeutic window for ROS-inducing agents.15 PKM2 overexpression ensures that rapidly proliferating cancer cells create enough NADPH to match oxidative metabolism-generating ATP production, protecting the cell from attack by oxidative damage.16,17,11
The tumor suppressor, p53, can also regulate flux into the oxidative PPP. During genotoxic stress, p53 induces TP53-induced glycolysis and apoptosis regulator (TIGAR), which encodes a protein that degrades fructose-2,6-bisphosphate (Figure 1).18 Low fructose-2,6-bisphosphate levels inhibit the activity of phosphofructokinase 1 (PFK1), a rate-limiting enzyme in glycolysis that leads to shuttling of earlier glycolytic metabolites into the oxidative PPP to generate NADPH. Overexpression of TIGAR was observed in colon, breast, and glioblastoma cancers.19–21 Consisten with the enzyme’s role in redox balance, TIGAR knockdown dramatically sensitized glioma cells to ionizing radiation.22
In cancers that overexpress PKM2, activating PKM2 with ML202, ML203, or other PKM2 activators blocks inhibition of PKM2 from ROS-inducing agents and decreases the flux of glucose through the oxidative PPP. This attenuates production of NADPH during oxidative damage, thereby sensitizing cancer cells to ROS-inducing agents. In contrast, normal cells that have inactive PKM2 are not sensitized to ROS-inducing agents.15,23,24 Alternatively, the FDA-approved G6PD inhibitor, 6-aminonicotinamide (6-AN), may be utilized in cancers with PKM2 or TIGAR overexpression, thus directly inhibiting NADPH production via the oxidative PPP pathway. This, in turn, also sensitizes cancer cells to ROS-inducing agents (Figures 1 and 2).25 The utility of this latter strategy needs to be empirically determined as G6PD is a major NADPH source in normal cells as well and toxicity concern will be a major factor in its efficacy.
Figure 2.

Agents targeting specific NADPH-biogenesis pathways.
Abbreviations: NADPH, nicotinamide adenine dinucleotide phosphate; BPTES, bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3; PPP, pentose phosphate pathway; PKM2, pyruvate kinase 2; ME, malic enzyme; GLS1, glutaminase 1; NAMPT, nicotinamide phosphoribosyltransferase; TS, thymidylate synthase; DHFR, dihydrofolate reductase; TIGAR, TP53-induced glycolysis and apoptosis regulator; PDAC, pancreatic ductal adenocarcinomas; SHMT2, serine hydroxymethyltransferase 2; MTHFD2, methylenetetrahydrofolate dehydrogenase 2.
Serine catabolism
Serine-driven, one-carbon metabolism was recently shown to be a major source of NADPH in dividing cells.26 Serine is metabolized in the cytoplasm or mitochondria to methylene-tetrahydrofolate (methylene-THF) by serine hydroxymethyltransferase (SHMT) 1 or 2 (cytoplasmic and mitochondrial, respectively), which then forms 10- formyl-THF via methylenetetrahydrofolate dehydrogenase (MTHFD) 1 or 2 (cytoplasmic and mitochondrial, respectively). The flux through MTHFD generates NADPH in the cytoplasm or mitochondria. 10-formyl-THF is an essential for purine biogenesis, and MTHFD’s most important function was thought to facilitate purine biosynthesis. However, the NADPH generated from this reaction is also an integral source of cellular reducing power in dividing cells (Figure 3), including normal tissues that turn over quickly, like the colon.26
Figure 3.

One-carbon serine catabolism pathway.
Notes: Mitochondrial one-carbon serine metabolism pathway highlighting the production of NADPH from serine and folic acid through mitochondrial MTHFD2. THF is produced from DHF via folic acid by DHFR, which is inhibited by PEM or MTX. Additionally, DHF can be generated from dUMP via TS, which is also inhibited by PEM.
Abbreviations: NADPH, nicotinamide adenine dinucleotide phosphate; MTHFD2, methylenetetrahydrofolate dehydrogenase; THF, tetrahydrofolate; DHF, dihydrofolate; DHFR, dihydrofolate reductase; PEM, pemetrexed; MTX, methotrexate; dUMP, deoxyuridine monophosphate; TS, thymidylate synthase; SHMT2, serine hydroxymethyltransferase 2.
Recently, it was reported that this serine catabolism pathway can regulate mitochondrial redox control during hypoxia in Myc-driven cancers.27 Specifically, SHMT2 was essential in maintaining mitochondrial NADPH and reduced GSH levels during hypoxia. SHMT2 expression was transcriptionally regulated by the coordinated activities of Myc and HIF-1α. Indeed, silencing SHMT2 in neuroblastoma cell lines significantly decreased growth in vitro under hypoxic conditions, and in a xenograft model of neuroblastoma (Figure 3).27 Additionally, the authors demonstrated that high SHMT2 levels correlated with a poorer prognostic outcome in neuroblastoma patients, providing a clinical context for targeting this pathway. While SHMT2 is a source of NADPH in normal dividing cells, inhibiting SHMT2 in normal cells should not significantly alter NADPH-biogenesis, since normal cells have robust compensatory mechanisms for redox balance, unlike cancer cells.26
In Myc-driven neuroblastoma, inhibiting SHMT2 or MTHFD2 would decrease NADPH biogenesis derived from one-carbon serine catabolism. While there are currently no known specific inhibitors of SHMT2 or MTHFD2, targeting production of serine’s obligate reaction partner, THF, may offer a strategy to decrease NADPH production from serine catabolism in a tumor-selective manner. For example, inhibiting dihydrofolate reductase (DHFR) with the anti-folate methotrexate (MTX) will decrease THF production, thereby decreasing the flux through SHMT2 and MTHFD2. This would attenuate NADPH levels specifically in cancer cells with elevated SHMT2 expression (Figures 2 and 3). Indeed, MTX exposure leads to cytostasis in overactive inflammatory cells seen in autoimmune diseases by decreasing GSH production, presumably due to decreased NADPH-biogenesis.28,29 Alternatively, the new-generation anti-folate, pemetrexed (PEM), can also attenuate NADPH production from THF/serine catabolism by inhibiting both thymidylate synthase (TS) and DHFR, enzymes essential in THF synthesis (Figures 2 and 3).30 Utilizing anti-folate drugs in SHMT2 overexpressing cancers in combination with ROS-inducing agents may provide a robust antitumor therapeutic window to exploit using these agents.
Malic enzymes
Another source of cellular NADPH is the NADP-dependent family of malic enzymes. This family of enzymes catalyzes the oxidative decarboxylation of malate to generate CO2 and pyruvate, while reducing NAD+ or NADP+ to NADH or NADPH in the process (Figure 4).31 Three isoforms were identified in mammalian systems: cytosolic NADP+- dependent (ME1), mitochondrial NAD(P)+-dependent (ME2), and mitochondrial NADP+-dependent malic enzyme (ME3).32
Figure 4.

KRAS-reprogrammed glutamine metabolism in pancreatic cancer.
Notes: In KRAS-mutant pancreatic cancer, mitochondrial glutamine flux is reprogrammed to predominantly flux through GOT2, instead of the canonical GLUD1 pathway. The Asp produced from this reaction is shuttled to the cytoplasm to produce pyruvate and NADPH from malate via cytosolic ME1. TCA cycle-derived malate can also produce NADPH from mitochondrial ME2. Wild-type p53 inhibits both ME1 and ME2. BPTES and CB-839 are non-competitive small-molecule inhibitors of mitochondrial GLS1.
Abbreviations: GOT2, mitochondrial aspartate transaminase; GLUD1, glutamate dehydrogenase 1; Asp, aspartate; NADPH, nicotinamide adenine dinucleotide phosphate; ME1, malic enzyme 1; ME2, malic enzyme 2; BPTES, bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3; GLS1, glutaminase 1; MDH1, malate dehydrogenase 1; GOT1, cytosolic aspartate transaminase; OAA, oxaloacetate; αKG, α-ketoglutarate; TCA, tricarboxylic acid cycle.
A recent report demonstrated the requirement of the cytosolic malic enzyme (ME1) in utilizing glutamine as an upstream metabolite to generate NADPH and to maintain redox balance in KRAS-mutated pancreatic ductal adenocarcinomas (PDAC), but not in normal pancreatic tissue (Figure 4).33 The canonical metabolism of glutamine generates α-ketoglutarate (αKG) via the upstream activity of glutamate dehydrogenase 1 (GLUD1) to drive anaplerosis to replenish the tricarboxylic acid cycle.34 However, in KRAS-mutated PDAC, glutamine flux is primarily driven through mitochondrial aspartate transaminase (GOT2) to generate mitochondrial αKG and aspartate from glutamate and oxaloacetate (OAA). Aspartate is then shuttled to the cytoplasm and then acted on by cytosolic aspartate transaminase (GOT1), which is converted back to OAA (Figure 4). OAA is then converted to malate by malate-dehydrogenase 1 (MDH1) and then to pyruvate and NADPH by ME1 (Figure 4). The depletion of ME1 in these PDAC cancer cells suppressed cell line growth and tumor growth in vivo by ROS accumulation from loss of NADPH.33 Furthermore, the inhibition of these enzymes in normal pancreatic cells did not significantly alter NADPH concentrations. Intriguingly, KRAS-mutated PDACs have dramatically decreased glucose flux into the oxidative PPP, suggesting that this non-canonical glutamine pathway is compensated for decreased NADPH production from the oxidative PPP.35 In PDAC with activating KRAS mutations (which is ~90% of all PDACs), inhibiting ME1 decreases the utilization of glutamine for NADPH production and sensitizes cells to oxidative damage. While there are currently no known ME1 inhibitors, inhibiting the upstream utilization of glutamine via glutaminase 1 (GLS1) with bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES), Compound 968, CB-839, or other GLS1 inhibitors would sensitize KRAS-mutated PDAC to ROS-inducing agents in a tumor-specific manner (Figures 2 and 4).36–38
A recent report demonstrated that a subset of lung tumors overexpress ME2 relative to normal lung tissue. A similar overexpression of ME2 was observed in melanoma vs normal skin, suggesting an important role for ME2 in these cancer-types.39 Indeed, when ME2 was knocked down in the A549 lung cancer cell line, the cellular NADPH/NADP+ ratio decreased three-fold compared to non-targeting control cells, indicative of a pro-oxidant state in the absence of ME2.
It was recently shown that ME1 and ME2 are negatively regulated by wild-type p53, and that the absence of a functional p53 led to a dramatic up-regulation of ME1/2 expression.40 Consistent with this finding, the authors demonstrated that ME1/2 enzymes were essential for NADPH maintenance in the absence of functional p53.40 In the context of cancer, this is an important observation as p53 is a commonly mutated tumor suppressor and the loss of its function may lead to a cancer cell-specific mechanism of NADPH biogenesis via ME1/2 de-repression.
Isocitrate dehydrogenases
NADPH production can also be driven by the conversion of isocitrate to αKG by NADP+-dependent cytosolic isocitrate dehydrogenase 1 (IDH1) and mitochondrial isocitrate dehydrogenase 2 (IDH2) (Figure 5).41,42 While NADPH generation has well known roles in the reduction of ROS, αKG can also detoxify ROS by scavenging hydrogen peroxide (H2O2) through non-enzymatic decarboxylation to form water and succinate.43,44
Figure 5.

NADPH biogenesis via IDH1.
Note: Cytoplasmic NADPH generated by IDH1 and mitochondrial NADPH generated by IDH2.
Abbreviations: IDH1, isocitrate dehydrogenase 1; NADPH, nicotinamide adenine dinucleotide phosphate; IDH2, isocitrate dehydrogenase 2; αKG, α-ketoglutarate.
IDH1 and IDH2 are mutated in ~80% of cases of adult glioma and secondary glioblastoma, and in 30% of cases of acute myeloid leukemia (AML).45,46 It was originally believed that these mutants led to enzymatic loss of function through dominant-negative inhibition of wild-type IDH1 and IDH2.46–48 However, it is now believed that IDH1 and IDH2 mutants confer to these enzymes the ability to convert αKG to the novel oncometabolite, 2-hydroxyglutarate (2-HG).49 This change causes mutated IDH1 and IDH2 enzymes to consume rather than produce NADPH, altering the cellular redox balance and leading to a pro-oxidant state in the cancer cell.50,51 Additionally, overexpression of the IDH1 mutant protein in glioma cell lines sensitizes these cells to the ROS-inducing effects of ionizing radiation.52
Glioma patients with IDH1R132 mutations have prolonged survival compared to patients with wild-type IDH1.47,50,53 A hypothesis for this observation could be that IDH1 mutants are defective in generating protective concentrations of NADPH to maintain reduced GSH and thus are more sensitive to oxidative damage. Thus, glioma or AML patients with a R132 mutation in IDH1 might benefit from ROS-inducing agents early during the course of treatment.
Nicotinamide phosphoribosyltransferase (NAMPT)
NADPH generation can be driven through the NAD+ salvage pathway via NAMPT, which catalyzes the transfer of the phosphoribosyl group from 5-phosphoribosyl-1-pyrophosphate to nicotinamide, forming nicotinamide mononucleotide (NMN), and pyrophosphate (Figure 1).54 NAD+ generation can then be coupled with NAD+ kinase (NADK) activity to generate NADP+ that can then be reduced to NADPH through the enzymes discussed above (Figure 1).55
Increased NAMPT expression was reported in colorectal, non-small cell lung (NSCL), prostate, and pancreatic cancers.56–60 In these contexts, NAMPT has been shown to be an important source of reducing equivalents for redox balance within the cancer cell.56 In fact, knockdown of NAMPT sensitized prostate and head and neck cancer cell lines to ROS induction from ionizing radiation.56,61–63
NAMPT inhibitors are undergoing clinical trials as single-agent therapies, but recent results have, unfortunately, not been promising (Figure 2).64 The NAMPT inhibitors, FK866, and GMX1778, may have the greatest efficacy when combined with ROS-inducing agents that take advantage of the pro-oxidant state of NAMPT-inhibited tumors. Indeed, pre-clinical studies have validated this strategy utilizing GMX1778 to sensitize breast cancer cells against the ROS production from ionizing radiation therapy, FK866 to sensitize prostate cancer cell against H2O2, and FK866 to sensitize neuroblastoma cells against cisplatin.56,61,65 To enhance the selectivities of NAMPT inhibitors, NADPH:quinone oxidoreductase 1 (NQO1) bioactivatable drugs can be used in combination treatments. This results in cancer-specific lethality of cells that overexpress NQO1,66 such as in pancreatic, NSCL, breast, prostate, and head and neck cancers.
Predicting tumor-specific NADPH-biogenesis profiles from publicly available datasets
Known NADPH-biogenesis pathways can be useful when combined with publically available cancer gene expression and patient outcomes data to generate hypotheses for tumor-specific NADPH-biogenesis profiles.
Here, we present a conservative set of criteria for determining candidate genes (Figure 6) using the following approach:
Determine if the genes of interest are significantly up-regulated in patient tumor tissue vs associated normal tissue in two or more independent datasets for the cancer-type in question. Only genes with P-values of <1×10−4 in each dataset will be considered for further analyses.
Of the up-regulated genes found in #1, determine which genes significantly predict poor outcomes in patients after radiation treatment.
Of the candidates from #2, determine if genes in the same NADPH biogenesis pathway as the candidate gene are coordinately up-regulated. Only positive correlation values of 0.5 and above will be considered for further analysis.
Determine if common oncogenic drivers of the tumor type in question drive genes from #3.
Figure 6.
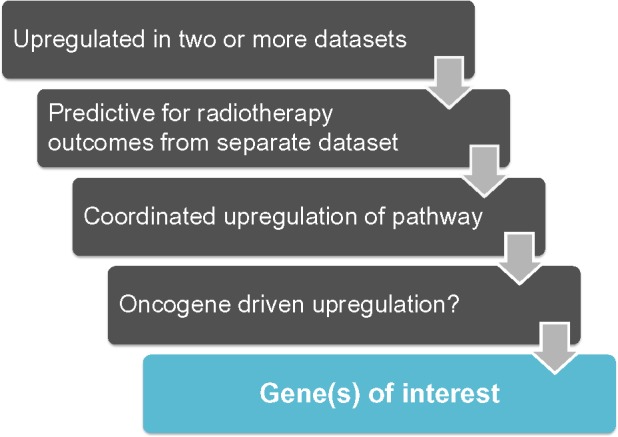
Work flow to identify NSCLC-specific NADPH-biogenesis genes in a cancer-specific NADPH-biogenesis screen.
Abbreviations: NSCLC, non-small cell lung cancer; NADPH, nicotinamide adenine dinucleotide phosphate.
Utilizing these criteria, we attempted to assess the NADPH-biogenesis profile of non-small cell lung cancer (NSCLC). Using the Oncomine webtool (http://www.oncomine.com), we first determined which NADPH-biogenesis related genes were overexpressed in NSCLC tissue vs associated normal tissue from patient samples. To ensure the robustness of our results, a gene was considered a potential hit only if it was found to be up-regulated in two or more datasets with a P-value <1×10−4 in each dataset. This narrowed our initial list of ten genes down to four potential genes (Figures 7 and 8A). Next, to determine if our potential targets were clinically relevant in the context of ROS-inducing therapy, we used the KMPLOT software and stratified NSCLC patients into high- and low-expressers of the genes of interest, and compared overall survival outcomes between these groups after radiation therapy (Figures 8A and 9).67
Figure 7.

A cancer-specific NADPH-biogenesis screen.
Notes: Overexpression status of NADPH-biogenesis genes of interest assessed using Oncomine in NSCLC patients. Datasets used are described.82–84
Abbreviations: NADPH, nicotinamide adenine dinucleotide phosphate; NSCLC, non-small cell lung cancer; G6PD, glucose-6-phosphate dehydrogenase; PKM2, pyruvate kinase 2; TIGAR, TP53-induced glycolysis and apoptosis regulator; SHMT2, serine hydroxymethyltransferase 2; MTHFD2, methylenetetrahydrofolate dehydrogenase; ME1, malic enzyme 1; ME2, malic enzyme 2; IDH1, isocitrate dehydrogenase 1; IDH2, isocitrate dehydrogenase 2; NAMPT, nicotinamide phosphoribosyltransferase.
Figure 8.

Cancer-specific NADPH-biogenesis screen continued.
Notes: (A) Prognosis after radiation therapy in NSCLC patients. Dataset used is described;67 (B) Correlation of genes of interest with upstream NADPH-biogenesis pathway members; (C) Mutant KRAS-dependence of MTHFD2 in a mouse model of NSCLC.
Abbreviations: NADPH, nicotinamide adenine dinucleotide phosphate; NSCLC, non-small cell lung cancer; MTHFD2, methylenetetrahydrofolate dehydrogenase; PKM2, pyruvate kinase 2; SHMT2, serine hydroxymethyltransferase 2; IDH2, isocitrate dehydrogenase 2; G6PD, glucose-6-phosphate dehydrogenase; TS, thymidylate synthase.
Figure 9.

Screen results for MTHFD2 in NSCLC patients.
Notes: (A and B) mRNA expression of MTHFD2 in normal lung tissue vs NSCLC tissue in two different datasets from Oncomine; (C) Overall survival after radiation therapy in NSCLC patients segregated in high and low MTHFD2 expressers. Dataset used is described;67 (D) Correlation of MTHFD2 with upstream NADPH-biogenesis pathway members; (E) Mutant KRAS-dependence of MTHFD2 in a mouse model of NSCLC (GSE40606).
Abbreviations: MTHFD2, methylenetetrahydrofolate dehydrogenase; NSCLC, non-small cell lung cancer; NADPH, nicotinamide adenine dinucleotide phosphate; SHMT2, serine hydroxymethyltransferase 2; TS, thymidylate synthase; HR, hazard ratio.
From this analysis, we selected the genes whose high expression in NSCLC patients led to significantly decreased survival after radiation therapy, suggesting that these genes may confer tumor protection from radiation-induced ROS, presumably through enhanced NADPH biogenesis.68 Using this cutoff, we were able to narrow down the gene list to PKM2, which regulates NADPH biogenesis via the oxidative PPP, and MTHFD2, which generates NADPH from serine catabolism (Figure 8B). We then determined if these genes were coordinately up-regulated with other enzymes in their respective NADPH-biogenesis pathways. Utilizing the r2 Genomics Analysis platform’s (r2.amc.nl) co-expression analysis feature in NSCLC patient samples, we found that G6PD was coordinately up-regulated with PKM2 in patient samples with a correlation value of 0.26, and that TS and SHMT2 were co-expressed with MTHFD2 with correlation values of 0.63 and 0.64, respectively (Figures 8B and 9D). TS and SHMT2 also appear to be co-expressed with a correlation value of 0.65. Given that we defined our correlation cutoff at 0.5 and above, we considered MTHFD2 as the top candidate for NSCLC (Figures 8B and 9D).
Next, we investigated whether mutant-KRAS, the most common oncogenic driver in NSCLC,69 might regulate MTHFD2. Such an association would provide insight into regulatory mechanisms of MTHFD2 and additional therapeutic targets. For this, we utilized publically available gene expression datasets from the National Center for Biotechnology Information Gene Expression Omnibus (GEO). To determine if mutant-KRAS regulates MTHFD2, we analyzed the mRNA expression-profiling data from a transgenic mouse model of NSCLC expressing doxycycline-inducible KRASG12D in the respiratory epithelium (GSE40606).70 When administered doxycycline, the mice develop lung tumors that are dependent on constitutive KRASG12D expression. Within 48 hours of doxycycline withdrawal, KRASG12D expression was extinguished and whole-genome gene expression analyses of lung tumors were performed. The expression levels of MTHFD2 were significantly up-regulated when KRASG12D was induced vs 48 hours after KRAS extinction with doxycycline withdrawal, indicating a positive regulatory role for mutant-KRAS in MTHDF2 expression (Figures 8C and 9E).
If this hypothesis is validated through RNAi and redox balance studies, it may suggest that the utilization of serine catabolism inhibitors, such as MTX or PEM, may provide an effective therapeutic strategy to target NADPH biogenesis, specifically in KRAS-mutated NSCLC. This analysis also reveals that more than one NADPH-biogenesis pathway may also be regulating NADPH as observed from the PKM2 data. However, validation experiments will need to be conducted to determine which pathway predominates in NSCLC.
Modulating NADPH biogenesis as a mechanism to potentiate NQO1-bioactivatable drugs
To exploit the metabolic vulnerabilities within a cancer as mentioned above, ROS-inducing agents with the capacity to specifically target tumors would be ideal. We believe that NQO1-bioactivatable drugs represent an ideal class of agents to combine with metabolic inhibitors. NQO1 is an inducible phase II detoxifying enzyme overexpressed in breast, lung, pancreatic, and colon cancers. The two-electron oxidoreductase is capable of reducing quinones by forming stable hydroquinones of the parent quinone.71,72 β-Lapachone (β-lap, in clinical trials as ARQ761), IB-DNQ, and other NQO1-bioactivatable drugs are unique quinones that are metabolized by NQO1 into an unstable hydroquinone that spontaneously oxidizes back to the parental compound, generating a futile redox cycle in which 1 mole of β-lap generates ~120 moles of superoxide within 2 minutes, consuming >60 moles of NADH and/or NADPH.73,74 The superoxide (O2−) radicals formed are quickly metabolized by superoxide dismutase (SOD) into H2O2.73,75 The massive levels of H2O2 formed causes extensive oxidative DNA damage that hyperactivates poly(ADP-ribosyl) polymerase 1 (PARP1), resulting in a dramatic loss of the intracellular NAD+/ATP pools and an inability to repair DNA damage.76–78 Cell death is stimulated by caspase-independent NAD+-Keresis, a form of programmed necrosis.66,73 Cancer cells with >100 units of NQO1 enzyme activity are sensitive to β-lap lethality, while normal tissues that lack, or express low levels of, NQO1 are spared.79 While this class of drugs represents an attractive antitumor strategy, dose-limiting methemoglobinemia caused by non-specific ROS generation at high doses may limit its efficacy in monotherapy.80 Strategies for increasing cancer cell- cytotoxicity while maintaining NQO1 specificity could greatly enhance the efficacy of β-lap for use in solid cancers that overexpress NQO1. Thus, combining β-lap with metabolic inhibitors that target cancer-specific NADPH-biogenesis pathways, such as FK866, CB-839, 6-AN, PEM or MTX, may synergistically expand the antitumor therapeutic window for NQO1 bioactivatable drugs, while increasing the tumor selectivity of metabolic inhibitors.
Conclusion
Cancer cells need to tightly regulate NADPH biogenesis to protect themselves against oxidative damage.81 To sustain protective levels of NADPH, cancer cells rely on various NADPH-biogenesis pathways, including oxidative PPP, serine catabolism, glutamine metabolism, and NAD+ salvage pathways (Figures 1, 3, and 4). Strategies to inhibit NADPH biogenesis may dramatically alter the ROS scavenging abilities of cancer cells and sensitize them to oxidative damage. However, to achieve therapeutic selectivity, NADPH must be modulated through tumor-specific NADPH-biogenesis pathways that are necessary for cancer cells but are expendable in normal cells. Thus, by rigorously studying these unique pathways in the context of a specific cancer, we will be able to create novel patient-specific antitumor therapeutic strategies that exploit the ROS balances of tumor tissue while sparing normal tissue in the process. Here, we summarized our current understanding of known cancer-specific NADPH-biogenesis pathways, drugs to specifically target these pathways, and an example of using publically available databases to predict cancer-type specific NADPH-biogenesis genes. It is our belief that studying these pathways and comprehensively profiling tumors based on this understanding will be an essential step forward in designing cancer-specific ROS combination therapies, such as the use of NQO1-bioactivatable drugs.
Acknowledgments
The authors thank Dr Ralph J DeBerardinis for his assistance in reviewing this work. Support for use of Shared Resources within the Simmons Comprehensive Cancer Center was funded by NIH/NCI grant 5P30CA142543. Support for these studies was provided by grant NIH/NCI R01 CA102972, and a grant from the Pancreatic Cancer Action Network (PanCan) to DAB. This work is dedicated to the memories of Mrs Sarah Hildebrand and Rosemary Bouley for their strong support of our research.
Footnotes
Disclosure
The authors declare no conflicts of interest in this work.
References
- 1.Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 2.Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31:1441–1444. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- 3.Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25:695–705. doi: 10.1007/s10555-006-9037-8. [DOI] [PubMed] [Google Scholar]
- 4.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 5.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 6.Fruehauf JP, Meyskens FL., Jr Reactive oxygen species: a breath of life or death? Clin Cancer Res. 2007;13:789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- 7.Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct. 2004;22:343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 8.Traverso N, Ricciarelli R, Nitti M, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2013;2013:972913. doi: 10.1155/2013/972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahmood DF, Abderrazak A, El Hadri K, Simmet T, Rouis M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid Redox Signal. 2013;19:1266–1303. doi: 10.1089/ars.2012.4757. [DOI] [PubMed] [Google Scholar]
- 10.Kruger NJ, von Schaewen A. The oxidative pentose phosphate pathway: structure and organisation. Curr Opin Plant Biol. 2003;6:236–246. doi: 10.1016/s1369-5266(03)00039-6. [DOI] [PubMed] [Google Scholar]
- 11.Nathan C, Ding A. SnapShot: reactive oxygen intermediates (ROI) Cell. 2010;140(951–951):e952. doi: 10.1016/j.cell.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Tian WN, Braunstein LD, Pang J, et al. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem. 1998;273:10609–10617. doi: 10.1074/jbc.273.17.10609. [DOI] [PubMed] [Google Scholar]
- 13.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39(8):347–354. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zwerschke W, Mazurek S, Massimi P, Banks L, Eigenbrodt E, Jansen-Durr P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc Natl Acad Sci U S A. 1999;96:1291–1296. doi: 10.1073/pnas.96.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anastasiou D, Poulogiannis G, Asara JM, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider J, Neu K, Grimm H, Velcovsky HG, Weisse G, Eigenbrodt E. Tumor M2-pyruvate kinase in lung cancer patients: immunohistochemical detection and disease monitoring. Anticancer Res. 2002;22:311–318. [PubMed] [Google Scholar]
- 17.Cerwenka H, Aigner R, Bacher H, et al. TUM2-PK (pyruvate kinase type tumor M2), CA19-9 and CEA in patients with benign, malignant and metastasizing pancreatic lesions. Anticancer Res. 1999;19:849–851. [PubMed] [Google Scholar]
- 18.Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 19.Cheung EC, Athineos D, Lee P, et al. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev Cell. 2013;25:463–477. doi: 10.1016/j.devcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinha S, Ghildiyal R, Mehta VS, Sen E. ATM-NFkappaB axis-driven TIGAR regulates sensitivity of glioma cells to radiomimetics in the presence of TNFalpha. Cell Death Dis. 2013;4:e615. doi: 10.1038/cddis.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Won KY, Lim SJ, Kim GY, et al. Regulatory role of p53 in cancer metabolism via SCO2 and TIGAR in human breast cancer. Hum Pathol. 2012;43:221–228. doi: 10.1016/j.humpath.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 22.Pena-Rico MA, Calvo-Vidal MN, Villalonga-Planells R, et al. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother Oncol. 2011;101:132–139. doi: 10.1016/j.radonc.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Walsh MJ, Brimacombe KR, Anastasiou D, et al. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information; 2010. ML265: a potent PKM2 activator induces tetramerization and reduces tumor formation and size in a mouse xenograft model. [PubMed] [Google Scholar]
- 24.Anastasiou D, Yu Y, Israelsen WJ, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belfi CA, Chatterjee S, Gosky DM, Berger SJ, Berger NA. Increased sensitivity of human colon cancer cells to DNA cross-linking agents after GRP78 up-regulation. Biochem Biophys Res Commun. 1999;257:361–368. doi: 10.1006/bbrc.1999.0472. [DOI] [PubMed] [Google Scholar]
- 26.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye J, Fan J, Venneti S, et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014;4(12):1406–1417. doi: 10.1158/2159-8290.CD-14-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips DC, Woollard KJ, Griffiths HR. The anti-inflammatory actions of methotrexate are critically dependent upon the production of reactive oxygen species. Br J Pharmacol. 2003;138:501–511. doi: 10.1038/sj.bjp.0705054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sung JY, Hong JH, Kang HS, et al. Methotrexate suppresses the interleukin-6 induced generation of reactive oxygen species in the synoviocytes of rheumatoid arthritis. Immunopharmacology. 2000;47:35–44. doi: 10.1016/s0162-3109(99)00185-x. [DOI] [PubMed] [Google Scholar]
- 30.Adjei AA. Pemetrexed (ALIMTA), a novel multitargeted antineoplastic agent. Clin Cancer Res. 2004;10:4276s–4280s. doi: 10.1158/1078-0432.CCR-040010. [DOI] [PubMed] [Google Scholar]
- 31.Baggetto LG. Deviant energetic metabolism of glycolytic cancer cells. Biochimie. 1992;74:959–974. doi: 10.1016/0300-9084(92)90016-8. [DOI] [PubMed] [Google Scholar]
- 32.Pongratz RL, Kibbey RG, Shulman GI, Cline GW. Cytosolic and mitochondrial malic enzyme isoforms differentially control insulin secretion. J Biol Chem. 2007;282:200–207. doi: 10.1074/jbc.M602954200. [DOI] [PubMed] [Google Scholar]
- 33.Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elhammali A, Ippolito JE, Collins L, Crowley J, Marasa J, Piwnica- Worms D. A high-throughput fluorimetric assay for 2-hydroxyglutarate identifies zaprinast as a glutaminase inhibitor. Cancer Discov. 2014;4:828–839. doi: 10.1158/2159-8290.CD-13-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shukla K, Ferraris DV, Thomas AG, et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012;55:10551–10563. doi: 10.1021/jm301191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13:890–901. doi: 10.1158/1535-7163.MCT-13-0870. [DOI] [PubMed] [Google Scholar]
- 39.Ren JG, Seth P, Clish CB, et al. Knockdown of malic enzyme 2 suppresses lung tumor growth, induces differentiation and impacts PI3K/AKT signaling. Sci Rep. 2014;4:5414. doi: 10.1038/srep05414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. 2013;493:689–693. doi: 10.1038/nature11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nekrutenko A, Hillis DM, Patton JC, Bradley RD, Baker RJ. Cytosolic isocitrate dehydrogenase in humans, mice, and voles and phylogenetic analysis of the enzyme family. Mol Biol Evol. 1998;15:1674–1684. doi: 10.1093/oxfordjournals.molbev.a025894. [DOI] [PubMed] [Google Scholar]
- 42.Leonardi R, Subramanian C, Jackowski S, Rock CO. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J Biol Chem. 2012;287:14615–14620. doi: 10.1074/jbc.C112.353946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mailloux RJ, Singh R, Brewer G, Auger C, Lemire J, Appanna VD. Alpha-ketoglutarate dehydrogenase and glutamate dehydrogenase work in tandem to modulate the antioxidant alpha-ketoglutarate during oxidative stress in Pseudomonas fluorescens. J Bacteriol. 2009;191:3804–3810. doi: 10.1128/JB.00046-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fedotcheva NI, Sokolov AP, Kondrashova MN. Nonezymatic formation of succinate in mitochondria under oxidative stress. Free Radic Biol Med. 2006;41:56–64. doi: 10.1016/j.freeradbiomed.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 45.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ichimura K, Pearson DM, Kocialkowski S, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009;11:341–347. doi: 10.1215/15228517-2009-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116:597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 48.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bleeker FE, Atai NA, Lamba S, et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010;119:487–494. doi: 10.1007/s00401-010-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atai NA, Renkema-Mills NA, Bosman J, et al. Differential activity of NADPH-producing dehydrogenases renders rodents unsuitable models to study IDH1R132 mutation effects in human glioblastoma. J Histochem Cytochem. 2011;59:489–503. doi: 10.1369/0022155411400606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li S, Chou AP, Chen W, et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol. 2013;15:57–68. doi: 10.1093/neuonc/nos261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garten A, Petzold S, Korner A, Imai S, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009;20:130–138. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 56.Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, Chen WY. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene. 2011;30:907–921. doi: 10.1038/onc.2010.468. [DOI] [PubMed] [Google Scholar]
- 57.Srivastava M, Khurana P, Sugadev R. Lung cancer signature biomarkers: tissue specific semantic similarity based clustering of digital differential display (DDD) data. BMC Res Notes. 2012;5:617. doi: 10.1186/1756-0500-5-617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hufton SE, Moerkerk PT, Brandwijk R, de Bruine AP, Arends JW, Hoogenboom HR. A profile of differentially expressed genes in primary colorectal cancer using suppression subtractive hybridization. FEBS Lett. 1999;463:77–82. doi: 10.1016/s0014-5793(99)01578-1. [DOI] [PubMed] [Google Scholar]
- 59.Bi TQ, Che XM, Liao XH, et al. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol Rep. 2011;26:1251–1257. doi: 10.3892/or.2011.1378. [DOI] [PubMed] [Google Scholar]
- 60.Chini CC, Guerrico AM, Nin V, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res. 2014;20:120–130. doi: 10.1158/1078-0432.CCR-13-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cerna D, Li H, Flaherty S, Takebe N, Coleman CN, Yoo SS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribo-syltransferase1 (NAPRT1)-dependent manner. J Biol Chem. 2012;287:22408–22417. doi: 10.1074/jbc.M112.357301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kato H, Ito E, Shi W, et al. Efficacy of combining GMX1777 with radiation therapy for human head and neck carcinoma. Clin Cancer Res. 2010;16:898–911. doi: 10.1158/1078-0432.CCR-09-1945. [DOI] [PubMed] [Google Scholar]
- 63.Okumura S, Sasaki T, Minami Y, Ohsaki Y. Nicotinamide phosphoribo-syltransferase: a potent therapeutic target in non-small cell lung cancer with epidermal growth factor receptor-gene mutation. J Thorac Oncol. 2012;7:49–56. doi: 10.1097/JTO.0b013e318233d686. [DOI] [PubMed] [Google Scholar]
- 64.von Heideman A, Berglund A, Larsson R, Nygren P. Safety and efficacy of NAD depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother Pharmacol. 2010;65:1165–1172. doi: 10.1007/s00280-009-1125-3. [DOI] [PubMed] [Google Scholar]
- 65.Travelli C, Drago V, Maldi E, et al. Reciprocal potentiation of the antitumoral activities of FK866, an inhibitor of nicotinamide phospho-ribosyltransferase, and etoposide or cisplatin in neuroblastoma cells. J Pharmacol Exp Ther. 2011;338:829–840. doi: 10.1124/jpet.111.184630. [DOI] [PubMed] [Google Scholar]
- 66.Moore Z, Chakrabarti G, Luo X, et al. NAMPT inhibition sensitizes pancreatic adenocarcinoma cells to tumor-selective, PAR-independent metabolic catastrophe and cell death induced by beta-lapachone. Cell Death Dis. 2015;6:e1599. doi: 10.1038/cddis.2014.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcript-tomic data in non-small-cell lung cancer. PLoS One. 2013;8:e82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roberts PJ, Stinchcombe TE. KRAS mutation: should we test for it, and does it matter? J Clin Oncol. 2013;31:1112–1121. doi: 10.1200/JCO.2012.43.0454. [DOI] [PubMed] [Google Scholar]
- 70.Fisher GH, Wellen SL, Klimstra D, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cao L, Li LS, Spruell C, et al. Tumor-selective, futile redox cycle-induced bystander effects elicited by NQO1 bioactivatable radiosensitizing drugs in triple-negative breast cancers. Antioxid Redox Signal. 2014;21:237–250. doi: 10.1089/ars.2013.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Awadallah NS, Dehn D, Shah RJ, et al. NQO1 expression in pancreatic cancer and its potential use as a biomarker. Appl Immunohistochem Mol Morphol. 2008;16:24–31. doi: 10.1097/PAI.0b013e31802e91d0. [DOI] [PubMed] [Google Scholar]
- 73.Bey EA, Reinicke KE, Srougi MC, et al. Catalase abrogates beta-lapachone-induced PARP1 hyperactivation-directed programmed necrosis in NQO1-positive breast cancers. Mol Cancer Ther. 2013;12:2110–2120. doi: 10.1158/1535-7163.MCT-12-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pink JJ, Wuerzberger-Davis S, Tagliarino C, et al. Activation of a cysteine protease in MCF-7 and T47D breast cancer cells during beta-lapachone-mediated apoptosis. Exp Cell Res. 2000;255:144–155. doi: 10.1006/excr.1999.4790. [DOI] [PubMed] [Google Scholar]
- 75.Tagliarino C, Pink JJ, Dubyak GR, Nieminen AL, Boothman DA. Calcium is a key signaling molecule in beta-lapachone-mediated cell death. J Biol Chem. 2001;276:19150–19159. doi: 10.1074/jbc.M100730200. [DOI] [PubMed] [Google Scholar]
- 76.Bentle MS, Reinicke KE, Dong Y, Bey EA, Boothman DA. Nonhomologous end joining is essential for cellular resistance to the novel antitumor agent, beta-lapachone. Cancer Res. 2007;67:6936–6945. doi: 10.1158/0008-5472.CAN-07-0935. [DOI] [PubMed] [Google Scholar]
- 77.Boothman DA, Greer S, Pardee AB. Potentiation of halogenated pyrimidine radiosensitizers in human carcinoma cells by beta-lapachone (3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyran- 5,6-dione), a novel DNA repair inhibitor. Cancer Res. 1987;47:5361–5366. [PubMed] [Google Scholar]
- 78.Boothman DA, Trask DK, Pardee AB. Inhibition of potentially lethal DNA damage repair in human tumor cells by beta-lapachone, an activator of topoisomerase I. Cancer Res. 1989;49:605–612. [PubMed] [Google Scholar]
- 79.Li LS, Bey EA, Dong Y, et al. Modulating endogenous NQO1 levels identifies key regulatory mechanisms of action of beta-lapachone for pancreatic cancer therapy. Clin Cancer Res. 2011;17:275–285. doi: 10.1158/1078-0432.CCR-10-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hartner LP, LR MH, Mendelson D, et al. Phase 2 dose multi-center, open-label study of ARQ 501, a checkpoint activator, in adult patients with persistent, recurrent or metastatic leiomyosarcoma (LMS) J Clin Oncol. 2007;25:20521. [Google Scholar]
- 81.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Landi MT, Dracheva T, Rotunno M, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3:e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hou J, Aerts J, den Hamer B, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Talbot SG, Estilo C, Maghami E, et al. Gene expression profiling allows distinction between primary and metastatic squamous cell carcinomas in the lung. Cancer Res. 2005;65:3063–3071. doi: 10.1158/0008-5472.CAN-04-1985. [DOI] [PubMed] [Google Scholar]